

ABSTRACT
We previously reported that MORC3, a protein associated with promyelocytic leukemia nuclear bodies (PML NBs), is a target of herpes simplex virus 1 (HSV-1) ICP0-mediated degradation (E. Sloan, et al., PLoS Pathog 11:e1005059, 2015, http://dx.doi.org/10.1371/journal.ppat.1005059). Since it is well known that certain other components of the PML NB complex play an important role during an intrinsic immune response to HSV-1 and are also degraded or inactivated by ICP0, here we further investigate the role of MORC3 during HSV-1 infection. We demonstrate that MORC3 has antiviral activity during HSV-1 infection and that this antiviral role is counteracted by ICP0. In addition, MORC3's antiviral role extends to wild-type (wt) human cytomegalovirus (HCMV) infection, as its plaque-forming efficiency increased in MORC3-depleted cells. We found that MORC3 is recruited to sites associated with HSV-1 genomes after their entry into the nucleus of an infected cell, and in wt infections this is followed by its association with ICP0 foci prior to its degradation. The RING finger domain of ICP0 was required for degradation of MORC3, and we confirmed that no other HSV-1 protein is required for the loss of MORC3. We also found that MORC3 is required for fully efficient recruitment of PML, Sp100, hDaxx, and γH2AX to sites associated with HSV-1 genomes entering the host cell nucleus. This study further unravels the intricate ways in which HSV-1 has evolved to counteract the host immune response and reveals a novel function for MORC3 during the host intrinsic immune response.
IMPORTANCE Herpesviruses have devised ways to manipulate the host intrinsic immune response to promote their own survival and persistence within the human population. One way in which this is achieved is through degradation or functional inactivation of PML NB proteins, which are recruited to viral genomes in order to repress viral transcription. Because MORC3 associates with PML NBs in uninfected cells and is a target for HSV-1-mediated degradation, we investigated the role of MORC3 during HSV-1 infection. We found that MORC3 is also recruited to viral HSV-1 genomes, and importantly it contributes to the fully efficient recruitment of PML, hDaxx, Sp100, and γH2AX to these sites. Depletion of MORC3 resulted in an increase in ICP0-null HSV-1 and wt HCMV replication and plaque formation; therefore, this study reveals that MORC3 is an antiviral factor which plays an important role during HSV-1 and HCMV infection.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is prevalent in populations throughout the world and is responsible for a number of clinically important diseases that range from facial and genital lesions to encephalitis (1, 2). This alphaherpesvirus establishes lifelong persistence within the host, remaining latent within sensory ganglia after the primary infection is resolved. Periodically the virus is reactivated from its latent state, resulting in recurrent lesions. HSV-1 has the capacity to remain persistent within the host and allow transmission within the population due to a variety of immune evasion strategies which it encodes.
Upon initial infection there is activation of an intrinsic immune response involving constitutively expressed proteins, such as the promyelocytic leukemia (PML) protein and other components of the PML nuclear body (PML NB) complex (e.g., Sp100 and hDaxx), which restrict viral gene replication (3–6). Wild-type (wt) HSV-1 overcomes this aspect of restriction though expression of the viral ubiquitin E3 ligase protein, ICP0, that preferentially targets specific SUMO (small ubiquitin-like modifier)-modified proteins for proteasome-mediated degradation. These include PML and certain other components of the PML NB complex (7, 8). In addition to these PML NB-associated proteins, HSV-1 infection results in an extensive reduction of high-molecular-weight SUMO-conjugated proteins at late times of infection. We recently used stable isotope labeling with amino acids in cell culture (SILAC) proteomics and mass spectrometry to identify a number of these SUMO2-modified proteins whose abundance is altered during HSV-1 infection (9). MORC3 (microrchidia family CW-type zinc finger 3, also known as NXP-2) was one such sumoylated protein which we discovered was decrease in abundance by 5.6-fold during HSV-1 infection. We went on to confirm that MORC3 was indeed sumoylated and that both sumoylated and unmodified forms were degraded during HSV-1 infection in an ICP0-dependent manner (9).
MORC3 is a nuclear matrix protein whose functional domains are highly conserved between prokaryotes and eukaryotes (10, 11); however, the function of MORC3 has not been studied in great detail. Interestingly, previous reports showed that MORC3 can associate with PML NBs (12). The localization of MORC3 to PML NBs is dependent on a SUMO-SIM (SUMO interaction motif) interaction with PML isoform I (PML.I) (13). In addition, Takahashi and colleagues found that Sp100 and p53 were recruited to PML NBs in a MORC3-dependent manner, providing some insight into the function of MORC3 within these complexes. MORC3 was also found to form nuclear body complexes in a PML-independent manner after transient overexpression, with the function of these structures being unknown (13).
In humans there are five members of the MORC family, MORC1-4 and the divergent SMCHD1 protein (structural maintenance of chromosome flexible hinge domain containing 1). There are three conserved domains within MORC3: the GHL (gyrase B, Hsp90, and MutL) ATPase domain (14), a CW-type zinc finger domain (15), and a coiled-coil dimerization domain (16, 17). The GHL-ATPase domain is thought to be involved in gene silencing and regulation of chromatin structure in response to DNA damage signals (18–20) and is required for localization of MORC3 to PML NBs and the recruitment of Sp100 and p53 to these structures (12). The CW-type zinc finger domain contains a histone H3 binding motif which binds predominantly methylated lysine 4 of histone H3 (21–23). The function of the coiled-coil domain within MORC proteins is unknown, although this domain in other proteins has been suggested to regulate protein-protein and protein-DNA interactions, protein localization, gene transcription, the DNA damage response, and signal transduction (24–43).
Therefore, it has been suggested that MORC3 is an epigenetic regulator that may play roles within a wide range of biological functions, such as transcription regulation, chromatin condensation and remodeling, and DNA break repair (44). Members of the MORC family of proteins have been associated with a number of types of cancers (45–49). However, to date there is little known about the role that MORC3 may play during a virus infection, although one recent study reported that MORC3 is required for efficient influenza replication (50). Until now, MORC3 has not been previously reported to have an antiviral role. Following our discovery that sumoylated MORC3 is targeted by ICP0 (9), we examined here the function of MORC3 during infection with HSV-1 and found that it is efficiently recruited to incoming HSV-1 genomes at early times postinfection (p.i.). We also discovered that during ICP0-null mutant infection of MORC3-depleted cells, the recruitment of Sp100, hDaxx, PML, and γH2AX to incoming viral genomes was less efficient. In addition, we observed that MORC3 colocalized with ICP0 early during infection, prior to its degradation in a RING finger-dependent manner. Importantly, MORC3 was found to have antiviral activity, which is counteracted in the presence of ICP0, and that MORC3 restricts wt human cytomegalovirus (HCMV) plaque formation efficiency, suggesting that MORC3 has an antiviral role that extends beyond HSV-1.
MATERIALS AND METHODS
Viruses and cells.
HSV-1 wild-type (wt) strain 17+ was used, from which the ICP0 null mutant dl1403 was derived (51). Wild-type HSV-1, in1863, and the derivative dl1403/CMVlacZ, containing the lacZ gene under the control of the HCMV promoter/enhancer inserted into the tk gene, were gifts from Chris Preston. HSV-1 dl0Y4 expresses enhanced yellow fluorescent protein (EYFP)-linked ICP4 and was derived from dl1403 (52). Viruses were propagated in baby hamster kidney (BHK) cells grown in Glasgow modified Eagles' medium (Gibco Life Technologies) supplemented with 10% newborn calf serum (Gibco Life Technologies) and 10% tryptose phosphate broth (Gibco Life Technologies). Virus titers were determined by titration on U2OS cells in the presence of 1% human serum (MP Biomedicals). Viral plaques were visualized using Giemsa stain (VWR). Human foreskin diploid human fibroblasts (HFs; a gift from Thomas Stamminger), telomerase-immortalized HFs (HFTs; a gift from Chris Boutell), HEK-293T cells, and U2OS cells were all grown in Dulbecco's modified Eagles' medium (Gibco Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Gibco Life Technologies), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Gibco Life Technologies). HepaRG cells (53) were grown in William's medium E (Gibco Life Technologies) supplemented with 10% FBS, 2 mM glutamine (Gibco Life Technologies), 5 μg/ml insulin (Sigma-Aldrich), and 500 nM hydrocortisone (Sigma-Aldrich). HepaRG cells that can be induced to express ICP0 (termed HA-cICP0) or ICP0 mutants (HA-FXE, HA-mSLS4, HA-mSLS457, and HA-E52X) in the presence of doxycycline (0.1 μg/ml) (Clontech) were described previously (7, 54). Lentivirus-transduced cells were maintained with the appropriate antibiotic selection.
Plasmids and lentiviral vectors.
Lentivirus vectors expressing anti-MORC3 short hairpin RNA (shRNA) (shMORC3) were obtained from Sigma-Aldrich (shMORC3-1335, CCGGGCTTAATACGTGTCGGTCATACTCGAGTATGACCGACACGTATTAAGCTTTTT; shMORC3-1337, CCGGGCCAATTACAAGAACTGAGAACTCGAGTTCTCAGTTCTTGTAATTGGCTTTTT; shMORC3-1339, CCGGGTGAGGTTGAATTGCTGGAAACTCGAGTTTCCAGCAATTCAACCTCACTTTTT). Lentivirus transduction of cells was as described previously (55). Briefly, pLKO plasmids expressing the gene of interest were cotransfected along with pVSV-G and pCMV.DR.8.91 (a gift from Didier Trono) into HEK-293T cells. Lentivirus supernatants were collected and HFT and HepaRG cells transduced. Selection during routine culture used puromycin at 500 ng/ml, which was omitted from cells seeded for and during experimentation. HA-cICP0 cells and their derivatives were maintained in medium containing G418 (500 μg/ml) and puromycin.
Virus plaque assays.
The relative plaque-forming efficiencies of wt and ICP0 null mutant HSV-1 were assessed as described previously (56), with HFT-shMORC3 cells seeded for plaque assays into 24-well dishes at 1 × 105 cells per well and infected the following day with appropriate sequential 3-fold dilutions of dl1403/CMVlacZ or wt in1863. After virus adsorption, the cells were overlaid with medium containing 1% human serum. The cells were stained for β-galactosidase-positive plaques 24 h later. Relative plaque formation efficiencies are expressed as fold changes in plaque numbers at a given dilution compared to the control. This approach gives a more robust and reliable comparison than apparent titers averaged over a range of dilutions because the plaque-forming efficiency of ICP0 null mutant HSV-1 varies in a nonlinear manner with respect to dilution. For assay of HCMV plaque formation, HFT-shMORC3 and control cells were seeded into 24-well dishes and infected with HCMV at appropriate multiplicities the following day. At 3 h after virus adsorption, the virus inoculum was removed and replaced with fresh medium. Plaques were stained at 10 days after infection by immunological detection of UL44 (ab6502; Abcam). The cells were fixed with formaldehyde and treated with NP-40 as for immunofluorescence staining and then washed twice with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST). The cells were treated with PBST containing 5% dried milk for 30 min and then incubated for 2 h at room temperature with anti-UL44 monoclonal antibody (MAb). The cells were then washed three times with PBST before being incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody for 1 h. The cells were washed with PBST three times and incubated with 0.2 ml True Blue solution (50-7802; Insight Biotechnology) for 10 min.
Western blot analysis.
Cells were seeded into 24-well dishes at 1 × 105 cells per well. After the relevant experimental manipulations, the cells were washed twice with PBS before harvesting in SDS-PAGE loading buffer. Proteins were resolved on 7.5% SDS-PAGE and then transferred to nitrocellulose membranes by Western blotting. The following primary antibodies were used: anti-actin MAb (AC-40) (Sigma-Aldrich) (1:10,000), anti-PML MAb (5E10) (57) (1:100), anti-tubulin MAb (T4026) (Sigma-Aldrich) (1:5,000), anti-ICP0 MAb (11060) (58) (1:1,000), anti-ICP4 MAb (58S) (59) (1:1,000), anti-UL42 MAb (Z1F11) (60) (1:1,000), anti-Sp100 rabbit antibody (RAb; SpGH) (61) (1:2,000), anti-MORC3 RAb (NBP1-83036) (Novus Biologicals) (1:300), and anti-RanGAP1 MAb (33-0800) (Invitrogen) (1:1,000). Secondary antibodies included anti-mouse-HRP (A4416) (Sigma-Aldrich) (1:1,500) and anti-rabbit-HRP (A4914) (Sigma-Aldrich) (1:20,000).
Immunofluorescence and confocal microscopy.
Cells were seeded onto 13-mm glass coverslips in 24-well plates at 1 × 105 cells per well, fixed, and prepared for immunofluorescence as described previously (62). Antibodies used were anti-PML MAb (5E10) (57) (1:20), anti-Sp100 rat serum (Sp26) (63) (1:2,000), anti-MORC3 RAb (NBP1-83036) (Novus Biologicals) (1:400), anti-ICP0 MAb (11060) (58) (1:1,000), anti-hDaxx MAb (MCA2143) (AbD Serotech) (1:1,000), and anti-phospho-H2AX (Ser139) RAb (clone JBW301; Upstate) (1:1,000), and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). The secondary antibodies used were Alexa 555-conjugated goat anti-mouse IgG (Life Technologies) (1:5,000), Alexa 633-conjugated goat anti-rabbit IgG (Life Technologies) (1:1,000), Alexa 488-conjugated goat anti-mouse IgG (Life Technologies) (1:1,000), Alexa 488-conjugated goat anti-rat IgG (Life Technologies) (1:1,000), and Alexa 555-conjugated goat anti-rat IgG (Life Technologies) (1:5,000). Immunofluorescence assays were done in replicate, and complete coverslips were examined in detail using a Zeiss LSM 710 confocal microscope with 488-nm, 561-nm, and 633-nm laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap.
RESULTS
MORC3 colocalizes with ICP0 prior to its degradation.
MORC3 can be detected within PML NBs and in one reported instance was found to be required for recruitment of Sp100 into these nuclear complexes (12, 13). Our study also observed MORC3 in association with PML NBs, although following examination of a large number of cells we noted that not every PML NB complex had detectable MORC3 (Fig. 1; see also Fig. 4 and 5). This difference may be due to differences in cell types analyzed and methods used or a limit of antibody detection efficiency. At the early stages of HSV-1 infection, ICP0 colocalizes with PML NB protein components such as PML and Sp100 prior to their degradation (8, 64–67). Since we have previously shown that both sumoylated and unmodified MORC3 is degraded during wt HSV-1 infection (9), we investigated whether MORC3 also colocalizes with ICP0 prior to its degradation. Human diploid fibroblasts (HFs) were infected for 1 h with wt HSV-1 at a multiplicity of infection (MOI) of 2 PFU per cell. The localizations of ICP0 and MORC3 were then visualized using immunofluorescence staining and confocal microscopy, with mock-infected cells included as a control (Fig. 1). Complete coverslips were examined, of which 21 and 38 cells were imaged at 1 and 2 h postinfection (p.i.), respectively. We found that by 1 h after wt HSV-1 infection, MORC3 colocalizes with ICP0 (cell indicated with an arrow) prior to the loss of the MORC3 signal in cells where ICP0 expression is more intense. By 2 h p.i. all cells expressing ICP0 contained no detectable MORC3 protein, with MORC3 only found in uninfected cells within this population. Some colocalization of MORC3 with an ICP0 RING-finger mutant was also observed (data not shown), suggesting this association is independent of the RING finger domain of ICP0. Therefore, MORC3 colocalizes with ICP0 very early during infection prior to its degradation.
FIG 1.
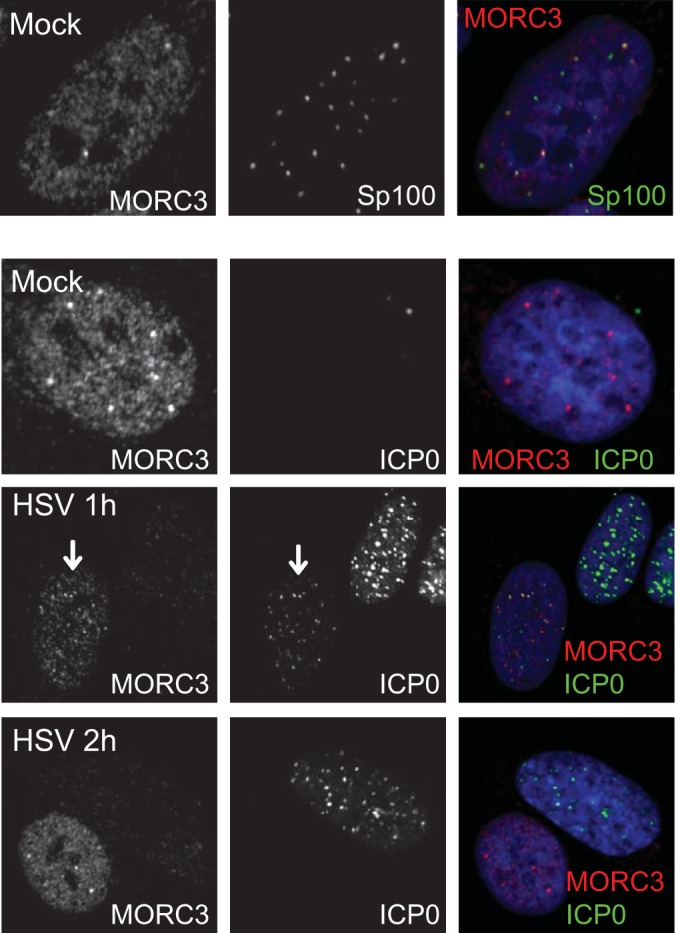
MORC3 associates with Sp100 in PML NBs and ICP0 early during wt HSV-1 infection and is subsequently degraded. HFs were either mock infected or infected with wt HSV-1 (MOI of 2), fixed, and permeabilized at 1 or 2 h postinfection. Cells were analyzed for association of MORC3 (Novus Biologicals) (red) with Sp100 (top row, showing concentrations of MORC3 associated with Sp100 in a variable manner; see also Fig. 4) and with ICP0 (11060) (green) in infected cells using confocal microscopy. The blue signal in the merged channel is DAPI. The arrow indicates a cell with ICP0 in association with MORC3. The MORC3 signal diminishes as infection progresses.
FIG 4.
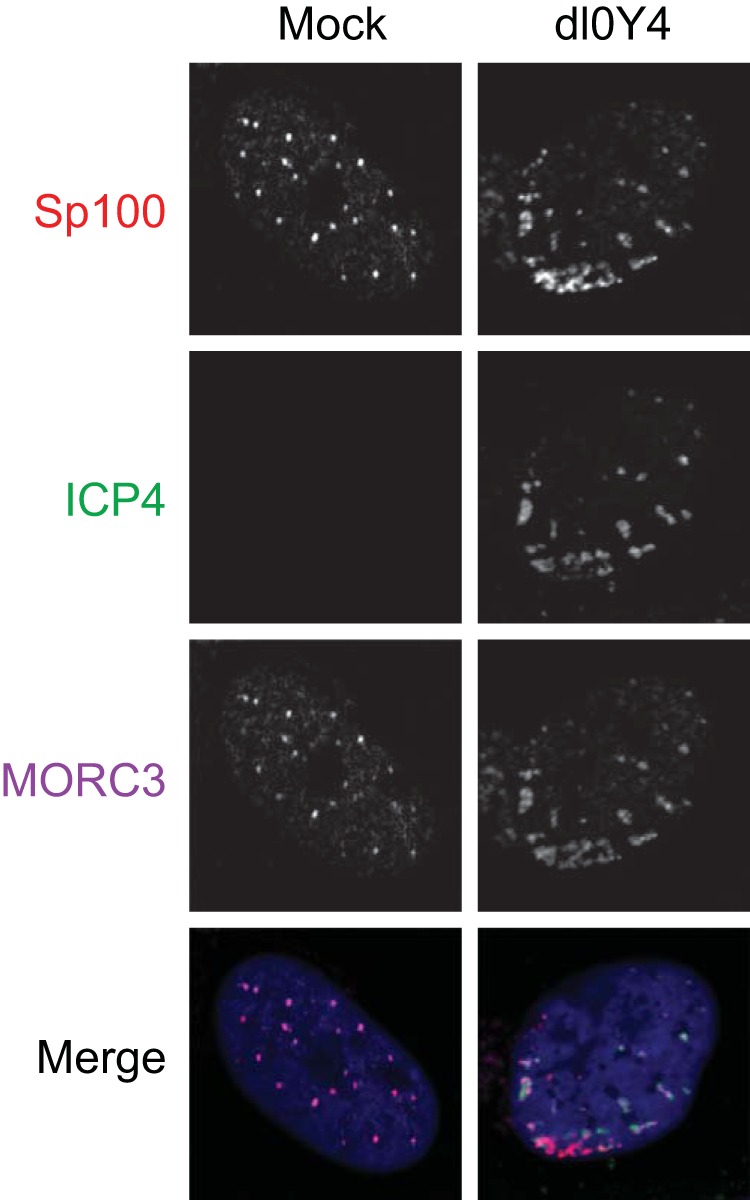
MORC3 is recruited to HSV-1 genomes during the initial stage of infection. HFs were infected at low MOI with dl0Y4 for 24 h, fixed, permeabilized, and probed with anti-MORC3 antibody (Novus Biologicals) (purple). Anti-Sp100 (Sp26) (red) was included as a positive control for recruitment to viral genomes, with nuclei stained with DAPI (blue). Recruitment to ICP4 was visualized by confocal microscopy (right column) and compared to that of a typical uninfected cell (left column).
FIG 5.

Characterization of MORC3-depleted cells. (A) Generation of MORC3-depleted HFT (HFT-shMORC3-1335 and -1339 cells) and HepaRG cells (HA-shMORC3-1335, -1337, and -1339 cells) using shRNAs expressed from lentiviral vectors. Total protein lysates were analyzed by Western blotting to determine the level of MORC3 depletion with HFT-shNeg and HepaRG cells included as controls. Tubulin was included as a loading control. (B and C) Characterization of MORC3-depleted HFT cells (B) and HepaRG cells (C). Mock HFT- and HA-shMORC3-1339 and control cells were immunostained for PML (red) and MORC3 (green) (left) as well as Sp100 (green) and PML (red) (right) to confirm MORC3 depletion and assess effects on PML NBs. DAPI staining of the nuclei is shown in blue in the merged panels.
MORC3 is degraded in an ICP0-dependent manner during HSV-1 infection.
Our previous study found that ICP0 expressed on its own is sufficient to cause a reduction in MORC3 protein abundance (9). However, it remains possible that other HSV-1 proteins display similar activity. Therefore, to confirm that no other HSV-1 proteins are capable of causing the degradation of MORC3, we infected telomerase-immortalized HFs with ICP0-null mutant HSV-1 (MOI of 20) over a time course infection, with wt HSV-1 (MOI of 2) included as a control. The different multiplicities of wt and mutant were used to enable similar rates of infection progression. Western blot analysis indeed confirmed that wt but not ICP0-null mutant HSV-1 infection results in a loss of MORC3 (Fig. 2). Therefore, we have shown through expression of ICP0 alone (9) and infection with an ICP0-null HSV-1 that ICP0 is the only HSV-1 protein required for the observed reduction in MORC3.
FIG 2.
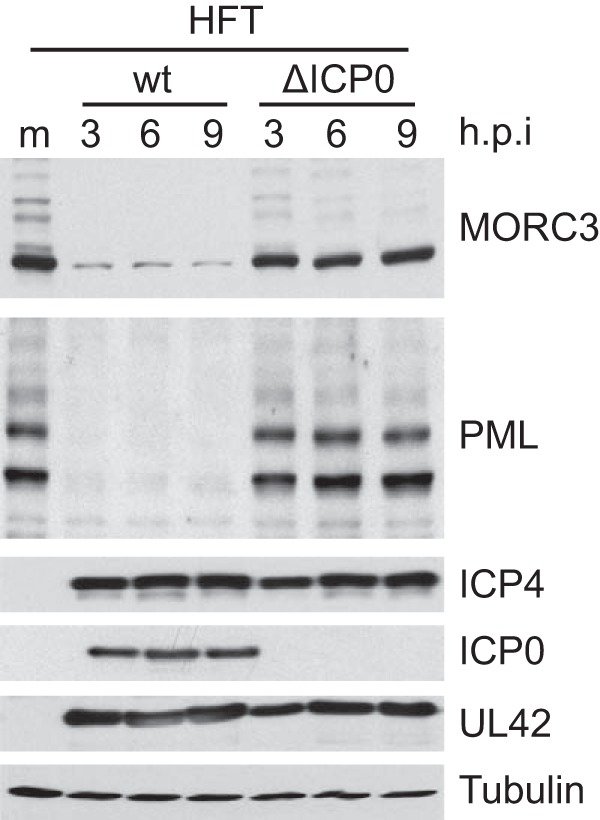
MORC3 is degraded in an ICP0-dependent manner. HFT cells were mock infected (m) or infected at an MOI of 20 with an ICP0 null HSV-1 (ΔICP0) and lysed at 3, 6, and 9 h p.i. for Western blot analysis. Analysis of PML abundance and wt HSV-1-infected (MOI of 2) lysates prepared in parallel were included as positive controls for degradation. Membranes were probed with anti-MORC3 (Novus Biologicals) and anti-PML (5E10) antibodies, with anti-tubulin (Sigma-Aldrich) used as a loading control. Viral proteins ICP4 and UL42 were included to show equivalent infection between wt and ΔICP0 HSV-1.
The RING finger domain of ICP0 is required for MORC3 degradation.
The region of ICP0 required for the degradation of MORC3 was assessed by utilizing cells that can be induced to express wt or mutant forms of ICP0 (54, 68). In this study, we utilized mutants of ICP0 that lack the RING finger domain, one or more of the motifs resembling SIMs, termed SIM-like sequences (SLS1-7), and a C-terminal deletion (Fig. 3A). Each region has individually been shown to be involved in the efficiency of degradation of selected proteins. The RING finger domain functions as a ubiquitin ligase which is required for degradation of many proteins, including unsumoylated PML.I, while the SLS motifs are likely to influence target specificity of the RING finger activity by binding to SUMO moieties which are conjugated to sumoylated proteins. We have found that the SLS4 motif is involved in targeting specific sumoylated proteins, including NACC1, ZBTB10, ZBTB4, ETV6, and specific PML isoforms (7, 9, 69 and data not shown). However, the SLS4 motif was not required for degradation of other sumoylated proteins, such as MBD1, BEND3, ZBTB12, NACC2, or ARID3a (9 and data not shown), demonstrating there is specificity in the SLS4-targeted group of proteins. A combination of mutations within these SLS motifs (mSLS457) diminishes ICP0 function and reduces the rate at which it causes the extensive reduction in high-molecular-weight SUMO conjugates (7). There are also motifs within the C-terminal region of ICP0 that have been implicated in binding to other proteins (70–72). To identify the regions of ICP0 that are involved in the degradation of MORC3, cells were induced to express either full-length ICP0, ICP0 RING finger deletion (FXE), ICP0 C-terminal deletion (E52X), ICP0 with a mutation in the SLS4 motif (mSLS4), or a combination of mutations in SLS4, SLS5, and SLS7 (mSLS457). Western blot analysis of these cell lysates prepared with or without ICP0 induction included PML as a control (Fig. 3B). As expected from previous results, expression of wt ICP0 and mutants E52X and mSLS4 caused substantial degradation of PML (7). The reduction in the PML band intensity in the RING finger mutant FXE sample in this particular gel was not reproducible and was not observed in a number of previous published reports (for example, see reference 54); therefore, we regard this as spurious. Degradation of sumoylated PML isoforms is evident in the cells expressing ICP0 mutant SLS457, which is consistent with previous observations (7). The role of the SLS motifs in degrading endogenous PML is complicated, because they are not absolutely required for degrading PML.I or its sumoylated forms (although their degradation by mSLS457 is slightly less efficient than that by wt), while SLS4 is required for degradation of all forms of PML.II when expressed in isolation (68, 73). Therefore, a major component of the PML band remaining in the right-most lane of Fig. 3 is likely to be unsumoylated PML.II. MORC3 was found to be degraded by all mutants analyzed except FXE, indicating the RING finger domain of ICP0, and not the C-terminal region or the SLS motifs, is required for this effect. In this respect the degradation of MORC3 resembles that of PML.I, which is RING finger dependent but SLS motif independent (73). In the latter instance, the degradation of PML.I could be attributed to a direct interaction between ICP0 and PML.I (73). Whether this is the case with MORC3 remains to be determined, although this would not be straightforward to investigate because of the rapidity of MORC3 degradation in the presence of ICP0.
FIG 3.
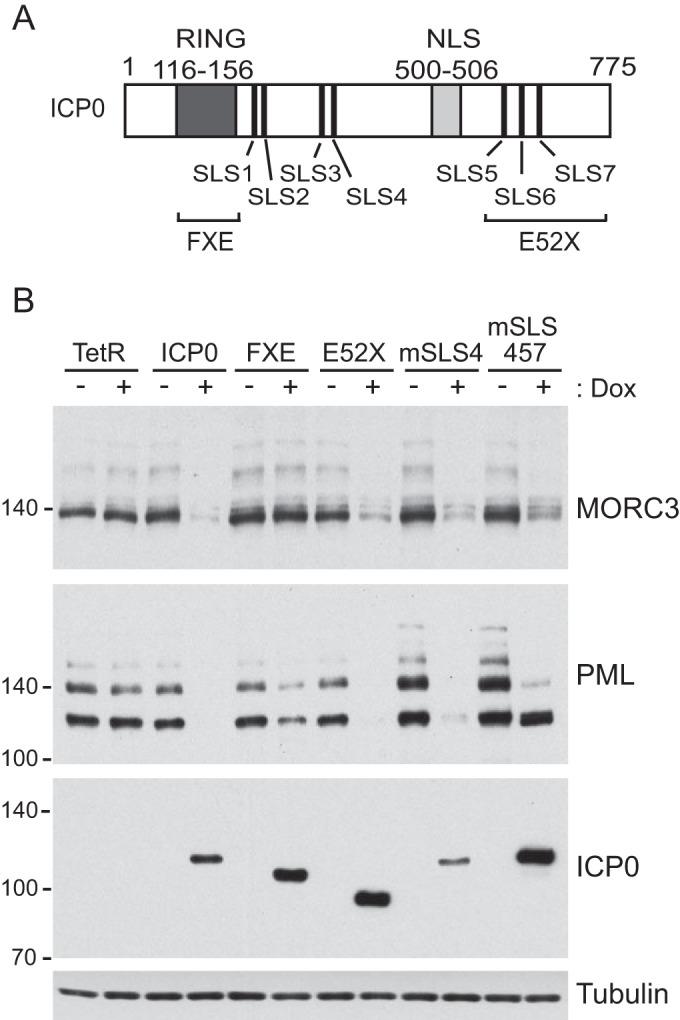
RING finger domain of ICP0 is required for MORC3 degradation. (A) Schematic diagram of ICP0 displaying internal domains and SIM-like sequences (SLS) as well as regions of mutations/deletions of the RING finger (FXE) and C-terminal (E52X) domains. NLS, nuclear localization signal. (B) HA-TetR (control) and HA-cICP0, -cICP0 FXE (Δ149-160), -cICP0 E52X (Δ594-775), -cICP0 mSLS4, and -cICP0 mSLS457 cells with the ability to express wt and mutant forms of ICP0 were treated with doxycycline (Dox) (+) or left untreated (−) and then analyzed by Western blotting. Membranes were probed with anti-MORC3 (Novus Biologicals), anti-ICP0 (11060), and anti-PML (5E10) antibodies, with anti-tubulin (Sigma-Aldrich) included as a loading control.
MORC3 is recruited to HSV-1 genomes.
Sp100, PML, and other PML NB protein components are recruited to HSV-1 genomes entering the nucleus of an infected cell in a process that contributes to the repression of viral gene transcription (54, 55, 62, 74–76). This repression is counteracted by ICP0-mediated degradation of selected PML NB protein components (reviewed in reference 3). The assay for the recruitment of PML NB proteins to HSV-1 genomes involves immunofluorescence staining of viral plaques on an infected monolayer of cells with an ICP4 antibody, which acts as a marker for HSV-1 genomes through its efficient association with the viral DNA. Cells at the edges of developing plaques receive a high number of viral genomes, visible as an arc-like pattern just inside the nucleus. This asymmetric pattern allows unambiguous visualization of recruitment of cellular proteins to sites associated with the viral genome foci. This recruitment is much more readily seen in ICP0 null mutant HSV-1 infections, as ICP0 very rapidly counteracts the recruitment process (62). Since MORC3 was reported to colocalize with PML NB proteins PML and Sp100 (12, 13) and has also been suggested as a DNA binding protein and transcriptional repressor (77), we set out to determine if MORC3 is also recruited to HSV-1 genomes. Due to the rapid degradation of MORC3 following infection, we utilized an ICP0-null mutant HSV-1 expressing EYFP-tagged ICP4 (dl0Y4) in order to visualize recruitment more clearly. We infected HFs with dl0Y4 for 24 h at a low MOI and imaged 10 representative cells at the periphery of developing plaques which exhibited ICP4 foci around the nuclear periphery. Immunofluorescence staining for Sp100 was included as a positive control for recruitment and uninfected (mock) cells as a negative control, which confirmed previous reports of MORC3 and Sp100 colocalization (12). As expected, we found close association of Sp100 with EYFP-ICP4 foci around the internal periphery of the nucleus of all newly infected cells, indicative of Sp100 recruitment to HSV-1 genomes entering the nucleus (a typical example is shown in Fig. 4). MORC3 was also in these recruited foci within these cells, demonstrating that MORC3 is indeed recruited to HSV-1 genomes during the initial stages of infection (Fig. 4).
MORC3 influences the recruitment of PML NB components to HSV-1 genomes.
The recruitment of the PML NB complex proteins (for example, PML, Sp100, and hDaxx) to HSV-1 genomes entering the nucleus of an infected cell occurs independently of each other (55, 74, 76, 78, 79). The precise mechanism responsible for the recruitment of these factors remains unknown, although it has been established that in each of these three cases the presence of a SIM is essential (75). Since MORC3 was recruited to HSV-1 genomes, directly interacts with PML.I, and in one situation is required for Sp100 localizing to PML NBs (12, 13), we investigated whether MORC3 influences the recruitment of these other factors to HSV-1 genomes. In addition, another MORC family protein, MORC2, has been reported to induce phosphorylation of H2AX and the subsequent formation of γH2AX foci and chromatin relaxation (20), which are steps in the pathway to double-strand break repair (80). Because it has been established that γH2AX forms regions surrounding replicating HSV-1 genomes in a double-strand break repair response (81–83), we included γH2AX in our panel of proteins to investigate whether their recruitment was influenced by MORC3.
To investigate this hypothesis, we established HFT and HepaRG cell lines depleted of MORC3 using independent shRNAs, namely, shMORC3-1335 and -1339, with the extent of depletion assessed by Western blot analysis (Fig. 5A). Depletion of MORC3 in HFT-shMORC3-1339 and HA-shMORC3-1339 cells was then confirmed by confocal microscopy, costaining with PML (Fig. 5B and C, left). Since a previous study found depletion of MORC3 from HeLa cells resulted in dispersal of Sp100 (12), we also examined the effect of MORC3 depletion on Sp100 localizing with PML in our cells (Fig. 5B and C, right). Surprisingly, in the HFTs examined here, Sp100 remained within PML NBs of uninfected cells when MORC3 was depleted. The difference we observe here may be due to differences in the cell types analyzed, the level of depletion of MORC3, or the antibodies utilized. The great majority of mock-infected HFT-shMORC3 cells, of which 95 were imaged, had no detectable MORC3 expression and showed Sp100, PML, hDaxx, and γH2AX with the typical nuclear dot formation distributed throughout the nucleus (data not shown). Uninfected HA-shMORC3-1339 cells also displayed Sp100 within PML NBs when MORC3 was more efficiently depleted (Fig. 5C).
Whether MORC3 was required for recruitment of PML, Sp100, hDaxx, or γH2AX to sites associated with HSV-1 genomes was assessed by infecting HFT-shMORC3-1339 cells with dl0Y4 (as explained above). Cells were then stained for either PML (Fig. 6A), Sp100, hDaxx, or γH2AX (data not shown) as well as MORC3 to confirm MORC3 depletion in these particular individual cells. Foci of EYFP-ICP4 indicate the localization of HSV-1 genomes, and nuclei were stained with DAPI. HFT-shNeg (where shNeg indicates a control shRNA) cells were included as a control in which MORC3 could be costained with either Sp100, PML, hDaxx, or γH2AX and tested for recruitment to HSV-1 genomes.
FIG 6.

Recruitment of PML NB proteins to viral DNA is diminished in the absence of MORC3. (A) HFT-shMORC3-1339 and HFT-shNeg cells were infected with dl0Y4 (MOI of 2) for 24 h. Cells were immunostained using PML (5E10) (red) and MORC3 (Novus Biologicals) (blue), and nuclei were stained with DAPI (far right) and visualized by confocal microscopy. Infected or presumed infected cells surrounding a plaque were counted and divided into three categories, which are represented here: category 1, ICP4+/PML+ (foci of ICP4 and PML in close association at the nuclear periphery of a cell); category 2, ICP4−/PML+ (foci of PML at the nuclear periphery of a cell in the pattern typical of an infected cell, but prior to detectable expression of ICP4); and category 3, ICP4+/PML− (ICP4 foci only at the nuclear periphery of a cell). (B) Percentages of cells within each category are presented in bar graphs. Sp100, hDaxx, and γH2AX were also assessed as described for PML, with results represented as bar graphs. (C) HFT-shMORC3-1339 and HFT-shNeg cells were infected with dl0Y4 (MOI of 2) for 24 h. Cells were immunostained using Sp100 (Sp26) (red) and PML (5E10) (blue) and nuclei were stained with DAPI (far right), while ICP4 was detected by the EYFP signal.
Cells that displayed EYFP-ICP4 around the internal nuclear periphery were characterized as newly infected cells with HSV-1 genomes entering the nucleus upon infection (Fig. 6A, ICP4+/PML+). We could also identify cells very early following infection due to the rapid recruitment of PML NB proteins to HSV-1 genomes prior to detectable levels of ICP4 expression; such cells have asymmetric PML foci of a type that are never seen in uninfected cells (Fig. 6A, ICP4−/PML+). Thus, a study that was blinded from the point of view of the observer was performed in which infected or presumed infected cells surrounding a plaque were identified and divided into the following categories: (i) those containing foci of both ICP4 and Sp100, PML, γH2AX, or hDaxx in close association at the nuclear periphery of a cell (referred to as ICP4+/X+); (ii) those containing foci of Sp100, PML, γH2AX, or hDaxx at the nuclear periphery of a cell in the pattern typical of an infected cell but prior to detectable expression of ICP4 (referred to as ICP4−/X+); and (iii) those containing ICP4 foci only at the nuclear periphery of a cell without accompanying foci of the cellular protein in question (referred to as ICP4+/X−) (Fig. 6A). Our analysis showed that in the absence of MORC3 the recruitment of PML was less efficient, with 48% of 69 counted cells described as ICP4+/PML− (Fig. 6B). Only 1% of counted cells were assigned as ICP4−/PML+, with the remaining 51% of cells being ICP4+/PML+, although visually the abundance of PML foci at the nuclear periphery appeared to be of a lesser degree than the equivalent shNeg control. Of note, some of these shMORC3 cells exhibiting PML recruitment also had low levels of MORC3 remaining; therefore, the percentage of cells which are defective for PML recruitment may be higher than that described here. Interestingly, MORC3-depleted cells that had no PML recruitment to ICP4 foci also no longer contained PML as punctate PML NB structures, suggesting that these structures are less stable in the absence of MORC3 in infected cells, even if ICP0 is not expressed. The corresponding data for the control HFT-shNeg cells indicated 60% of 40 counted cells were ICP4+/PML+ and 35% were ICP4−/PML+, indicating that in the presence of MORC3, PML can be recruited very early following infection, prior to detectable ICP4 expression. Only 5% of counted cells were described as ICP4+/PML−. We also investigated the reciprocal effect and found that recruitment of MORC3 was unaffected by depletion of PML (data not shown).
Sp100 recruitment to sites of incoming HSV-1 genomes was also assessed in these MORC3-depleted cells in a similar manner (Fig. 6B and data not shown). We found that in the absence of MORC3, Sp100 is recruited more slowly to viral genomes than MORC3-expressing cells. For example, 54% of the 68 counted control HFT-shNeg cells were ICP4+/Sp100+, and 46% were ICP4−/Sp100+. Unlike with PML, there were no cells that had ICP4 around the periphery of the nucleus that did not have Sp100 recruited to the same site. However, when MORC3 was depleted, only 4% of the 104 counted cells were observed as ICP4−/Sp100+, while 3% of counted cells were ICP4+/Sp100−. The remaining 93% of counted cells were designated ICP4+/Sp100+. Thus, the rate of recruitment of Sp100 is decreased in the MORC3-depleted cells. A phenotype similar to that of Sp100 was seen with γH2AX and hDaxx (Fig. 6B and data not shown), with 47% of the 43 counted HFT-shNeg cells displaying ICP4 and γH2AX foci associated at the nuclear periphery (ICP4+/γH2AX+) and 53% with rapid recruitment of γH2AX to the nuclear periphery prior to ICP4 expression (ICP4−/γH2AX+). In contrast, MORC3-depleted cells displayed 87% of 52 counted cells as ICP4+/γH2AX+, with only the remaining 13% as ICP4−/γH2AX+. Similarly, for hDaxx, 66% of 71 counted shNeg cells were described as ICP4+/hDaxx+, 32% as ICP4−/hDaxx+, and the remaining 1% as ICP4+/hDaxx+. There was evidence of hDaxx recruitment to viral genomes (ICP4+/hDaxx+) in 82% of 71 counted MORC3-depleted cells, while only 3% of counted cells were described as ICP4−/hDaxx+ and 15% as ICP4+/hDaxx−. Taken together, these results suggest that MORC3 plays a role in the speed or efficiency of recruitment of these PML NB proteins to incoming HSV-1 genomes.
Since PML is required for the formation of PML NBs (84, 85) and was observed as diffuse throughout the nucleus in many of these HFT-shMORC3-1339 dl0Y4-infected cells (Fig. 6A and B), it was perhaps surprising to observe some Sp100 remaining as nuclear dots in unrecruited foci. To further assess this observation, mock- or dl0Y4-infected HFT-shMORC3-1339 cells were dual stained with Sp100 and PML (Fig. 6C). Our analysis confirmed that when PML is nuclear diffuse, Sp100 remains as nuclear dots in both ICP4-associated and unassociated foci. We also observed that in some HFT-shMORC3 cells, PML staining was apparently less intense than in HFT-shNeg controls, which might be explained by a greater component of diffuse PML distribution.
MORC3 has antiviral activity during HSV-1 and HCMV infection.
In previous studies, we noted a correlation between the recruitment of PML NB components and their role in the restriction of virus gene replication (54, 55, 62, 75, 76, 79, 86), with their functions in this regard counteracted by ICP0 (reviewed in reference 3). Since we discovered that MORC3 is recruited to HSV-1 genomes, we wanted to determine if MORC3 also has an antiviral role during HSV-1 infection. For these experiments, both HFT and HepaRG (HA) cells that were depleted of MORC3 using shRNAs shMORC3-1335 and -1339 were assessed (Fig. 5A). Viral plaque assays were then performed to compare the plaque formation efficiencies of wt and ICP0-null HSV-1 in these cells (Fig. 7A). Cells expressing shNeg were included to normalize plaque numbers. As expected, because of the rapid degradation of MORC3 by ICP0, wt HSV-1 exhibited no change in plaque formation efficiency within HFT-shMORC3-1335, HFT-shMORC3-1339, HA-shMORC3-1335, and HA-shMORC3-1339 cells compared to the shNeg controls (Fig. 7A). However, ICP0-null HSV-1 infection of MORC3-depleted HFT and HA cells resulted in a marked increase in plaque formation efficiency compared to the respective shNeg controls, suggesting that, like several other components of PML NBs, MORC3 has antiviral activity which is counteracted by ICP0. These increases in plaque formation efficiency are at least as marked, even in the most conservative estimation, as the corresponding increases previously seen in cells depleted individually of PML, Sp100, or hDaxx (55, 74, 76).
FIG 7.

MORC3 has antiviral activity during ICP0-null mutant HSV-1 and wt HCMV infection. (A) Data from several independent viral plaque assays of both wt- and ΔICP0 HSV-1-infected HFT-shMORC3-1335, HFT-shMORC3-1339, HA-shMORC3-1335, and HA-shMORC3-1339 cells were averaged and normalized to the respective shNeg cell controls and plotted ± standard deviations. (B) Western blot analysis comparing infection efficiency of ΔICP0 HSV-1 within HFT-shMORC3-1339 and HFT-shNeg cells over a time course infection. Cells were infected at an MOI of 2 with ΔICP0 HSV-1, lysed at 2, 4, or 6 h p.i., and probed for ICP4, ICP0, UL42, and actin loading control. Mock-infected cells (m) were included as a control. (C) HCMV viral plaque-forming efficiencies using HFT-shMORC3-1335 and HFT-shMORC3-1339 cells normalized to HFT-shNeg cells and plotted ± standard deviations.
An increase in plaque formation efficiency would result in increased infectivity of the ICP0-null HSV-1. We therefore confirmed this effect using Western blot analysis to detect viral proteins ICP4, UL42, and ICP0 (control for ICP0-null HSV-1). HFT-shMORC3-1339 cells were infected with ICP0-null HSV-1 (dl1403; labeled ΔICP0) at an MOI of 2 with samples collected over a time course infection. ICP4 and UL42 were expressed at greater levels in the shMORC3 cells than the shNeg control, confirming MORC3-depleted cells are more readily infected (Fig. 7B).
HCMV is a beta-herpesvirus which is also restricted by the same set of PML NB components as HSV-1, with repression mediated through hDaxx being counteracted by pp71 and through PML being nullified by IE1 (87–93). Therefore, we analyzed these MORC3-depleted HFT cells for their ability to restrict HCMV infection using the same controls as those listed above (Fig. 7C). The plaque-forming efficiency of wt HCMV within HFT-shMORC3 cells was also increased compared to that of HFT-shNeg cells, illustrating that MORC3 has antiviral activity toward DNA viruses in addition to HSV-1. The increase in wt HCMV plaque formation in MORC3-depleted cells observed here is reminiscent of that seen in cells depleted of PML or Sp100, indicating that even wt HCMV is sensitive to PML NB component-mediated restriction (as described in the studies cited above).
DISCUSSION
Here, we report an antiviral function for the nuclear matrix protein MORC3. Our previous study identified sumoylated MORC3 as a degradation target of the HSV-1 E3 ubiquitin ligase protein ICP0 (9), which led us to further investigate the function of MORC3 during HSV-1 infection. Until now, the role of MORC3 during virus infections was relatively unknown, although one other report suggested that MORC3 was required for efficient influenza A virus (IAV) infection (50). MORC3 is a sumoylated nuclear matrix protein with RNA and DNA binding activities (13, 17) that associates with PML NBs. This association is through a SUMO-SIM interaction with PML.I, which requires a functional ATPase domain within MORC3 (12, 13). In our analysis of HFT cells, we found that MORC3 was detectable in only a subset of PML NBs, whereas others reported that in HeLa and Saos-2 cells MORC3 was colocalized to the majority of PML nuclear foci. This difference in detail may be due to cell type or efficiency of antibody detection, and indeed Ver et al. found no significant colocalization of MORC3 with PML in A549 cells (50). One reported role of MORC3 in PML NBs is to recruit Sp100 and p53 into these complexes (12), although in the HF-derived cells used here the former function was not evident. The discrepancy may be due to differences in cell types examined or the efficiency of MORC3 depletion.
Upon infection with HSV-1 there is activation of an intrinsic immune response, including recruitment of protein components of the PML NB complex, such as PML, Sp100, and hDaxx, independently to sites associated with HSV-1 genomes entering the infected cell nucleus. In the absence of ICP0, this recruitment contributes to cell-mediated repression of viral gene replication (54, 55, 62, 74, 76, 94). The DNA repair protein γH2AX also accumulates in regions surrounding the viral genomes (82, 83). The repression mediated through the recruitment of PML NB components is counteracted by the expression of ICP0, which preferentially degrades sumoylated forms of these proteins and causes dispersion of others (7, 8, 64–67). The association of MORC3 with PML NBs in uninfected cells led us to investigate the recruitment of MORC3 to HSV-1 genomes during the initial stages of infection, which led to the finding that MORC3 becomes associated with foci of ICP4 in a manner similar to that of the well-characterized behavior of PML NB components. This provoked some intriguing questions as to the role of MORC3 during an intrinsic immune response to HSV-1 infection. We found that PML recruitment was noticeably affected in MORC3-depleted cells and that Sp100, γH2AX, and hDaxx were also recruited less efficiently, although this defect was not as marked as that of PML. In control cells, there appears to be a temporal transfer of PML protein from PML NBs to ICP4-associated foci, such that foci of both types can frequently be observed in the infected cells. However, in a large number of MORC3-depleted ICP0 null-infected cells, the PML signal becomes disperse with neither PML NB-like nor recruited foci. This observation is very intriguing, since we still see Sp100 in nuclear foci despite the fact that PML is required for the formation of PML NB complexes (84, 85). Since these PML NB complexes are dynamic, the lack of detectable punctate nuclear PML foci in these cells may be due to a change in the equilibrium of PML shuttling in and out of these structures in a manner that is in some way influenced by MORC3.
Our HSV-1 viral plaque assays in MORC3-depleted cells indicated that MORC3 has antiviral functions that are counteracted in the presence of ICP0, thus providing the first report of evidence for an antiviral role for MORC3. To enhance the repertoire of viruses investigated for the role of MORC3, we also assessed the replication efficiency of the beta-herpesvirus human cytomegalovirus (HCMV). Like HSV-1, HCMV also counteracts the PML NB-mediated repression of its viral gene transcription (87–93, 95). Infection of our MORC3-depleted cells with wt HCMV resulted in an increase in plaque formation efficiency compared to the control cells. Therefore, MORC3 has antiviral activity toward DNA viruses in addition to HSV-1. It would be of interest to examine other herpesviruses in the future to determine if this response is common to all herpesviruses.
In summary, we report that MORC3 has an antiviral role during HSV-1 and HCMV infection. The antiviral effect of MORC3 during HSV-1 infection was counteracted by the viral ubiquitin ligase protein, ICP0, and we identified that depletion of MORC3 reduces the efficiency of recruitment of PML NB component proteins to HSV-1 genomes. Because the recruitment of these PML NB proteins plays an important role during the intrinsic immune response to HSV-1, this role of MORC3 may underlie why restriction of ICP0-null HSV-1 is reduced in its absence. It is notable that our interest in MORC3 came through a general screen of changes in the SUMO2 proteome during HSV-1 infection which eventually and independently led to connections with PML NB-mediated restriction, thus underlining the biological relevance of virus-PML NB interactions. This study has provided further insight into the understanding of the intrinsic immune response to HSV-1 and opens a new avenue of research into MORC3 beyond herpesviruses.
ACKNOWLEDGMENTS
We are very grateful for the gifts of the indicated reagents from the following people: antibodies from Roel van Driel (PML 5E10) and Hans Will (SpGH and Sp26), HSV-1 in1863 and dl1403/CMVlacZ from Chris Preston, and plasmid pCMV.DR.8.91 from Didier Trono.
Funding Statement
The funder had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Knipe D, Howley P, Griffin D, Lamb R, Martin M, Roizman B, Strauss S. 2006. Fields Virology. Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Weller S. 2011. Alphaherpesviruses. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 3.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 4.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 5.Everett RD, Boutell C, Hale BG. 2013. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol 11:400–411. doi: 10.1038/nrmicro3015. [DOI] [PubMed] [Google Scholar]
- 6.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783:2207–2221. doi: 10.1016/j.bbamcr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol 72:6581–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sloan E, Tatham MH, Groslambert M, Glass M, Orr A, Hay RT, Everett RD. 2015. Analysis of the SUMO2 proteome during HSV-1 Infection. PLoS Pathog 11:e1005059. doi: 10.1371/journal.ppat.1005059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer LM, Abhiman S, Aravind L. 2008. MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biol Direct 3:8. doi: 10.1186/1745-6150-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer LM, Anantharaman V, Wolf MY, Aravind L. 2008. Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int J Parasitol 38:1–31. doi: 10.1016/j.ijpara.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi K, Yoshida N, Murakami N, Kawata K, Ishizaki H, Tanaka-Okamoto M, Miyoshi J, Zinn AR, Shime H, Inoue N. 2007. Dynamic regulation of p53 subnuclear localization and senescence by MORC3. Mol Biol Cell 18:1701–1709. doi: 10.1091/mbc.E06-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimura Y, Takahashi K, Kawata K, Akazawa T, Inoue N. 2010. Two-step colocalization of MORC3 with PML nuclear bodies. J Cell Sci 123:2014–2024. doi: 10.1242/jcs.063586. [DOI] [PubMed] [Google Scholar]
- 14.Dutta R, Inouye M. 2000. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 25:24–28. doi: 10.1016/S0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 15.Perry J, Zhao Y. 2003. The CW domain, a structural module shared amongst vertebrates, vertebrate-infecting parasites and higher plants. Trends Biochem Sci 28:576–580. doi: 10.1016/j.tibs.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 16.Inoue N, Hess KD, Moreadith RW, Richardson LL, Handel MA, Watson ML, Zinn AR. 1999. New gene family defined by MORC, a nuclear protein required for mouse spermatogenesis. Hum Mol Genet 8:1201–1207. doi: 10.1093/hmg/8.7.1201. [DOI] [PubMed] [Google Scholar]
- 17.Kimura Y, Sakai F, Nakano O, Kisaki O, Sugimoto H, Sawamura T, Sadano H, Osumi T. 2002. The newly identified human nuclear protein NXP-2 possesses three distinct domains, the nuclear matrix-binding, RNA-binding, and coiled-coil domains. J Biol Chem 277:20611–20617. doi: 10.1074/jbc.M201440200. [DOI] [PubMed] [Google Scholar]
- 18.Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, Hale CJ, Feng W, Michaels SD, Frand AR, Pellegrini M, Dekker J, Kim JK, Jacobsen SE. 2012. MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336:1448–1451. doi: 10.1126/science.1221472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorkovic ZJ, Naumann U, Matzke AJ, Matzke M. 2012. Involvement of a GHKL ATPase in RNA-directed DNA methylation in Arabidopsis thaliana. Curr Biol 22:933–938. doi: 10.1016/j.cub.2012.03.061. [DOI] [PubMed] [Google Scholar]
- 20.Li DQ, Nair SS, Ohshiro K, Kumar A, Nair VS, Pakala SB, Reddy SD, Gajula RP, Eswaran J, Aravind L, Kumar R. 2012. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep 2:1657–1669. doi: 10.1016/j.celrep.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He F, Umehara T, Saito K, Harada T, Watanabe S, Yabuki T, Kigawa T, Takahashi M, Kuwasako K, Tsuda K, Matsuda T, Aoki M, Seki E, Kobayashi N, Guntert P, Yokoyama S, Muto Y. 2010. Structural insight into the zinc finger CW domain as a histone modification reader. Structure 18:1127–1139. doi: 10.1016/j.str.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Hoppmann V, Thorstensen T, Kristiansen PE, Veiseth SV, Rahman MA, Finne K, Aalen RB, Aasland R. 2011. The CW domain, a new histone recognition module in chromatin proteins. EMBO J 30:1939–1952. doi: 10.1038/emboj.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Foley EA, Molloy KR, Li Y, Chait BT, Kapoor TM. 2012. Quantitative chemical proteomics approach to identify post-translational modification-mediated protein-protein interactions. J Am Chem Soc 134:1982–1985. doi: 10.1021/ja210528v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hurme R, Berndt KD, Namork E, Rhen M. 1996. DNA binding exerted by a bacterial gene regulator with an extensive coiled-coil domain. J Biol Chem 271:12626–12631. doi: 10.1074/jbc.271.21.12626. [DOI] [PubMed] [Google Scholar]
- 25.Nikolay R, Wiederkehr T, Rist W, Kramer G, Mayer MP, Bukau B. 2004. Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J Biol Chem 279:2673–2678. doi: 10.1074/jbc.M311112200. [DOI] [PubMed] [Google Scholar]
- 26.Parachoniak CA, Park M. 2009. Distinct recruitment of Eps15 via Its coiled-coil domain is required for efficient down-regulation of the met receptor tyrosine kinase. J Biol Chem 284:8382–8394. doi: 10.1074/jbc.M807607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rairdan GJ, Collier SM, Sacco MA, Baldwin TT, Boettrich T, Moffett P. 2008. The coiled-coil and nucleotide binding domains of the Potato Rx disease resistance protein function in pathogen recognition and signaling. Plant Cell 20:739–751. doi: 10.1105/tpc.107.056036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Y, Wertheim JA, Xu L, Miller JP, Karnell FG, Choi JK, Ren R, Pear WS. 2002. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood 99:2957–2968. doi: 10.1182/blood.V99.8.2957. [DOI] [PubMed] [Google Scholar]
- 29.Cheng HY, Schiavone AP, Smithgall TE. 2001. A point mutation in the N-terminal coiled-coil domain releases c-Fes tyrosine kinase activity and survival signaling in myeloid leukemia cells. Mol Cell Biol 21:6170–6180. doi: 10.1128/MCB.21.18.6170-6180.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Subrahmanyam R, Wong R, Gross AW, Ren R. 2001. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol Cell Biol 21:840–853. doi: 10.1128/MCB.21.3.840-853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lupas AN, Gruber M. 2005. The structure of alpha-helical coiled coils. Adv Protein Chem 70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 32.Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, Zhang M, Yue Z, Zhao Y. 2012. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun 3:662. doi: 10.1038/ncomms1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkhard P, Stetefeld J, Strelkov SV. 2001. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol 11:82–88. doi: 10.1016/S0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 34.Cheng P, Yang Y, Heintzen C, Liu Y. 2001. Coiled-coil domain-mediated FRQ-FRQ interaction is essential for its circadian clock function in Neurospora. EMBO J 20:101–108. doi: 10.1093/emboj/20.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng H, Begg GE, Schultz DC, Friedman JR, Jensen DE, Speicher DW, Rauscher FJ III. 2000. Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J Mol Biol 295:1139–1162. doi: 10.1006/jmbi.1999.3402. [DOI] [PubMed] [Google Scholar]
- 36.Buisson R, Masson JY. 2012. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res 40:10312–10323. doi: 10.1093/nar/gks807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hohl M, Kwon Y, Galvan SM, Xue X, Tous C, Aguilera A, Sung P, Petrini JH. 2011. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat Struct Mol Biol 18:1124–1131. doi: 10.1038/nsmb.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itakura E, Sawada I, Matsuura A. 2005. Dimerization of the ATRIP protein through the coiled-coil motif and its implication to the maintenance of stalled replication forks. Mol Biol Cell 16:5551–5562. doi: 10.1091/mbc.E05-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ball HL, Cortez D. 2005. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem 280:31390–31396. doi: 10.1074/jbc.M504961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma J, Zhang T, Novotny-Diermayr V, Tan AL, Cao X. 2003. A novel sequence in the coiled-coil domain of Stat3 essential for its nuclear translocation. J Biol Chem 278:29252–29260. doi: 10.1074/jbc.M304196200. [DOI] [PubMed] [Google Scholar]
- 41.Raiborg C, Bremnes B, Mehlum A, Gillooly DJ, D'Arrigo A, Stang E, Stenmark H. 2001. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J Cell Sci 114:2255–2263. [DOI] [PubMed] [Google Scholar]
- 42.Begitt A, Meyer T, van Rossum M, Vinkemeier U. 2000. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc Natl Acad Sci U S A 97:10418–10423. doi: 10.1073/pnas.190318397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blazek D, Barboric M, Kohoutek J, Oven I, Peterlin BM. 2005. Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Res 33:7000–7010. doi: 10.1093/nar/gki997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li DQ, Nair SS, Kumar R. 2013. The MORC family: new epigenetic regulators of transcription and DNA damage response. Epigenetics 8:685–693. doi: 10.4161/epi.24976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Condomines M, Hose D, Raynaud P, Hundemer M, De Vos J, Baudard M, Moehler T, Pantesco V, Moos M, Schved JF, Rossi JF, Reme T, Goldschmidt H, Klein B. 2007. Cancer/testis genes in multiple myeloma: expression patterns and prognosis value determined by microarray analysis. J Immunol 178:3307–3315. doi: 10.4049/jimmunol.178.5.3307. [DOI] [PubMed] [Google Scholar]
- 46.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, Severson T, Taylor GA, Teschendorff AE, Tse K, Turashvili G, Varhol R, Warren RL, Watson P, Zhao Y, Caldas C, Huntsman D, Hirst M, Marra MA, Aparicio S. 2009. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 47.Tripathi A, King C, de la Morenas A, Perry VK, Burke B, Antoine GA, Hirsch EF, Kavanah M, Mendez J, Stone M, Gerry NP, Lenburg ME, Rosenberg CL. 2008. Gene expression abnormalities in histologically normal breast epithelium of breast cancer patients. Int J Cancer 122:1557–1566. [DOI] [PubMed] [Google Scholar]
- 48.Chen LH, Kuo WH, Tsai MH, Chen PC, Hsiao CK, Chuang EY, Chang LY, Hsieh FJ, Lai LC, Chang KJ. 2011. Identification of prognostic genes for recurrent risk prediction in triple negative breast cancer patients in Taiwan. PLoS One 6:e28222. doi: 10.1371/journal.pone.0028222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liggins AP, Cooper CD, Lawrie CH, Brown PJ, Collins GP, Hatton CS, Pulford K, Banham AH. 2007. MORC4, a novel member of the MORC family, is highly expressed in a subset of diffuse large B-cell lymphomas. Br J Haematol 138:479–486. doi: 10.1111/j.1365-2141.2007.06680.x. [DOI] [PubMed] [Google Scholar]
- 50.Ver LS, Marcos-Villar L, Landeras-Bueno S, Nieto A, Ortin J. 2015. The Cellular Factor NXP2/MORC3 Is a Positive Regulator of Influenza Virus Multiplication. J Virol 89:10023–10030. doi: 10.1128/JVI.01530-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J Gen Virol 67 (Part 12):2571–2585. [DOI] [PubMed] [Google Scholar]
- 52.Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, Everett RD. 2013. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 87:13422–13432. doi: 10.1128/JVI.02474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A 99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Everett RD, Parsy ML, Orr A. 2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J Virol 83:4963–4977. doi: 10.1128/JVI.02593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jamieson DR, Robinson LH, Daksis JI, Nicholl MJ, Preston CM. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J Gen Virol 76(Part 6):1417–1431. [DOI] [PubMed] [Google Scholar]
- 57.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J Cell Sci 101(Part 4):773–784. [DOI] [PubMed] [Google Scholar]
- 58.Everett RD, Cross A, Orr A. 1993. A truncated form of herpes simplex virus type 1 immediate-early protein Vmw110 is expressed in a cell type dependent manner. Virology 197:751–756. doi: 10.1006/viro.1993.1651. [DOI] [PubMed] [Google Scholar]
- 59.Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect Immun 34:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schenk P, Ludwig H. 1988. The 65 K DNA binding protein appears early in HSV-1 replication. Arch Virol 102:119–123. doi: 10.1007/BF01315568. [DOI] [PubMed] [Google Scholar]
- 61.Guldner HH, Szostecki C, Schroder P, Matschl U, Jensen K, Luders C, Will H, Sternsdorf T. 1999. Splice variants of the nuclear dot-associated Sp100 protein contain homologies to HMG-1 and a human nuclear phosphoprotein-box motif. J Cell Sci 112 (Part 5):733–747. [DOI] [PubMed] [Google Scholar]
- 62.Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grotzinger T, Sternsdorf T, Jensen K, Will H. 1996. Interferon-modulated expression of genes encoding the nuclear-dot-associated proteins Sp100 and promyelocytic leukemia protein (PML). Eur J Biochem 238:554–560. doi: 10.1111/j.1432-1033.1996.0554z.x. [DOI] [PubMed] [Google Scholar]
- 64.Maul GG, Guldner HH, Spivack JG. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J Gen Virol 74(Part 12):2679–2690. [DOI] [PubMed] [Google Scholar]
- 65.Muller S, Dejean A. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J Virol 73:5137–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chelbi-Alix MK, de The H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 67.Parkinson J, Everett RD. 2000. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J Virol 74:10006–10017. doi: 10.1128/JVI.74.21.10006-10017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Everett RD, Boutell C, Pheasant K, Cuchet-Lourenco D, Orr A. 2014. Sequences related to SUMO interaction motifs in herpes simplex virus 1 protein ICP0 act cooperatively to stimulate virus infection. J Virol 88:2763–2774. doi: 10.1128/JVI.03417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fukuyo Y, Horikoshi N, Ishov AM, Silverstein SJ, Nakajima T. 2011. The herpes simplex virus immediate-early ubiquitin ligase ICP0 induces degradation of the ICP0 repressor protein E2FBP1. J Virol 85:3356–3366. doi: 10.1128/JVI.02105-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J 16:1519–1530. doi: 10.1093/emboj/16.7.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hagglund R, Roizman B. 2002. Characterization of the novel E3 ubiquitin ligase encoded in exon 3 of herpes simplex virus-1-infected cell protein 0. Proc Natl Acad Sci U S A 99:7889–7894. doi: 10.1073/pnas.122246999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J Virol 86:11209–11222. doi: 10.1128/JVI.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J Virol 84:4026–4040. doi: 10.1128/JVI.02597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cuchet-Lourenco D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog 7:e1002123. doi: 10.1371/journal.ppat.1002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82:2661–2672. doi: 10.1128/JVI.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, Shi Y, Shi Y, Gill G. 2006. NXP-2 association with SUMO-2 depends on lysines required for transcriptional repression. Proc Natl Acad Sci U S A 103:5308–5313. doi: 10.1073/pnas.0601066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol 81:10991–11004. doi: 10.1128/JVI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 80.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 81.Wilkinson DE, Weller SK. 2006. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J Cell Sci 119:2695–2703. doi: 10.1242/jcs.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weller SK. 2010. Herpes simplex virus reorganizes the cellular DNA repair and protein quality control machinery. PLoS Pathog 6:e1001105. doi: 10.1371/journal.ppat.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. 2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog 7:e1002084. doi: 10.1371/journal.ppat.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748–2752. [PubMed] [Google Scholar]
- 86.Saffert RT, Kalejta RF. 2008. Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both? Future Virol 3:265–277. doi: 10.2217/17460794.3.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Preston CM, Nicholl MJ. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J Gen Virol 87:1113–1121. doi: 10.1099/vir.0.81566-0. [DOI] [PubMed] [Google Scholar]
- 88.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem 281:37652–37660. doi: 10.1074/jbc.M604273200. [DOI] [PubMed] [Google Scholar]
- 90.Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn JH. 2011. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol 85:11928–11937. doi: 10.1128/JVI.00758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol 80:8006–8018. doi: 10.1128/JVI.00743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tavalai N, Papior P, Rechter S, Stamminger T. 2008. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J Virol 82:126–137. doi: 10.1128/JVI.01685-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tavalai N, Adler M, Scherer M, Riedl Y, Stamminger T. 2011. Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J Virol 85:9447–9458. doi: 10.1128/JVI.00870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ahn JH, Hayward GS. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39–55. doi: 10.1006/viro.2000.0448. [DOI] [PubMed] [Google Scholar]