

ABSTRACT
To investigate the molecular mechanism(s) by which herpes simplex virus 1 (HSV-1) tegument protein UL51 promotes viral replication, we screened for viral proteins that interact with UL51 in infected cells. Affinity purification of tagged UL51 in HSV-1-infected Vero cells was coupled with immunoblotting of the purified UL51 complexes with various antibodies to HSV-1 virion proteins. Subsequent analyses revealed that UL51 interacted with another tegument protein, UL14, in infected cells. Mutational analyses of UL51 showed that UL51 amino acid residues Leu-111, Ile-119, and Tyr-123 were required for interaction with UL14 in HSV-1-infected cells. Alanine substitutions of these UL51 amino acid residues reduced viral replication and produced an accumulation of unenveloped and partially enveloped nucleocapsids in the cytoplasm at levels comparable to those of UL51-null, UL14-null, and UL51/UL14 double-null mutations. In addition, although UL51 and UL14 colocalized at juxtanuclear domains in HSV-1-infected cells, the amino acid substitutions in UL51 produced aberrant localization of UL51 and UL14. The effects of these substitutions on localization of UL51 and UL14 were similar to those of the UL51-null and UL14-null mutations on localization of UL14 and UL51, respectively. These results suggested that the interaction between UL51 and UL14 was required for proper localization of these viral proteins in infected cells and that the UL51-UL14 complex regulated final viral envelopment for efficient viral replication.
IMPORTANCE Herpesviruses contain a unique virion structure designated the tegument, which is a protein layer between the nucleocapsid and the envelope. HSV-1 has dozens of viral proteins in the tegument, which are thought to facilitate viral envelopment by interacting with other virion components. However, although numerous interactions among virion proteins have been reported, data on how these interactions facilitate viral envelopment is limited. In this study, we have presented data showing that the interaction of HSV-1 tegument proteins UL51 and UL14 promoted viral final envelopment for efficient viral replication. In particular, prevention of this interaction induced aberrant accumulation of partially enveloped capsids in the cytoplasm, suggesting that the UL51-UL14 complex acted in the envelopment process but not in an upstream event, such as transport of capsids to the site for envelopment. This is the first report showing that an interaction between HSV-1 tegument proteins directly regulated final virion envelopment.
INTRODUCTION
Herpesviruses have a unique virion structure designated the tegument, which is a proteinaceous layer consisting of a number of different viral proteins and is located between the nucleocapsid and the envelope (1). Herpes simplex virus 1 (HSV-1), classified in the Alphaherpesvirinae subfamily of the Herpesviridae family, is one of the best-studied members in the family and has at least 23 different viral proteins in the tegument (2).
In HSV-1-infected cells, packaging of nascent progeny virus genomes into preformed capsids takes place in the nucleus. The nascent progeny nucleocapsids acquire a primary envelope by budding through the inner nuclear membrane (INM) into the perinuclear space between the INM and outer nuclear membrane (ONM) (primary envelopment) (3, 4). The enveloped nucleocapsids then fuse with the ONM to release unenveloped nucleocapsids into the cytoplasm. Subsequently, the nucleocapsids acquire a final envelope by budding into cytoplasmic vesicles, probably membranes derived from the trans-Golgi network and/or endosomes (secondary envelopment), and mature virions are secreted from the infected cells by exocytosis (3, 4). Thus far, the site(s) for HSV-1 tegument acquisition remains to be determined. It has been proposed that HSV-1 nucleocapsids primarily acquire tegument proteins in the cytoplasm (5). In contrast, it has been reported that some HSV-1 tegument proteins were detected in both primary and secondary enveloped virions (i.e., VP16, VP22, US3, UL11, and VP13/14) and some only in primary enveloped virions (i.e., UL31 and UL34), suggesting that these tegument proteins were incorporated into nascent virus particles in the nucleus and/or during nuclear egress of the nucleocapsids (2, 6–10).
The location of the tegument in herpesvirus virions indicates that this structure bridges the nucleocapsid and the envelope to maintain the structural integrity of virions by interacting directly with capsid proteins, other tegument proteins, and the cytoplasmic domains of envelope glycoproteins and/or membrane-associated tegument proteins. In agreement with this, numerous protein-protein interactions between tegument proteins and between tegument proteins, capsid proteins, and envelope proteins have been reported (5). These data also suggest that, during virion morphogenesis, viral tegument proteins facilitate viral envelopment by establishing the protein-protein network found in enveloped virions. In support of this hypothesis, a number of HSV-1 tegument proteins (i.e., UL31, UL34, UL36, UL37, UL14, UL47, UL48, UL11, and UL16) were reported to be required for efficient viral primary or secondary envelopment (7, 11–17). In particular, defective particles, designated L-particles, that contain most of the tegument proteins and all of the envelope glycoproteins but lack capsids, are produced in HSV-infected cells (18, 19). These results indicate that the tegument and envelope proteins are sufficient for viral secondary envelopment, and that interactions between tegument proteins and/or between tegument proteins and envelope proteins are important for viral envelopment. However, although numerous reports have shown various interactions among these virion proteins as described above, data confirming that these interactions facilitate viral envelopment are limited. Thus far, only interactions between HSV-1 tegument proteins UL31 and UL34 and between tegument proteins UL36 and UL37 have been shown to be required for HSV-1 primary and secondary envelopment, respectively (17, 20).
HSV-1 tegument protein UL51 is conserved in the Herpesviridae family (1). Like HSV-1 UL51, UL51 homologs in other members of the Alpha-, Beta-, and Gammaherpesvirinae subfamilies were also shown to be incorporated into the tegument of virions (2, 21–24), suggesting that UL51 homologs are conserved tegument proteins in herpesviruses. UL51 is an important positive regulator for HSV-1 replication in cell cultures based on the observation that recombinant HSV-1 UL51-null mutants show significantly impaired plaque formation and have a significant reduction in progeny virus titers in cell cultures (25). Palmitoylation of UL51 has been suggested to have a role in UL51 association with cytoplasmic membranes, with UL51 topology indicating that it should be displayed on the exterior surface of cytoplasmic membranes and, therefore, on the interior surface of virions (26). UL51 homologs of porcine alphaherpesvirus pseudorabies virus (PRV) and human betaherpesvirus cytomegalovirus (HCMV) have been reported to participate in viral secondary envelopment (27, 28). Although HSV-1 UL51 was shown to interact with UL7 (29) and UL7 homologs in PRV and HCMV were shown to facilitate viral secondary envelopment (30, 31), it remained to be determined whether HSV-1 UL51 and UL7 are involved in viral secondary envelopment. HSV-1 UL51 has also been reported to interact with viral envelope glycoprotein E (gE) (25). However, the biological significance of these interactions in HSV-1 replication remains to be addressed.
To investigate the mechanism by which UL51 acts in HSV-1 replication, we screened for HSV-1 virion proteins that interact with UL51 in HSV-1-infected cells. We then focused on UL14, which was identified as a putative UL51-interacting HSV-1 virion protein. HSV-1 UL14 is a conserved herpesvirus tegument protein, like UL51 and UL7 (1), and was reported to promote viral replication and secondary envelopment (16). In the present study, we investigated the effects of the interaction between UL51 and UL14 in HSV-1-infected cells, especially effects on viral replication and virion morphogenesis.
MATERIALS AND METHODS
Cells and viruses.
Vero, COS-7, and rabbit skin cells have been described previously (32, 33). The HSV-1 wild-type strain HSV-1(F) and the UL13-null mutant virus R7356 (ΔUL13) (a kind gift from B. Roizman) have been described previously (32, 34).
Plasmids.
To construct the transfer plasmid pBS-UL51, used for generating recombinant viruses YK5012 (ΔUL51-repair) and YK5018 (ΔUL51/ΔUL14-repair) in which the UL51-null mutation in YK5011 (ΔUL51) and the UL51/UL14-null mutations in YK5017 (ΔUL51/ΔUL14), respectively, were repaired (Fig. 1), we amplified the domain containing the UL51 open reading frame (ORF), the 894-bp upstream sequence flanking the UL51 start codon, and the 1-kbp downstream sequence flanking the UL51 stop codon from pYEbac5002, a full-length infectious HSV-1(F) clone (35), using primers 5′-GCGAGCTCCGAATGGCTATGCCGGCTGA-3′ and 5′-CGGAATTCGACGGTGTCGGGGCGGGGGG-3′, followed by cloning into pBluescript II KS(+) (Stratagene).
FIG 1.
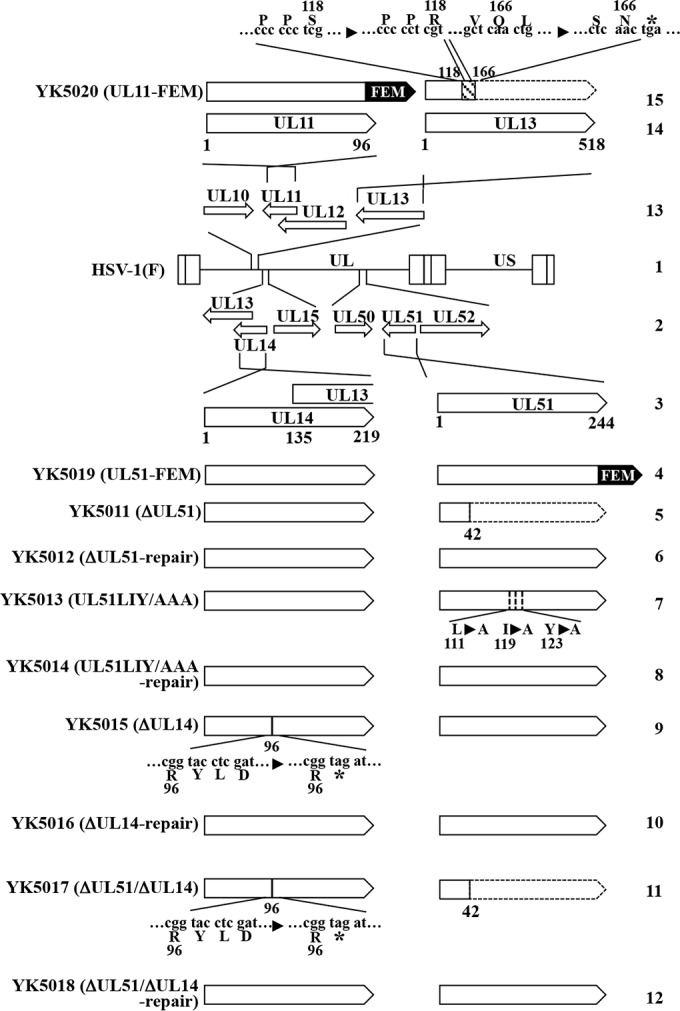
Schematic diagrams of the genome structures of wild-type HSV-1(F) and the relevant domains of the recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domains of the UL13 to UL15 and UL50 to UL52 genes; line 3, domains of the UL14 and UL51 genes; lines 4 to 12, recombinant viruses with mutations in the UL14 and/or UL51 genes; line 13, domains of the UL10 to UL13 genes; line 14, domains of the UL11 and UL13 genes; line 15, recombinant viruses with mutations in UL11 and UL13. An asterisk denotes a stop codon.
Plasmid pBS-FEM-kan, for generating recombinant viruses YK5019 (UL51-FEM) and YK5020 (UL11-FEM), expressing UL51 and UL11, respectively, tagged with an FEM tag, with Flag and myc epitopes, and a tobacco etch virus (TEV) protease cleavage site (Flag-TEV-myc), was constructed as follows. The domain containing the FEM tag, I-SceI site, and kanamycin resistance gene was amplified using primers 5′-ATAAGAATGCGGCCGCATGGACTACAAGGACGACG-3′ and 5′-GAGAATTCTCACAAGTCCTCTTCAGAAATGAGC-3′, which were from PCR fragments that were amplified from pEP-KanS (36) using primers 5′-ATGGACTACAAGGACGACGATGACAAAGATGACCGTGATTATGATATTCCAACTACTGCTAGCGAGAATTTGTATTTTCAGGGTGAAGGATGACGACGATAAGTAGGG-3′ and 5′-TCACAAGTCCTCTTCAGAAATGAGCTTTTGCTCCATGGTGGCGGATCCGAGCTCACCCTGAAAATACAAATTCTCGCTAGCAGTAGTTGGAACAACCAATTAACCAATTCTGATTAG-3′ and then cloned into pBluescript II KS(+).
To generate a fusion protein of the maltose binding protein (MBP) and a domain of UL15 or UL52, pMAL-UL15:344-745 and pMAL-UL52:150-349 were constructed by cloning the UL15 domain, encoding codons 344 to 745, and the UL52 domain, encoding codons 150 to 349, respectively, that had been amplified by PCR from the HSV-1(F) genome into pMAL-c (New England BioLabs) in frame with MBP.
To generate a fusion protein of glutathione S-transferase (GST) and UL51 and fusion proteins of GST and each of the UL51 domains (see Fig. 4A), pGEX-UL51, pGEX-UL51:12-170, pGEX-UL51:164-244, pGEX-UL51:12-130, pGEX-UL51:47-170, pGEX-UL51:47-130, pGEX-UL51:47-94, and pGEX-UL51:90-130 were constructed by cloning the UL51 domains encoding codons 1 to 244, 12 to 170, 164 to 244, 12 to 130, 47 to 170, 47 to 130, 47 to 94, and 90 to 130, respectively, that had been amplified by PCR from the HSV-1(F) genome into pGEX-4T1 (GE Healthcare) in frame with GST. To generate a fusion protein of GST and the UL51 domain (UL51:90-130) with alanine replacements of leucine 111 (Leu-111), isoleucine 119 (Ile-119), and tyrosine 123 (Tyr-123) (UL51:90-130LIY/AAA), pGEX-UL51:90-130LIY/AAA was generated by site-directed PCR mutagenesis. Primers flanking the UL51 gene along with primers in the middle of the gene that contained the mutations and introduced a restriction site were used to amplify the gene in two pieces from pGEX-UL51:90-130. After cutting with a restriction enzyme, the two halves of the gene were ligated and amplified by other primers flanking the gene. The primer sets used were 5′-GATGCGGCGCGCCGCGGACACGTGTATGGCCACCGCCCTGCAGATGGCCATGTCCGTG-3′ and 5′-CCGGGAGCTGCATGTGTCAGAGG-3′, 5′-CACGGACATGGCCATCTGCAGGGCGGTGGCCATACACGTGTCCGCGGCGCGCCGCATC-3′ and 5′-GGGCTGGCAAGCCACGTTTGGTG-3′, and 5′-GCGAATTCCCCGGGCTCGAGGCCCCCAC-3′ and 5′-GCGTCGACGTCAGCCGCCCCCACGGACA-3′.
FIG 4.

Mapping of UL51 domains required for UL51 interaction with UL14. (A) Schematic diagram of the UL51 gene product (top line), with its putative α-helix domains (black boxes), and diagrams of GST-UL51, the GST-UL51 deletion mutants, and the GST-UL51:90-130LIY/AAA mutant. The level of binding of each GST fusion protein to UL14 determined in the pulldown experiments shown in panels B and C is shown at the right of each protein. (B and C) The GST fusion proteins shown in panel A were immobilized on glutathione-Sepharose beads and mock reacted or reacted with lysates of Vero cells that had been infected with wild-type HSV-1(F) at an MOI of 5 for 18 h. The beads were washed extensively and divided into two parts. One part was analyzed by immunoblotting with anti-UL14 antibody (top gels), and the other was electrophoretically analyzed in a denaturing gel and CBB stained (bottom gels). (D) Amount of UL14 pulled down by GST, GST-UL51:90-130, or GST-UL51:90-130LIY/AAA shown in the top of panel C relative to those of the GST fusion protein shown in the bottom of panel C. Each value is the mean ± standard error from three independent experiments and is expressed relative to the mean value of GST, which was normalized to 100. Asterisks indicate significant differences: *, P < 0.05 (by two-tailed Student t test).
Production and purification of MBP and GST fusion proteins in Escherichia coli.
MBP fusion proteins MBP-UL15:344-745 and MBP-UL52:150-349 were expressed in E. coli that had been transformed with pMAL-UL15:344-745 and pMAL-UL52:150-349, respectively, and purified using amylose resin (New England BioLabs) as described previously (33). GST fusion proteins GST-UL51, GST-UL51:12-170, GST-UL51:164-244, GST-UL51:12-130, GST-UL51:47-170, GST-UL51:47-130, GST-UL51:47-94, GST-UL51:90-130, and GST-UL51:90-130L111A/I119A/Y123A were expressed in E. coli that had been transformed with pGEX-UL51, pGEX-UL51:12-170, pGEX-UL51:164-244, pGEX-UL51:12-130, pGEX-UL51:47-170, pGEX-UL51:47-130, pGEX-UL51:47-94, pGEX-UL51:90-130, and pGEX-UL51:90-130L111A/I119A/Y123A, respectively, and purified using glutathione-Sepharose resin (GE Healthcare Life Science) as described previously (37).
Antibodies.
Commercial mouse monoclonal antibodies to Myc (PL14; MBL), α-tubulin (DM1A; Sigma), ICP0 (1112; Goodwin Institute), ICP4 (58S; ATCC), VP5 (3B6; Virusys), gB (H1817; Virusys), gD (DL6; Danta Cruz Biotechnology), gH (52-S; ATCC), gC (H1A022; Virusys), gG (7F5; Virusys), and gE (9H3; Abcam) and commercial rabbit polyclonal antibodies to VP23 (CAC-CT-HSV-UL18; CosmoBio) and UL11 (CAC-CT-HSV-UL11; CosmoBio) were used. Rabbit polyclonal antibodies to UL51, UL14, UL7, UL31, UL34, VP26, UL41, UL46, VP13/14, VP16, VP22, UL50, Us3, and Us9, a mouse monoclonal antibody to UL13, and a mouse polyclonal antibody to gN were described previously (7, 26, 35, 38–42). Rabbit polyclonal antibody to gM (43) was kindly provided by J. Baines. To generate mouse polyclonal antibodies to HSV-1 UL15, UL51, and UL52, BALB/c mice were immunized with purified MBP-fusion protein MBP-UL15:344-745, GST-UL51, or MBP-UL52:150-349 as described previously (38). The rabbit and mouse polyclonal antibodies to UL51 were used for immunoblotting and immunofluorescence, respectively.
Immunoblotting, immunofluorescence, and immunoprecipitation.
Immunoblotting, immunofluorescence, and immunoprecipitation were performed as described previously (44, 45).
Mutagenesis of viral genomes in E. coli and generation of recombinant HSV-1.
To generate recombinant viruses YK5019 (UL51-FEM) and YK5020 (UL11-FEM), expressing FEM-tagged UL51 and UL11, respectively (Fig. 1), a two-step Red-mediated mutagenesis procedure was carried out using E. coli GS1783 containing pYEbac5002 (35) as described previously (46), except using pBS-FEM-kan and the primers listed in Table 1. Recombinant viruses YK5011 (ΔUL51), in which the UL51 gene was disrupted by deleting UL51 codons 43 to 244, YK5015 (ΔUL14), in which a deletion of 4 bp from nucleotides 291 to 294 in the UL14 gene (relative to the first nucleotide of the UL14 gene) replaced UL14 Tyr-97 with a stop codon, and YK5013 (UL51LIY/AAA), carrying the L111A/I119A/Y123A mutations in UL51 (Fig. 1), were constructed by the two-step Red-mediated mutagenesis procedure using E. coli GS1783 carrying pYEbac5002 as described previously (46), except using the primers listed in Table 1. The 4-bp deletion in UL14 has previously been used to generate an HSV-1 UL14-null mutant (16). Recombinant virus YK5017 (ΔUL51/ΔUL14), carrying mutations in YK5011 (ΔUL51) and YK5015 (ΔUL14) (Fig. 1), was constructed by the two-step Red-mediated mutagenesis procedure using E. coli GS1783 carrying the YK5011 (ΔUL51) genome as described previously (46), except using the primers listed in Table 1. Recombinant viruses YK5016 (ΔUL14-repair) and YK5014 (UL51LIY/AAA-repair), in which the mutations in YK5015 (ΔUL14) and YK5013 (UL51LIY/AAA) were repaired (Fig. 1), were generated as described previously (38), except using the primers listed in Table 1. Recombinant virus YK5012 (ΔUL51-repair), in which the mutation in YK5011 (ΔUL51) was repaired (Fig. 1), was generated by transfection of COS-7 cells with pBS-UL51 using Lipofectamine 2000 (Invitrogen) and subsequent infection with YK5011 (ΔUL51). Plaques of progeny viruses from the transfections and infections were isolated three times on Vero cells, and restoration of original sequences was confirmed by sequencing. Recombinant virus YK5018 (ΔUL51/ΔUL14-repair), in which the mutations in YK5017 (ΔUL51/ΔUL14) were repaired (Fig. 1), was generated as follows. A viral genome with a mutation in UL14 in YK5017 (ΔUL51/ΔUL14) was repaired by the same procedure as that used to generate YK5016 (ΔUL14-repair). The resultant viral genome was isolated and cotransfected into rabbit skin cells with pBS-UL51. Plaques of progeny viruses from the transfections and infections were isolated three times on Vero cells, and restoration of the original sequences of UL51 and UL14 was confirmed by sequencing.
TABLE 1.
Primer sequences used for the construction of recombinant viruses in this study
| Recombinant virus | Primer sequence (5′-3′) |
|---|---|
| YK5019 (UL51-FEM) | 5′-CCCTGGCCCATCTCGAGCAGCTCCGTGTGTTTTGGGTCAAATGGACTACAAGGACGACGA-3′ |
| 5′-CCACAAACACAGAACACACGGGTTGGATGAAAACACGCATTCACAAGTCCTCTTCAGAAATGA-3′ | |
| YK5020 (UL11-FEM) | 5′-GAGGCGCCTGCCCCGCCACCCAGTTTCCACCCCCCATGTCCGATAGCGAAGACTACAAGGACGACGATGA-3′ |
| 5′-TTAAAAACGACACGCGTGCGACCGTATACAGAACATTATTTTGGTTTTTACAAGTCCTCTTCAGAAATGA-3′ | |
| YK5011 (ΔUL51) | 5′-GGAGGCGGAGCCGCGGCTGCAGGAGGCCCTGGTGGTCGTTTAAATGCGTGTTTTCATCCAAGGATGACGACGATAAGTAGGG-3′ |
| 5′-CAAACACAGAACACACGGGTTGGATGAAAACACGCATTTAAACGACCACCAGGGCCTCAACCAATTAACCAATTCTGATTAG-3′ | |
| YK5015 (ΔUL14) | 5′-AAGCGGCGCGTCGTGACTTTCTAACCGCACACCGACGGTAGATCCGGCGCTTGGTGAGCGAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GATCGTCCACGGCGTCCAGGCGCTCACCAAGCGCCGGATCTACCGTCGGTGTGCGGTTAGCAACCAATTAACCAATTCTGATTAG-3′ | |
| YK5016 (ΔUL14-repair) | 5′-AAGCGGCGCGTCGTGACTTTCTAACCGCACACCGACGGTACCTCGATCCGGCGCTTGGTGAGCGAGGATGACGACGATAAGTAGGG-3′ |
| 5′-GATCGTCCACGGCGTCCAGGCGCTCACCAAGCGCCGGATCGAGGTACCGTCGGTGTGCGGTTAGCAACCAATTAACCAATTCTGATTAG-3′ | |
| YK5013 (UL51LIY/AAA) | 5′-GACGGGGCCGTGGCGGCCCATCAGGACAAGATGCGGCGCGCCGCGGACACGTGTATGGCCACCGCCCTGCAGATGGCCAGGATGACGACGATAAGTAGGG-3′ |
| 5′-ACATCCGCGGACTTGTCAGCCGCCCCCACGGACATGGCCATCTGCAGGGCGGTGGCCATACACGTGTCCGCGGCGCAACCAATTAACCAATTCTGATTAG-3′ | |
| YK5014 (UL51LIY/AAA-repair) | 5′-GACGGGGCCGTGGCGGCCCATCAGGACAAGATGCGGCGCCTGGCGGACACGTGTATGGCCACCATCCTGCAGATGTACAGGATGACGACGATAAGTAGGG-3′ |
| 5′-ATCCGCGGACTTGTCAGCCGCCCCCACGGACATGTACATCTGCAGGATGGTGGCCATACACGTGTCCGCCAGGCAACCAATTAACCAATTCTGATTAG-3′ |
GST pulldown assays.
GST pulldown assays were carried out essentially as described previously (37). Briefly, Vero cells were infected with wild-type HSV-1(F) at a multiplicity of infection (MOI) of 5 for 18 h and then lysed in NP-40 buffer (120 mM NaCl, 50 mM Tris-HCl [pH 8.0], 0.5% NP-40, 50 mM NaF) containing a proteinase inhibitor cocktail. The infected cell lysates were precleared by incubation with glutathione-Sepharose resin and then reacted with purified GST-UL51 and its conjugated glutathione-Sepharose resin derivatives. After extensive washing of the resin with NP-40 buffer, the resin was divided into two parts. One part was analyzed by immunoblotting with anti-UL14 antibody and the other was electrophoretically separated in a denaturing gel and stained with Coomassie brilliant blue (CBB). The amount of protein in the bands detected by immunoblotting and in the CBB-stained bands after denaturing gel electrophoresis were quantitated using the Dolphin Doc system with Dolphin1D software.
Electron microscopic analysis.
Vero cells infected with each of the indicated viruses at an MOI of 5 for 18 h were examined by ultrathin section electron microscopy as described previously (47), except using 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.4).
RESULTS
Identification of a novel HSV-1 protein that interacts with UL51 in HSV-1-infected cells.
To identify HSV-1 proteins that interact with UL51 in HSV-1-infected cells, we carried out affinity purification of FEM-tagged UL51 from an HSV-1-infected cell lysate followed by immunoblotting of the purified UL51 complexes with antibodies to 27 different HSV-1 virion proteins (ICP0, ICP4, UL7, UL11, UL13, UL14, UL31, UL34, UL41, UL46, UL50, Us3, Us9, VP13/14, VP16, VP22, VP5, VP23, VP26, gB, gC, gD, gG, gE, gH, gN, and gM). For this study, we constructed recombinant virus YK5019 (UL51-FEM), in which the UL51 C-terminal nucleotide was tagged with FEM (Fig. 1). We also constructed recombinant virus YK5020 (UL11-FEM), expressing FEM-tagged UL11, which was used as a control for YK5019 (UL51-FEM). As shown in Fig. 2A and B, Vero cells infected with YK5019 (UL51-FEM) or YK5020 (UL11-FEM) expressed FEM-tagged UL51 or UL11, respectively. Progeny virus titers in Vero cells infected with YK5019 (UL51-FEM) or YK5020 (UL11-FEM) at an MOI of 5 or 0.01 and assayed at 18 or 48 h postinfection, respectively, were similar to those in cells infected with wild-type HSV-1(F) (Fig. 2C and D), indicating that tagging these viral proteins with FEM had no effect on viral replication in Vero cells.
FIG 2.
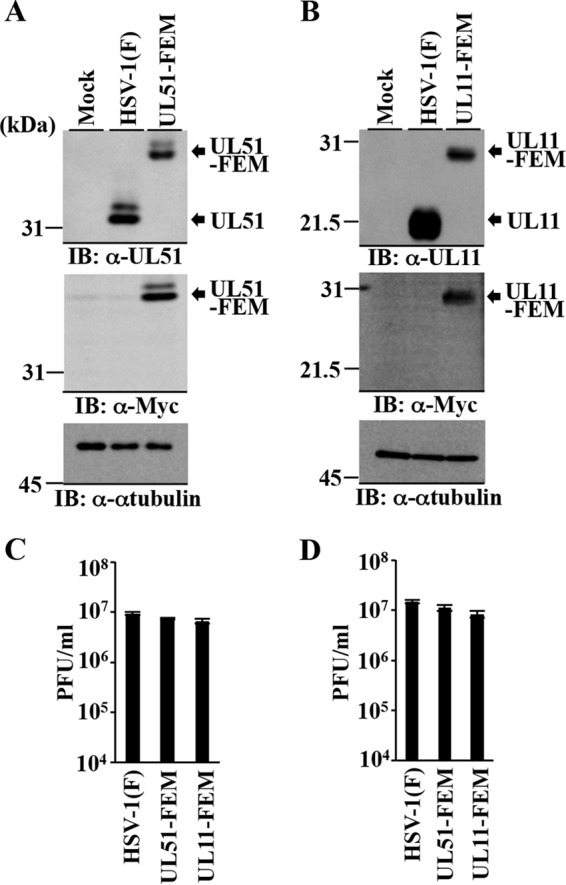
Characterization of the recombinant viruses expressing FEM-tagged UL51 and UL11. (A) Vero cells were mock infected or infected with wild-type HSV-1(F) or YK5019 (UL51-FEM) at an MOI of 5 for 18 h and then analyzed by immunoblotting (IB) with the indicated antibodies. (B) Vero cells were mock infected or infected with wild-type HSV-1(F) or YK5020 (UL11-FEM) at an MOI of 5 for 18 h and then analyzed by immunoblotting with the indicated antibodies. (C and D) Vero cells were infected with wild-type HSV-1(F), YK5019 (UL51-FEM), or YK5020 (UL11-FEM) at an MOI of 5 (C) or 0.01 (D). Total virus from cell culture supernatants and infected cells was harvested at 18 h (C) or 48 h (D) postinfection and assayed on Vero cells. Each data point is the mean ± standard error from the results of triplicate samples. Differences in viral yields between HSV-1(F) and YK5019 (UL51-FEM) and between HSV-1(F) and YK5020 (UL11-FEM) were not statistically significant by analysis of variance (ANOVA) and Dunnett's test. Data are representative of three independent experiments.
Vero cells were then infected with YK5019 (UL51-FEM) or YK5020 (UL11-FEM) at an MOI of 5 for 18 h, lysed, and immunoprecipitated with anti-myc antibody, and the immunoprecipitates were immunoblotted with antibodies to various HSV-1 virion proteins. These experiments identified two HSV-1 virion proteins, UL14 (Fig. 3A) and UL7 (data not shown), that coimmunoprecipitated with FEM-tagged UL51 but not with FEM-tagged UL11 in HSV-1-infected cells. As described above, UL7 has been reported to interact with UL51 (29), and therefore we focused on UL14 in this study. We noted that viral gE coimmunoprecipitated with both FEM-tagged UL51 and FEM-tagged UL11 (data not shown), which was in agreement with previous reports (25, 48).
FIG 3.
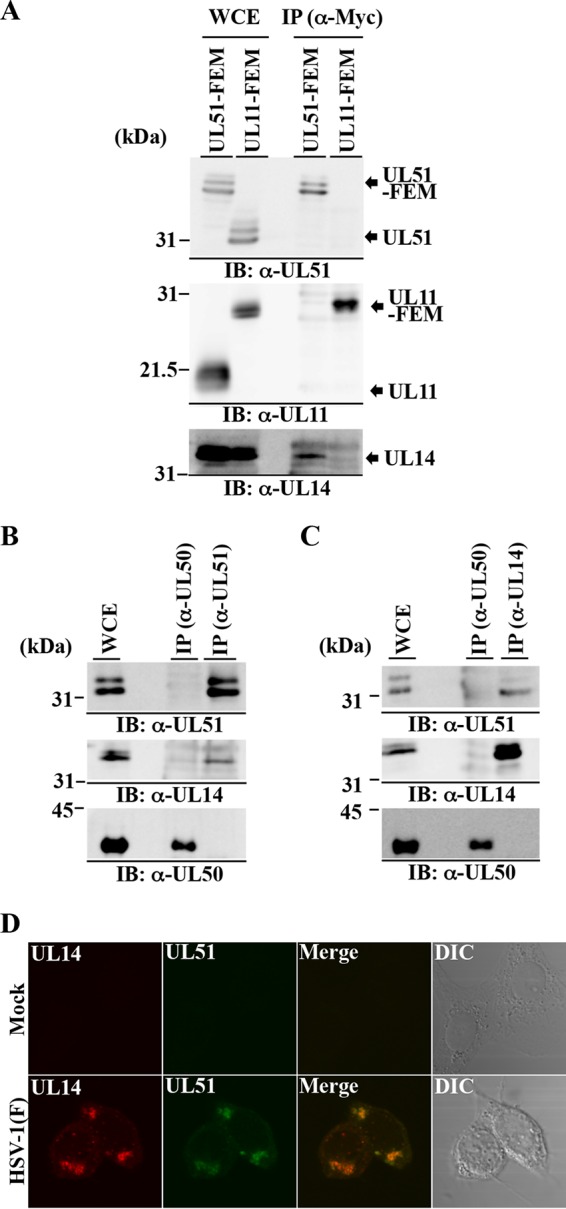
Interaction between UL51 and UL14 in HSV-1-infected cells. (A) Vero cells were infected with YK5019 (UL51-FEM) or UK5020 (UL11-FEM) at an MOI of 5 for 18 h, harvested, immunoprecipitated (IP) with anti-Myc antibody (α-Myc), and analyzed by immunoblotting (IB) with the indicated antibodies. WCE, whole-cell extract. (B and C) Vero cells were infected with wild-type HSV-1(F) at an MOI of 5 for 18 h, harvested, immunoprecipitated with anti-UL50 or anti-UL51 antibody (B) or with anti-UL50 or anti-UL14 antibody (C), and analyzed by immunoblotting with the indicated antibodies. (D) Vero cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5, fixed at 18 h postinfection, permeabilized, stained with anti-UL51 and anti-UL14 antibodies, and examined by confocal microscopy.
To verify the interaction of UL51 with UL14 in wild-type HSV-1-infected cells, two series of experiments were carried out. In the first series of experiments, Vero cells were infected with wild-type HSV-1(F) at an MOI of 5 for 18 h, lysed, immunoprecipitated with anti-UL51, anti-UL50, or anti-UL14 antibody, and immunoblotted with anti-UL14, anti-UL50, and anti-UL51 antibodies. As shown in Fig. 3B and C, anti-UL51 antibody coprecipitated both UL14 and UL51, and in a reciprocal experiment, anti-UL14 antibody coprecipitated UL51 and UL14. In contrast, anti-UL50 precipitated UL50 but not UL14 or UL51. In the second series of experiments, Vero cells infected with wild-type HSV-1(F) at an MOI of 5 for 18 h were fixed and analyzed by confocal microscopy with anti-UL14 and anti-UL51 antibodies. In agreement with previous reports (16, 26), both UL51 and UL14 were predominantly localized in juxtanuclear domains and colocalized in these domains (Fig. 3D). Taken together, these results indicated that UL51 interacted with UL14 in wild-type HSV-1-infected cells.
Identification of UL51 residues responsible for the interaction with UL14.
To map UL51 residues required for the interaction with UL14, we carried out GST pulldown experiments using various UL51 mutants (Fig. 4A). In agreement with the result described above (Fig. 3A to C) that UL51 coprecipitated with UL14 in lysates of HSV-1-infected cells, GST-UL51 was able to pull down UL14 in the lysates of wild-type HSV-1(F)-infected cells, but GST could not (Fig. 4B). As shown in Fig. 4A, UL51 contains seven putative α-helixes (I to VII) linked by random coils (PSIPRED; bioinf.cs.ucl.ac.uk/psipred/); therefore, we constructed a series of GST-UL51 deletion mutants to examine the effect of each α-helix on UL51 interaction with UL14: GST-UL51:12-170 containing α-helixes II to VII, GST-UL51:12-130 containing α-helixes II to VI, GST-UL51:47-170 containing α-helixes IV to VII, and GST-UL51:90-130 containing α-helix VI pulled down UL14 (Fig. 4C). In contrast, GST-UL51:47-94 containing α-helixes IV and V and GST-UL51:164-244 containing the C-terminal domain of UL51 with no α-helix did not pull down UL14 (Fig. 4C). These results indicated that the UL51 domain containing α-helix VI (UL51:90-130) was necessary and sufficient for UL51 interaction with UL14 in these GST pulldown experiments.
In general, solvent-exposed hydrophobic and charged residues are important for protein-protein interactions (49). Therefore, we focused on three hydrophobic residues (Leu-111, Ile-119, and Tyr-123) in UL51 α-helix VI. These residues were predicted to be in the same face of α-helix VI (http://heliquest.ipmc.cnrs.fr/). Therefore, we constructed mutant GST-UL51:90-130LIY/AAA, in which Leu-111, Ile-119, and Tyr-123 in GST-UL51:90-130 were replaced with alanines (LIY/AAA) (Fig. 4A), and tested this mutant in GST pulldown experiments. We noted that the LIY/AAA mutation in UL51 was predicted to have no obvious effect on the secondary structure of α-helix VI. As shown in Fig. 4C and D, the LIY/AAA mutation in GST-UL51:90-130 significantly reduced the interaction between UL51:90-130 and UL14 in these experiments. In addition, alanine substitutions of three UL51 charged amino acid residues (Asp-106, Arg-110, and Asp-113), which were predicted to be in the other face of α-helix VI (http://heliquest.ipmc.cnrs.fr/), had no effect on UL51 interaction with UL14 in GST pulldown experiments (data not shown). These results indicated that UL51 residues Leu-111, Ile-119, and Tyr-123 were required for UL51 interaction with UL14.
Characterization of recombinant viruses carrying mutation(s) in UL51 and/or UL14.
To investigate the effects of the interaction between UL51 and UL14 in HSV-1-infected cells, we constructed and characterized UL51-null mutant virus YK5011 (ΔUL51), UL14-null mutant virus YK5015 (ΔUL14), UL51/UL14-double null mutant virus YK5017 (ΔUL51/ΔUL14), recombinant virus YK5013 (UL51LIY/AAA) carrying the LIY/AAA mutations in UL51, and repaired viruses YK5012 (ΔUL51-repair), YK5016 (ΔUL14-repair), YK5018 (ΔUL51/ΔUL14-repair), and YK5014 (UL51LIY/AAA-repair) (Fig. 1). As expected, Vero cells infected with wild-type HSV-1(F), YK5012 (ΔUL51-repair), or YK5018 (ΔUL51/ΔUL14-repair) expressed UL51, but cells infected with YK5011 (ΔUL51) or YK5017 (ΔUL51/ΔUL14) did not (Fig. 5A and C). Vero cells infected with wild-type HSV-1(F), YK5016 (ΔUL14-repair), or YK5018 (ΔUL51/ΔUL14-repair) expressed UL14, but those infected with YK5015 (ΔUL14) or YK5017 (ΔUL51/ΔUL14) did not (Fig. 5B and C). Vero cells infected with YK5013 (UL51LIY/AAA) produced UL51 as efficiently as those infected with wild-type HSV-1(F) or YK5013 (UL51LIY/AAA-repair) (Fig. 5A), indicating that the LIY/AAA mutations in UL51 had no obvious effect on expression of UL51 in HSV-1-infected cells.
FIG 5.
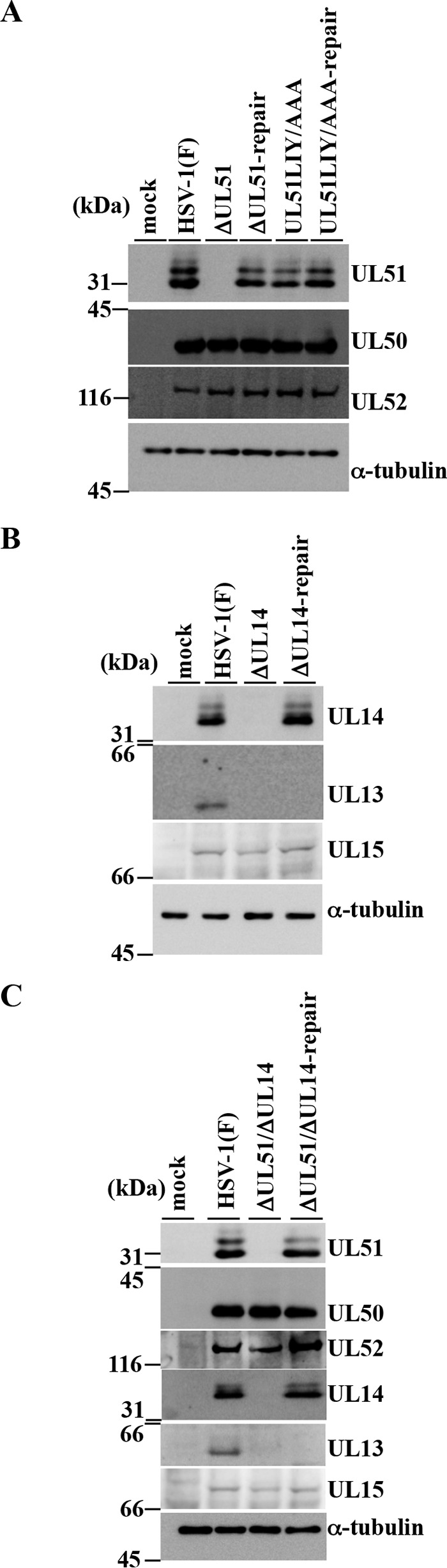
Effects of mutation(s) in UL51 and/or UL14 on expression of neighboring genes. (A) Vero cells were mock infected or infected with wild-type HSV-1(F), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), or YK5014 (UL51LIY/AAA-repair) at an MOI of 5 for 18 h and then analyzed by immunoblotting with the indicated antibodies. (B) Vero cells were mock infected or infected with wild-type HSV-1(F), YK5015 (ΔUL14), or YK5016 (ΔUL14-repair) at an MOI of 5 for 18 h and then analyzed by immunoblotting with the indicated antibodies. (C) Vero cells were mock infected or infected with wild-type HSV-1(F), YK5017 (ΔUL51/ΔUL14), or YK5018 (ΔUL51/ΔUL14-repair) (C) at an MOI of 5 for 18 h and then analyzed by immunoblotting with the indicated antibodies.
We next examined the effects of the mutation(s) in UL51 and/or UL14 on expression of their neighboring genes. The results were as follows.
First, Vero cells infected with wild-type HSV-1(F), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), or YK5014 (UL51LIY/AAA-repair) produced similar levels of UL50 and UL52 (Fig. 5A).
Second, Vero cells infected with wild-type HSV-1(F), YK5015 (ΔUL14), or YK5016 (ΔUL14-repair) produced similar levels of UL15 (Fig. 5B).
Third, UL13 was detected in Vero cells infected with wild-type HSV-1(F) but not in Vero cells infected with YK5015 (ΔUL14) or YK5016 (ΔUL14-repair) (Fig. 5B). It has been reported that wild-type HSV-1(F) is a mixture of at least two species: one carrying a UL13 gene that expresses a UL13 polypeptide containing 518 amino acids and one carrying a UL13 gene with one C deletion in a mononucleotide sequence of six Cs (50). This frameshift changes the UL13 amino acid sequence downstream of amino acid 118 and introduces a stop codon that truncates the protein at residue 166 (50). Sequence analysis showed that HSV-1(F)-BAC, pYEbac5002, which was cloned from wild-type HSV-1(F) and used for construction of the recombinant viruses in this study, has the same C deletion in UL13 described above (data not shown), indicating that pYEbac5002 was derived from a virus species with deletion in UL13 and that all recombinant viruses constructed in this study have the same deletion in UL13 (Fig. 1). The monoclonal anti-UL13 antibody used in this study was not able to recognize the truncated UL13 expressed by the recombinant viruses constructed for this study, because the anti-UL13 antibody was raised against the UL13 domain that was predicted to be truncated (40). Therefore, we were not able to investigate whether the UL14-null mutation had an effect on UL13 expression in cells infected with each of the recombinant viruses constructed in this study.
Fourth, Vero cells infected with wild-type HSV-1(F), YK5017 (ΔUL51/ΔUL14), or YK5018 (ΔUL51/ΔUL14-repair) produced similar levels of UL50, UL52, and UL15 (Fig. 5C). In agreement with the results described above (Fig. 5B), UL13 was not detected in Vero cells infected with YK5017 (ΔUL51/ΔUL14) or YK5018 (ΔUL51/ΔUL14-repair) (Fig. 5C).
Collectively, these results indicated that the mutations in UL51 and/or UL14 had no obvious effect on expression of their neighboring genes (i.e., UL50, UL52, and UL15), but the effects of the null mutation in UL14 on UL13 expression was not determined. We should note that, in our previous report (35), YK5002 reconstituted from pYEbac5002 was shown to retain wild-type HSV-1(F) levels of replication in Vero cells. Similarly, after infection of Vero cells with UL13-null mutant virus R7356 (ΔUL13) at an MOI of 5 or 0.01, at 18 or 48 h postinfection, respectively, progeny virus production was at levels similar to that of wild-type HSV-1(F) (data not shown). These observations suggested that full-length and truncated UL13 expressed by two HSV-1 virus species played no obvious role in viral replication in Vero cells.
Effect of the UL51 residues Leu-111, Ile-119, and Tyr-123 on interaction between UL51 and UL14 in HSV-1-infected cells.
To confirm whether UL51 residues Leu-111, Ile-119, and Tyr-123 are involved in UL51 interaction with UL14 in HSV-1-infected cells, Vero cells were infected with wild-type HSV-1(F), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), YK5011 (ΔUL51), or YK5015 (ΔUL14), lysed at 18 h postinfection, immunoprecipitated with anti-UL51 or anti-UL14 antibody, and immunoblotted with anti-UL14 and anti-UL51 antibodies. In agreement with the results shown in Fig. 3B, anti-UL51 antibody precipitated UL51 and UL14 from lysates of Vero cells infected with wild-type HSV-1(F) or YK5014 (UL51LIY/AAA-repair) (Fig. 6A). In contrast, although anti-UL51 antibody precipitated UL51 from lysates of cells infected with YK5013 (UL51LIY/AAA) as efficiently as from lysates of cells infected with wild-type HSV-1(F) or YK5014 (UL51LIY/AAA-repair), anti-UL51 antibody did not precipitate UL14 in lysates of cells infected with YK5013 (UL51LIY/AAA) (Fig. 6A). In a reciprocal experiment, anti-UL14 antibody precipitated UL14 and UL51 from lysates of cells infected with wild-type HSV-1(F) or YK5014 (UL51LIY/AAA-repair), in agreement with the results in Fig. 3C, but did not precipitate UL51LIY/AAA from lysates of cells infected with YK5013 (UL51LIY/AAA) (Fig. 6B). Anti-UL14 antibody precipitated UL14 from lysates of cells infected with YK5014 (UL51LIY/AAA-repair) as efficiently as from lysates of cells infected with wild-type HSV-1(F) or YK5013 (UL51LIY/AAA) (Fig. 6B). Anti-UL51 antibody did not precipitate UL51 or UL14 from lysates of cells infected with YK5011 (ΔUL51) and anti-UL14 did not precipitate UL51 or UL14 from lysates of cells infected with YK5015 (ΔUL14), although these infected cells produced UL14 and UL51 at levels comparable to those in cells infected with wild-type HSV-1(F) (Fig. 6). These results confirmed that anti-UL51 and anti-UL14 antibodies specifically precipitated UL14 or UL51, respectively, that were in a UL51-UL14 complex in lysates of wild-type HSV-1(F)-infected cells. Also, in agreement with the results of the GST pulldown experiments, UL51 residues Leu-111, Ile-119, and Tyr-123 were required for the interaction between UL51 and UL14 in HSV-1-infected cells.
FIG 6.
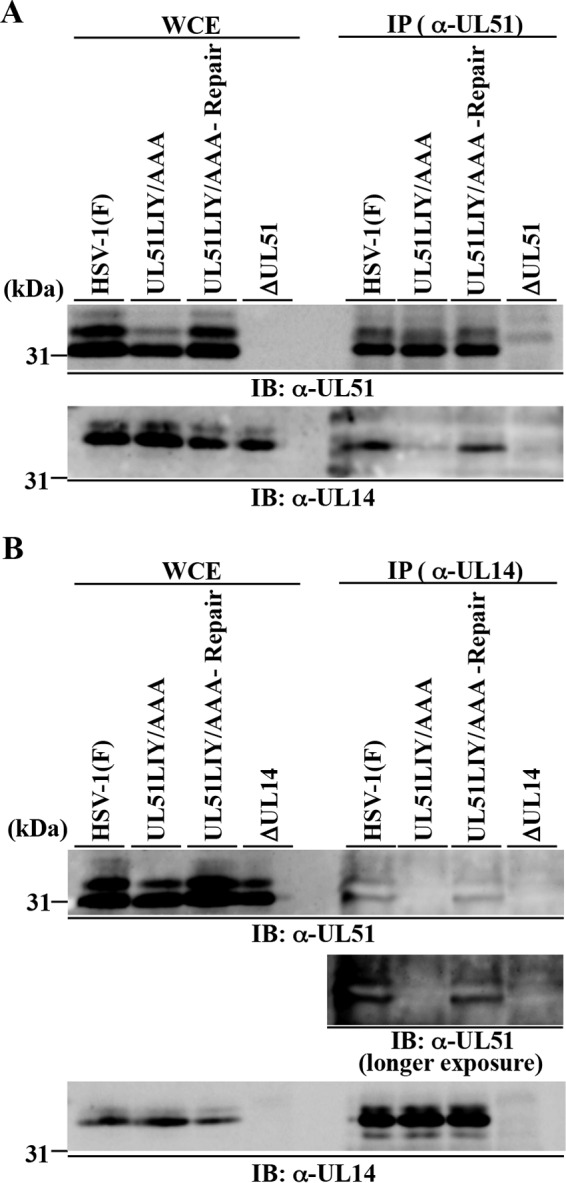
Effects of the UL51LIY/AAA mutations in UL51 on the interaction of UL51 with UL14 in HSV-1-infected cells. Vero cells were infected with wild-type HSV-1(F), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), YK5011 (ΔUL51), or YK5015 (ΔUL14) at an MOI of 5 for 18 h, harvested, immunoprecipitated with anti-UL51 (A) or anti-UL14 (B) antibody, and analyzed by immunoblotting with anti-UL51 and anti-UL14 antibodies.
Effects of mutations in UL51 and/or UL14 on their localization in HSV-1-infected cells.
To investigate the effects of the interaction between UL51 and UL14 on their localization in HSV-1-infected cells, we examined localization of UL51, UL14, and UL51LIY/AAA in Vero cells infected with wild-type HSV-1(F), YK5015 (ΔUL14), YK5016 (ΔUL14-repair), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), or YK5014 (UL51LIY/AAA-repair) at an MOI of 5 for 18 h by confocal microscopy. As shown above (Fig. 3D), UL51 and UL14 were predominantly localized at juxtanuclear domains in cells infected with wild-type HSV-1(F), YK5016 (ΔUL14-repair), YK5012 (ΔUL51-repair), or YK5014 (UL51LIY/AAA-repair) (Fig. 7A). In cells infected with YK5015 (ΔUL14), YK5011 (ΔUL51), or YK5013 (UL51LIY/AAA), UL51 and UL14 were difficult to detect by confocal microscopy (Fig. 7A), although these proteins were easily detected by immunoblotting (Fig. 5 and 6). However, a closer inspection showed that, in cells infected with YK5015 (ΔUL14) or YK5011 (ΔUL51), UL51 and UL14 were dispersed throughout the cytoplasm, and the UL14 and UL51 aggregates in the juxtanuclear domains observed in wild-type HSV-1(F)-infected cells were not detected (Fig. 7A). In cells infected with YK5013 (UL51LIY/AAA), the localization patterns of UL51LIY/AAA and UL14 were similar to those of UL51 and UL14 in cells infected with YK5015 (ΔUL14) or YK5011 (ΔUL51), respectively (Fig. 7A). The UL14-null, UL51-null, and UL51LIY/AAA mutations had no effect on the localization of UL11, another HSV-1 tegument protein, in infected Vero cells (Fig. 7B). In addition, the UL13-null mutation had no effect on localization of UL51 and UL14 in HSV-1-infected cells (Fig. 7A). These results indicated that UL51 and UL14 were required for proper localization of each other, and that UL51 residues Leu-111, Ile-119, and Tyr-123 were necessary for both UL51 and UL14 to regulate proper localization of each other in HSV-1-infected cells.
FIG 7.
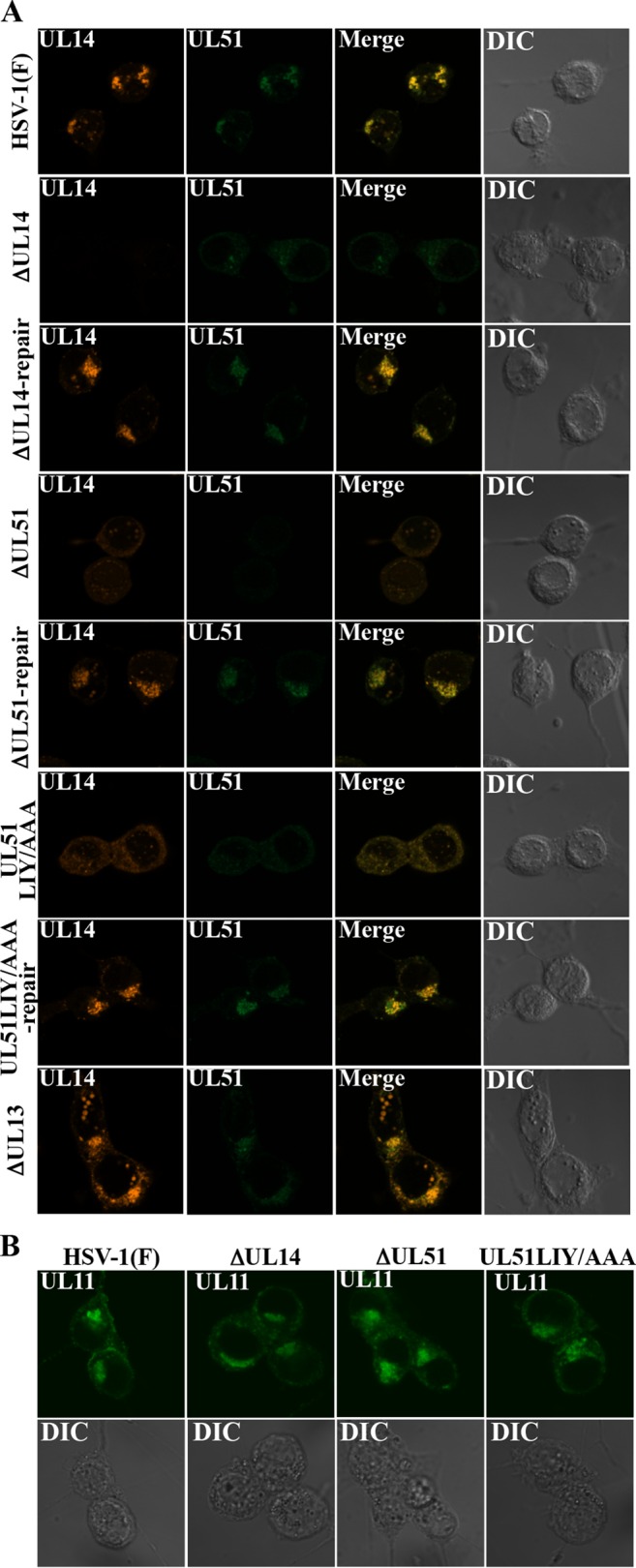
Effects of mutation(s) in UL51 and/or UL14 on localization of UL51 and UL14 in HSV-1-infected cells. Vero cells were infected with wild-type HSV-1(F), YK5015 (ΔUL14), YK5016 (ΔUL14-repair), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), or R7356 (ΔUL13) at an MOI of 5, fixed at 18 h postinfection, permeabilized, stained with anti-UL51 and anti-UL14 (A) or anti-UL11 (B) antibody, and examined by confocal microscopy.
Effects of UL51, UL14, and UL51 residues Leu-111, Ile-119, and Tyr-123 on HSV-1 replication.
In earlier reports, the effects of UL51 and UL14 on HSV-1 replication were investigated using different HSV-1 strains (16, 25). To clarify the role of the interaction between UL51 and UL14 in HSV-1 replication in infected cells, we simultaneously examined the effect of the UL14-null, UL51-null, and UL51LIY/AAA mutations on progeny virus yields in Vero cells infected with wild-type HSV-1(F), YK5015 (ΔUL14), YK5016 (ΔUL14-repair), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), YK5017 (ΔUL51/ΔUL14), or YK5018 (ΔUL51/ΔUL14-repair) at an MOI of 5 or 0.01 for 18 h or 48 h, respectively. The progeny virus yields in cells infected with YK5015 (ΔUL14), YK5011 (ΔUL51), YK5013 (UL51LIY/AAA), or YK5017 (ΔUL51/ΔUL14) at an MOI of 5 (Fig. 8A) or 0.01 (Fig. 8B) were markedly reduced compared to the yields in cells infected with wild-type HSV-1(F) or the corresponding repaired viruses. In particular, all of these mutations in UL51 and/or UL14 reduced progeny virus yields at comparable levels (Fig. 8A and B).
FIG 8.
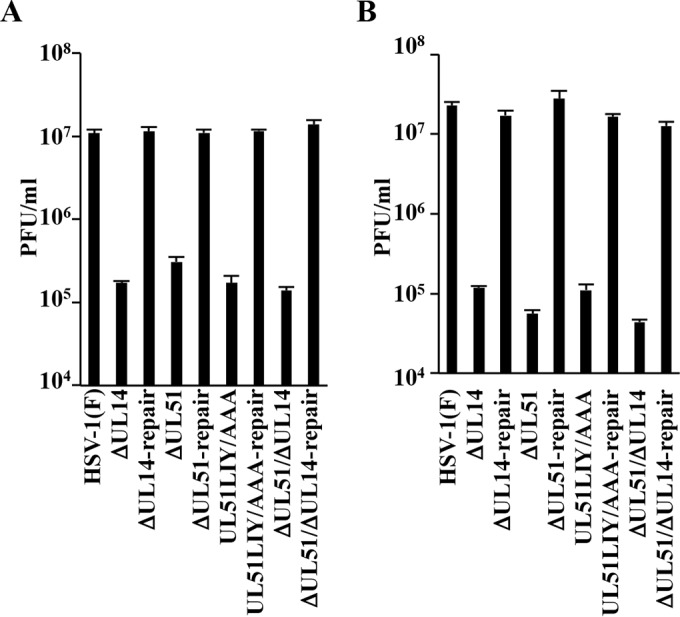
Effects of mutation(s) in UL51 and/or UL14 on progeny virus yields. (A and B) Vero cells were infected with HSV-1(F), YK5015 (ΔUL14), YK5016 (ΔUL14-repair), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), YK5017 (ΔUL51/ΔUL14), or YK5018 (ΔUL51/ΔUL14-repair) at an MOI of 5 (A) or 0.01 (B). Total virus from cell culture supernatants and infected cells was harvested at 18 h (A) or 48 h (B) postinfection and assayed on Vero cells. Each data point is the mean ± standard error from the results for triplicate samples. Differences in viral yields between HSV-1(F) and each of the mutant viruses were statistically significant by ANOVA and Dunnett's test (P < 0.05). Differences in viral yields between HSV-1(F) and each of the repaired viruses were not statistically significant by ANOVA and Dunnett's test. Data are representative of three independent experiments.
These results indicated that UL51, UL14, and UL51 residues Leu-111, Ile-119, and Tyr-123 were required for efficient HSV-1 replication in Vero cells. In particular, the level of the effects of these factors on HSV-1 replication in Vero cells appeared to be comparable; therefore, UL51 and UL14 showed no synergistic effect.
Effects of mutations in UL51 and/or UL14 and UL51 residues Leu-111, Ile-119, and Tyr-123 on HSV-1 morphogenesis.
To determine the step(s) at which UL51, UL14, and their interaction act during HSV-1 replication, we investigated viral morphogenesis by quantitating the number of virus particles at different morphogenetic stages by electron microscopy of Vero cells infected with wild-type HSV-1(F), YK5015 (ΔUL14), YK5016 (ΔUL14-repair), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), YK5017 (ΔUL51/ΔUL14), YK5018 (ΔUL51/ΔUL14-repair), or R7356 (ΔUL13) at an MOI of 5 for 18 h. As shown in Fig. 9 and Table 2, in cells infected with YK5015 (ΔUL14), YK5011 (ΔUL51), YK5013 (UL51LIY/AAA), or YK5017 (ΔUL51/ΔUL14), 11 to 12% of virus particles were partially enveloped nucleocapsids in the cytoplasm. However, only 2 to 4% of virus particles were partially enveloped nucleocapsids in the cytoplasm of cells infected with wild-type HSV-1(F) or the corresponding repaired viruses. In cells infected with wild-type HSV-1(F), 9.3% of virus particles were unenveloped or partially enveloped nucleocapsids in the cytoplasm, but in cells infected with YK5015 (ΔUL14), YK5011 (ΔUL51), YK5013 (UL51LIY/AAA), or YK5017 (ΔUL51/ΔUL14), 38 to 44% of virus particles were unenveloped or partially enveloped nucleocapsids in the cytoplasm, which was 4 to 5 times more than in cells infected with wild-type HSV-1(F) (Table 2). Wild-type levels of unenveloped and partially enveloped nucleocapsids in the cytoplasm were restored in cells infected with each of the corresponding repaired viruses (Table 2). In contrast, in cells infected with wild-type HSV-1(F), 40% of virus particles were enveloped virions in the cytoplasm and extracellular space, but in cells infected with YK5015 (ΔUL14), YK5011 (ΔUL51), YK5013 (UL51LIY/AAA), or YK5017 (ΔUL51/ΔUL14), the percentage of virus particles that were enveloped virions in the cytoplasm and extracellular space decreased to 9 to 15%, which was 2.6- to 4.4-fold less than that in cells infected with wild-type HSV-1(F) (Table 2). Wild-type levels of enveloped virions in the cytoplasm and extracellular space were restored in cells infected with the corresponding repaired viruses (Table 2). The fraction of total virus particles in the nucleus and perinuclear space in cells infected with wild-type HSV-1(F), YK5015 (ΔUL14), YK5011 (ΔUL51), YK5013 (UL51LIY/AAA), or YK5017 (ΔUL51/ΔUL14) was comparable (i.e., 43 to 51%) (Table 2). In addition, these data confirmed that the UL13-null mutation had no effect on virion morphogenesis in Vero cells (Fig. 9 and Table 2). These results indicated that the mutations in UL51 and/or UL14 induced comparable levels of aberrant accumulation of unenveloped capsids and partially enveloped nucleocapsids in the cytoplasm.
FIG 9.
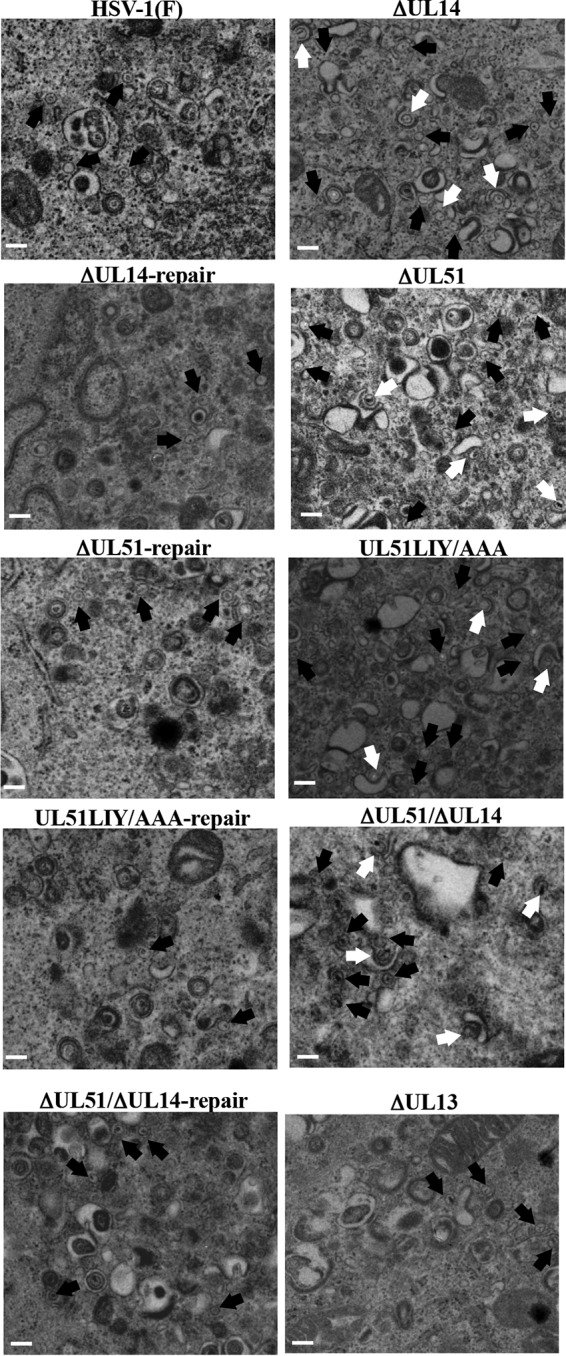
Effects of mutation(s) in UL51 and/or UL14 on HSV-1 secondary envelopment. Vero cells were infected with wild-type HSV-1(F), YK5015 (ΔUL14), YK5016 (ΔUL14-repair), YK5011 (ΔUL51), YK5012 (ΔUL51-repair), YK5013 (UL51LIY/AAA), YK5014 (UL51LIY/AAA-repair), YK5017 (ΔUL51/ΔUL14), YK5018 (ΔUL51/ΔUL14-repair), or R7356 (ΔUL13) at an MOI of 5, fixed at 18 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. Black arrows indicate unenveloped capsids. White arrows indicate partially enveloped capsids. Scale bar, 200 nm.
TABLE 2.
Effects of the mutations in UL51 and/or UL14 on the distribution of viral particles in HSV-1-infected Vero cells
| Virus | % of virus particles in morphogenetic stage (no. of particles in stage) |
Total no. counted (particles/cells) | |||||
|---|---|---|---|---|---|---|---|
| Capsids in nucleus | Enveloped virions in perinuclear space | Unenveloped capsids in cytoplasm | Partially enveloped capsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 44.0 (521) | 6.8 (81) | 5.2 (62) | 4.1 (49) | 15.8 (187) | 24.1 (285) | 1,185/8 |
| ΔUL14 | 38.4 (308) | 4.6 (37) | 29.8 (239) | 11.8 (95) | 8.3 (67) | 7.1 (57) | 803/8 |
| ΔUL14-repair | 39.8 (449) | 4.5 (51) | 11.0 (124) | 3.5 (39) | 21.3 (240) | 19.9 (224) | 1,127/8 |
| ΔUL51 | 38.0 (353) | 7.3 (68) | 30.0 (278) | 12.1 (112) | 4.2 (39) | 8.4 (78) | 928/8 |
| ΔUL51-repair | 42.9 (438) | 7.1 (73) | 8.9 (91) | 1.7 (17) | 17.1 (175) | 22.2 (227) | 1,021/8 |
| UL51LIY/AAA | 45.8 (352) | 3.0 (23) | 26.3 (202) | 12.0 (92) | 5.5 (42) | 7.5 (58) | 769/8 |
| UL51LIY/AAA-repair | 36.9 (404) | 9.1 (100) | 10.0 (109) | 3.7 (41) | 18.4 (202) | 21.8 (239) | 1,095/8 |
| ΔUL51/ΔUL14 | 43.2 (328) | 3.6 (27) | 32.8 (249) | 11.3 (86) | 3.6 (27) | 5.5 (42) | 759/8 |
| ΔUL51/ΔUL14-repair | 39.8 (382) | 5.3 (51) | 8.3 (80) | 1.7 (16) | 18.2 (175) | 26.6 (255) | 959/8 |
| ΔUL13 | 48.1 (494) | 5.4 (55) | 5.2 (53) | 3.1 (32) | 17.7 (182) | 20.5 (210) | 1,026/8 |
DISCUSSION
Affinity purification of tagged UL51 from HSV-1-infected Vero cells coupled with immunoblotting of the purified UL51 complexes with antibodies to various HSV-1 virion proteins identified a putative interaction between tegument proteins UL51 and UL14 in HSV-1-infected cells. This interaction was confirmed by reciprocal coimmunoprecipitation and immunofluorescence microscopy studies of wild-type HSV-1(F)-infected cells and by GST pulldown experiments. Further GST pulldown and coimmunoprecipitation experiments using UL51 mutants identified Leu-111, Ile-119, and Tyr-123 in putative UL51 α-helix VI as the residues required for UL51 interaction with UL14 in HSV-1-infected cells. These results indicated that UL51 binding to UL14 in HSV-1-infected cells involved UL51 residues Leu-111, Ile-119, and Tyr-123. However, the mutation analyses in this study could not eliminate the possibility that UL51 residues Leu-111, Ile-119, and Tyr-123 were not the UL51 binding sites for UL14 but that the UL51LIY/AAA mutation caused global misfolding of UL51 that masked an unidentified binding site(s) for UL14, so that the UL51LIY/AAA mutant was not able to interact with UL14.
We have shown here that the UL51-null mutation reduced viral replication and affected virion morphogenesis in the cytoplasm of infected cells at levels comparable to those of the UL14-null mutation and the UL51/UL14 double null mutation. The phenotypes in virion morphogenesis, including aberrant accumulation of unenveloped and partially enveloped virions and the lack of enveloped virions in the cytoplasm and extracellular space, have been suggested to reflect a block in a process(es) for viral secondary envelopment (27, 28, 51). The results in this study indicated that UL51 and UL14 acted in the same HSV-1 secondary envelopment pathway during HSV-1 replication. In addition, we presented data that the UL51LIY/AAA mutation, which was shown to prevent the interaction of UL51 with UL14, exhibited a phenotype identical to that observed with the UL51-null and UL14-null mutations, and the effect of this phenotype was comparable to the null mutations in UL51 and UL14. Taken together, all of these results suggested that UL51 and UL14 formed a complex in HSV-1-infected cells, and this complex regulated viral secondary envelopment for efficient viral replication. The UL51-UL14 complex likely anchors the cytoplasmic membrane and interacts with viral envelope glycoprotein gE and capsid proteins VP26 and VP19C (25, 52, 53); therefore, the UL51-UL14 complex may facilitate viral secondary envelopment by bridging the nucleocapsid and the cytoplasmic membrane via these interactions. Since this conclusion was based on experiments with the UL51LIY/AAA mutation, we cannot eliminate the possibility that UL14 regulated viral secondary envelopment upstream or downstream of UL51 in the same pathway and/or that the phenotype observed with the UL51LIY/AAA mutation was due to global misfolding of UL51 caused by the amino acid substitutions that may have impaired the function of UL51 rather than preventing UL51 interaction with UL14.
It has been reported that interaction between UL36 and UL37 was required for HSV-1 secondary envelopment (20), like the interaction between UL51 and UL14 shown in this study. Thus far, the mechanism by which the UL36-UL37 complex acts in HSV-1 secondary envelopment has not been directly addressed. However, it is likely that the UL36-UL37 complex does not regulate viral secondary envelopment itself but promotes nucleocapsid transport to sites in the cytoplasm for HSV-1 secondary envelopment. This proposal is based on previous reports that: (i) cytoplasmic nucleocapsids were transported along microtubules to the sites for viral secondary envelopment (54, 55), (ii) the UL36-UL37 complex appeared to be a bridge between nucleocapsids and microtubule motors (56, 57), (iii) nucleocapsids of either the UL36-null or UL37-null mutant virus showed reduced ability to be transported in the cytoplasm (54, 55), and (iv) nucleocapsids of a UL36-null mutant virus, a UL37-null mutant virus, or a UL36 mutant virus with the deletion of a minimal domain required for UL37 binding did not associate with cytoplasmic membranes (11, 12, 20). In this study, we showed that mutations in UL51 and/or UL14 accumulated not only unenveloped nucleocapsids, as observed with the mutations in UL36 or UL37 (11, 12), but also partially enveloped nucleocapsids that appeared to be arrested at the membrane deformation stage during HSV-1 cytoplasmic budding. This was not observed with the mutations in UL36 and UL37 (11, 12). Therefore, unlike the UL36-UL37 complex, the UL51-UL14 complex appeared to directly regulate viral secondary envelopment at cytoplasmic membranes.
In this study, we found that UL14 became difficult to detect by confocal microscopy in the absence of UL51 in HSV-1-infected cells, and UL51 became difficult to detect in the absence of UL14 in HSV-1-infected cells, although these proteins were easily detected by immunoblotting. Similarly, the LIY/AAA mutation in UL51 made it difficult to detect both UL51 and UL14 in HSV-1-infected cells by confocal microscopy. These results indicated that UL51 and UL14 were required for proper antigenic properties of UL14 and UL51, respectively, and the UL51 residues Leu-111, Ile-119, and Tyr-123 were required for proper antigenic properties of both UL51 and UL14 in HSV-1-infected cells. Taking these results together, the interaction between UL51 and UL14 appeared to affect the folding properties of these viral proteins in HSV-1-infected cells. In addition, the UL14-null mutation and the UL51LIY/AAA mutation appeared to reduce the solubility of the slower migrating form of UL51 (Fig. 6). Collectively, these results suggested that the interaction between UL51 and UL14 was required for proper folding of these viral proteins in HSV-1-infected cells. In agreement with this possibility, it has been reported that interaction between HSV-1 envelope glycoproteins gH and gL was required for proper folding, posttranslational processing, and localization of gH (58).
In conclusion, we have analyzed the interaction between HSV-1 tegument proteins UL51 and UL14, which appeared to be important for viral secondary envelopment. This is the first report showing that an interaction between HSV-1 virion proteins directly regulated viral secondary envelopment. As described above, HSV-1 UL51 and UL14 are conserved throughout the Herpesviridae family (1), raising the interesting possibility that complexes of UL51 and UL14 homologues in other herpesviruses regulate viral secondary envelopment. In support of this possibility, it has been reported that UL51 homologs in alphaherpesvirus PRV and betaherpesvirus HCMV are involved in viral secondary envelopment (27, 28). However, the HSV-1 UL51 domain and amino acid residues required for interaction with UL14 are conserved in alphaherpesviruses but not in beta- and gammaherpesviruses (Fig. 10). Further studies will be needed to clarify whether interactions between herpesvirus UL51 and UL14 homologues regulate viral secondary envelopment, like the interaction between UL34 and UL31 homologues in viral primary envelopment (3, 4).
FIG 10.

Sequence alignment of the UL51 homologs in HSV-1 helix VI in members of the three herpesvirus subfamilies. HSV-2, herpes simplex virus 2; VZV, varicella-zoster virus; MCMV, murine cytomegalovirus; EBV, Epstein-Barr virus; KSHV, Kaposi's sarcoma-associated herpesvirus. HSV-1, HSV-2, VZV, and PRV are in the subfamily Alphaherpesvirinae, HCMV and MCMV are in the Betaherpesvirinae, and EBV and KSHV are in the Gammaherpesvirinae. The residues conserved in at least two herpesviruses are in light, medium, or dark shading. The HSV-1 residues required for interaction with UL14 and the corresponding residues of other herpesviruses are boxed.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Sachi Matsumoto for excellent technical assistance and Bernard Roizman and Joel Baines for providing us the UL13-null mutant virus R7356 and the rabbit polyclonal antibody to gM, respectively.
Funding for this work came from the following sources: Funding Program for Next Generation World-Leading Researchers from the Japan Society for the Promotion of Science (JSPS) to Y.K., grants for Scientific Research from JSPS to Y.K., A.K., and J.A., grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan (16H06433, 16H06429, and 16K21723) to Y.K., a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from MEXT and the Japan Agency for Medical Research and Development (AMED) to Y.K., grants from the Takeda Science Foundation to Y.K., A.K., and J.A., and a grant from the Mitsubishi Foundation to Y.K.
Funding Statement
This work, including the efforts of Yasushi Kawaguchi, was funded by Ministry of Education, Culture, Sports, Science, and Technology (MEXT) and Japan Agency for Medical Research and Development (AMED) (contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases [J-GRID]). This work, including the efforts of Yasushi Kawaguchi, was funded by Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (Grants for Scientific Research on Innovative Areas (16H06433 16H06429 16K21723)). This work, including the efforts of Yasushi Kawaguchi, Jun Arii, and Akihisa Kato, was funded by Japan Society for the Promotion of Science (JSPS) (Grants for Scientific Research). This work, including the efforts of Yasushi Kawaguchi, was funded by Japan Society for the Promotion of Science (JSPS) (funding program for Next-Generation World-Leading Researchers). This work, including the efforts of Jun Arii, Akihisa Kato, and Yasushi Kawaguchi, was funded by Takeda Science Foundation. This work, including the efforts of Yasushi Kawaguchi, was funded by Mitsubishi Foundation.
REFERENCES
- 1.Pellett PE, Roizman B. 2013. Herpesviridae, p 1802–1822. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 4.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 5.Owen DJ, Crump CM, Graham SC. 2015. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naldinho-Souto R, Browne H, Minson T. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J Virol 80:2582–2584. doi: 10.1128/JVI.80.5.2582-2584.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padula ME, Sydnor ML, Wilson DW. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J Virol 83:4757–4765. doi: 10.1128/JVI.01927-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baines JD, Jacob RJ, Simmerman L, Roizman B. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J Virol 69:825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74:11608–11618. doi: 10.1128/JVI.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J Virol 75:10259–10271. doi: 10.1128/JVI.75.21.10259-10271.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mossman KL, Sherburne R, Lavery C, Duncan J, Smiley JR. 2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J Virol 74:6287–6299. doi: 10.1128/JVI.74.14.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baines JD, Roizman B. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J Virol 66:5168–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. 2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88:110–119. doi: 10.1128/JVI.02555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham C, Davison AJ, MacLean AR, Taus NS, Baines JD. 2000. Herpes simplex virus type 1 gene UL14: phenotype of a null mutant and identification of the encoded protein. J Virol 74:33–41. doi: 10.1128/JVI.74.1.33-41.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szilagyi JF, Cunningham C. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J Gen Virol 72(Part 3):661–668. doi: 10.1099/0022-1317-72-3-661. [DOI] [PubMed] [Google Scholar]
- 19.Rixon FJ, Addison C, McLauchlan J. 1992. Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J Gen Virol 73(Part 2):277–284. doi: 10.1099/0022-1317-73-2-277. [DOI] [PubMed] [Google Scholar]
- 20.Kelly BJ, Bauerfeind R, Binz A, Sodeik B, Laimbacher AS, Fraefel C, Diefenbach RJ. 2014. The interaction of the HSV-1 tegument proteins pUL36 and pUL37 is essential for secondary envelopment during viral egress. Virology 454-455:67–77. [DOI] [PubMed] [Google Scholar]
- 21.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol 79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roller RJ, Haugo AC, Yang K, Baines JD. 2014. The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J Virol 88:4058–4068. doi: 10.1128/JVI.03707-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nozawa N, Daikoku T, Koshizuka T, Yamauchi Y, Yoshikawa T, Nishiyama Y. 2003. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J Virol 77:3204–3216. doi: 10.1128/JVI.77.5.3204-3216.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klupp BG, Granzow H, Klopfleisch R, Fuchs W, Kopp M, Lenk M, Mettenleiter TC. 2005. Functional analysis of the pseudorabies virus UL51 protein. J Virol 79:3831–3840. doi: 10.1128/JVI.79.6.3831-3840.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, von Einem J. 2011. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol 85:3821–3832. doi: 10.1128/JVI.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roller RJ, Fetters R. 2015. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J Virol 89:3112–3122. doi: 10.1128/JVI.02799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuchs W, Granzow H, Klopfleisch R, Klupp BG, Rosenkranz D, Mettenleiter TC. 2005. The UL7 gene of pseudorabies virus encodes a nonessential structural protein which is involved in virion formation and egress. J Virol 79:11291–11299. doi: 10.1128/JVI.79.17.11291-11299.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahlqvist J, Mocarski E. 2011. Cytomegalovirus UL103 controls virion and dense body egress. J Virol 85:5125–5135. doi: 10.1128/JVI.01682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawaguchi Y, Kato K, Tanaka M, Kanamori M, Nishiyama Y, Yamanashi Y. 2003. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J Virol 77:2359–2368. doi: 10.1128/JVI.77.4.2359-2368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purves FC, Ogle WO, Roizman B. 1993. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc Natl Acad Sci U S A 90:6701–6705. doi: 10.1073/pnas.90.14.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato A, Ando T, Oda S, Watanabe M, Koyanagi N, Arii J, Kawaguchi Y. 2016. Roles of Us8A and its phosphorylation mediated by Us3 in herpes simplex virus 1 pathogenesis. J Virol 90:5622–5635. doi: 10.1128/JVI.00446-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J Virol 71:1019–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88:655–666. doi: 10.1128/JVI.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujii H, Mugitani M, Koyanagi N, Liu Z, Tsuda S, Arii J, Kato A, Kawaguchi Y. 2014. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J Virol 88:2359–2364. doi: 10.1128/JVI.03621-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka M, Kato A, Satoh Y, Ide T, Sagou K, Kimura K, Hasegawa H, Kawaguchi Y. 2012. Herpes simplex virus 1 VP22 regulates translocation of multiple viral and cellular proteins and promotes neurovirulence. J Virol 86:5264–5277. doi: 10.1128/JVI.06913-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamauchi Y, Wada K, Goshima F, Takakuwa H, Daikoku T, Yamada M, Nishiyama Y. 2001. The UL14 protein of herpes simplex virus type 2 translocates the minor capsid protein VP26 and the DNA cleavage and packaging UL33 protein into the nucleus of coexpressing cells. J Gen Virol 82:321–330. doi: 10.1099/0022-1317-82-2-321. [DOI] [PubMed] [Google Scholar]
- 43.Baines JD, Wills E, Jacob RJ, Pennington J, Roizman B. 2007. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J Virol 81:800–812. doi: 10.1128/JVI.01756-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirohata Y, Arii J, Liu Z, Shindo K, Oyama M, Kozuka-Hata H, Sagara H, Kato A, Kawaguchi Y. 2015. Herpes simplex virus 1 recruits CD98 heavy chain and beta1 integrin to the nuclear membrane for viral de-envelopment. J Virol 89:7799–7812. doi: 10.1128/JVI.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J Virol 83:5773–5783. doi: 10.1128/JVI.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J Virol 81:319–331. doi: 10.1128/JVI.01842-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frankel AD, Young JA. 1998. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem 67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Szpara ML, Parsons L, Enquist LW. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol 84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson DC, Wisner TW, Wright CC. 2011. Herpes simplex virus glycoproteins gB and gD function in a redundant fashion to promote secondary envelopment. J Virol 85:4910–4926. doi: 10.1128/JVI.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 53.Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, Schmidt T, Kraus T, Stellberger T, Rutenberg C, Suthram S, Bandyopadhyay S, Rose D, von Brunn A, Uhlmann M, Zeretzke C, Dong YA, Boulet H, Koegl M, Bailer SM, Koszinowski U, Ideker T, Uetz P, Zimmer R, Haas J. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog 5:e1000570. doi: 10.1371/journal.ppat.1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandbaumhuter M, Dohner K, Schipke J, Binz A, Pohlmann A, Sodeik B, Bauerfeind R. 2013. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell Microbiol 15:248–269. doi: 10.1111/cmi.12075. [DOI] [PubMed] [Google Scholar]
- 55.Pasdeloup D, McElwee M, Beilstein F, Labetoulle M, Rixon FJ. 2013. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J Virol 87:2857–2867. doi: 10.1128/JVI.02676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6:e1000991. doi: 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]