

ABSTRACT
Aminoquinolines and piperazines, linked or not, have been used successfully to treat malaria, and some molecules of this family also exhibit antiviral properties. Here we tested several derivatives of 4-aminoquinolines and piperazines for their activity against hepatitis C virus (HCV). We screened 11 molecules from three different families of compounds, and we identified anti-HCV activity in cell culture for six of them. Of these, we selected a compound (B5) that is currently ending clinical phase I evaluation for neurodegenerative diseases. In hepatoma cells, B5 inhibited HCV infection in a pangenotypic and dose-dependent manner, and its antiviral activity was confirmed in primary hepatocytes. B5 also inhibited infection by pseudoparticles expressing HCV envelope glycoproteins E1 and E2, and we demonstrated that it affects a postattachment stage of the entry step. Virus with resistance to B5 was selected by sequential passage in the presence of the drug, and reverse genetics experiments indicated that resistance was conferred mainly by a single mutation in the putative fusion peptide of E1 envelope glycoprotein (F291I). Furthermore, analyses of the effects of other closely related compounds on the B5-resistant mutant suggest that B5 shares a mode of action with other 4-aminoquinoline-based molecules. Finally, mice with humanized liver that were treated with B5 showed a delay in the kinetics of the viral infection. In conclusion, B5 is a novel interesting anti-HCV molecule that could be used to decipher the early steps of the HCV life cycle.
IMPORTANCE In the last 4 years, HCV therapy has been profoundly improved with the approval of direct-acting antivirals in clinical practice. Nevertheless, the high costs of these drugs limit access to therapy in most countries. The present study reports the identification and characterization of a compound (B5) that inhibits HCV propagation in cell culture and is currently ending clinical phase I evaluation for neurodegenerative diseases. This molecule inhibits the HCV life cycle by blocking virus entry. Interestingly, after selection of drug-resistant virus, a resistance mutation in the putative fusion peptide of E1 envelope glycoprotein was identified, indicating that B5 could be used to further investigate the fusion mechanism. Furthermore, mice with humanized liver treated with B5 showed a delay in the kinetics of the viral infection. In conclusion, B5 is a novel interesting anti-HCV molecule that could be used to decipher the early steps of the HCV life cycle.
INTRODUCTION
Hepatitis C virus (HCV) infection remains a global epidemic. More than 185 million individuals are seropositive for HCV, and the large majority of them suffer from chronic hepatitis C, with increased risk of developing severe, and if untreated fatal, liver disease (1). This virus has a high propensity for establishing a chronic infection that can lead to liver fibrosis, cirrhosis, and hepatocellular carcinoma (2). Until recently, the standard anti-HCV treatment was based on a combination therapy using pegylated interferon alpha plus ribavirin (3). However, such treatment had significant side effects and variable efficacy depending on the viral genotype. In the last 4 years, HCV therapy has been profoundly improved with the approval of direct-acting antivirals in clinical practice. These new treatments lead to a higher sustained virological response rate with significant reduction of the treatment period and no major adverse effects (4). Nevertheless, several challenges remain: high costs limit access to therapy, and certain subgroups of difficult-to-treat patients may need adjunctive therapeutic approaches (5, 6). Furthermore, there is no vaccine available against this major pathogen.
HCV is a small enveloped virus with a positive-stranded RNA genome belonging to the Hepacivirus genus in the Flaviviridae family (7). The HCV genome encodes a single polyprotein that is processed by cellular and viral proteases to produce 10 polypeptides, which include three structural proteins, namely, core (capsid protein) and E1 and E2 (envelope glycoproteins), and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (8). HCV envelope glycoproteins are incorporated into viral particles and are the major viral determinants of HCV entry (9). The precise role of HCV envelope glycoproteins in virus entry remains poorly understood. However, recent crystallographic data suggest that E1, alone or in association with E2, is the fusion protein (10, 11). The major cell type supporting HCV replication is the hepatocyte, and the virus enters these cells using a multistep process dependent on four essential cellular factors: tetraspanin CD81, scavenger receptor class B member I (SR-B1), and tight-junction proteins claudin 1 and occludin (12). After its interaction with entry factors at the cell surface, the HCV particle is internalized by clathrin-mediated endocytosis (13), and fusion between the HCV envelope and a target cell membrane is thought to occur in early endosomes (14).
Recently, we identified ferroquine (FQ), a ferrocenic analog of chloroquine (CQ) (15) as a new inhibitor of HCV entry. Furthermore, CQ has also been shown to inhibit several steps of the HCV life cycle (13, 16, 17). We were therefore interested to test other derivatives of 4-aminoquinoline-based molecules for their anti-HCV activity. Here, we tested a series of molecules from three different families of antimalarial compounds (18–20), and we observed an anti-HCV activity for several of them. Based on its current development for another medical condition, we selected one of them (B5) for further characterization of its anti-HCV activity. Our data show that B5 inhibits HCV infection at a half-maximal inhibitory concentration (IC50) of close to 1 μM. This compound blocks HCV at a postattachment stage of the entry step. Furthermore, chimeric mice with humanized liver treated with B5 showed a delay in the kinetics of the viral infection.
MATERIALS AND METHODS
Chemicals.
CQ and amodiaquine-derived compounds were synthesized as previously described (18–22). The level of purity of the compounds was higher than 98%. CQ, alpha interferon (IFN-α), heparin, brefeldin A, and quinidine were purchased from Sigma-Aldrich. B5 was prepared as 1 mM stock solutions in phosphate-buffered saline (PBS). CQ was prepared as 1 mM stock solutions in water. Boceprevir and FQ were kindly provided by Philippe Halfon (Hôpital Ambroise Paré, Marseille, France) and Christophe Biot (Université de Lille), respectively.
Cell culture.
Huh-7 cells (23), HEK-293T cells (ATCC CRL-11268), and RFP-NLS-IPS-Huh7 cells (24) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS). Primary human hepatocytes were cultured as previously described (25). Drug toxicity on Huh-7 cells and primary hepatocytes was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay and the CytoTox 96 nonradioactive cytotoxicity assay, respectively, following the recommendations of the manufacturer (Promega).
Antibodies.
Monoclonal antibodies (MAbs) anti-HCV E1 glycoprotein (A4) (26), anti-HCV E2 glycoprotein (3/11; kindly provided by J. McKeating, University of Birmingham, United Kingdom) (27), anti-NS5A antibody (Austral Biologicals), anti-CD81 MAb JS-81 (BD Pharmingen), and anti-GFP (Roche) were used in this study. Cy3-, Alexa 488-, and phycoerythrin (PE)-conjugated secondary antibodies were from Jackson ImmunoResearch, Invitrogen, and BD Pharmingen, respectively.
Viruses.
To produce cell-cultured HCV (HCVcc), we used a modified version of the plasmid carrying the JFH1 genome (genotype 2a; GenBank accession number AB237837), kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) (28). Briefly, HCVcc was produced in Huh-7 cells electroporated with in vitro-transcribed RNA of JFH1 engineered to express the A4 epitope (29) and to bear titer-enhancing mutations (30). JFH1 stocks were produced by further amplification in Huh-7 cells. The JFH1-ΔE1E2-Luc plasmid, containing an in-frame deletion in the E1E2 region, and the JFH1-GND-Luc replication-defective mutant have been described previously (29, 31). Both constructs contain the Renilla luciferase gene fused with the viral open reading frame in a monocistronic configuration. The inhibitory effect of the drugs was determined by quantifying infectivity by indirect immunofluorescence with the anti-E1 MAb A4 (26) or by measuring viral titers with the anti-E1 MAb A4 or the anti-NS5A antibody. For quantitative binding experiments, purified virus was obtained by precipitation of supernatants of HCVcc-infected Huh-7 cells with 8% polyethylene glycol 6000. Pelleted virus was then loaded onto a continuous 10 to 40% iodixanol gradient. One-milliliter fractions were collected, and the most infectious fractions were pooled. The titer of the resulting stock was 5 × 106 focus-forming units (FFU)/ml. The intergenotypic HCVcc chimeras GT1b(Con1)/JFH1 (kindly provided by R. Bartenschlager, University of Heidelberg, Germany) and GT3a(S52)/JFH1, GT4a(ED43)/JFH1, GT5a(SA13)/JFH1, GT6a(HK6a)/JFH1 (kindly provided by J. Bukh, University of Copenhagen, Denmark) (32–35), were also used in some experiments. HCV-pseudotyped retroviral particles (HCVpp) harboring HCV envelope glycoproteins of the JFH1 isolate (genotype 2a) and expressing the firefly luciferase reporter gene were produced in HEK-293T as previously described (36).
Indirect immunofluorescence.
Infected cells grown on glass coverslips were processed for immunofluorescent detection of viral proteins as previously described (37). Nuclei were stained with 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI). Coverslips were observed with a Zeiss Axiophot microscope, and fluorescent signals were collected with a Coolsnap ES camera (Photometrix, Kew, Australia). For quantification of antigen-positive cells, images of randomly picked areas from each coverslip were recorded.
Acridine orange staining.
Huh-7 cells seeded in Lab-Tek wells were either left untreated or treated for 90 min with medium containing B5 (10 μM) or monensin (1 μM). Cells were then incubated for 20 min at 37°C with complete medium supplemented with 5 μg/ml acridine orange, and stains were analyzed immediately by confocal microscopy.
Western blotting.
Cells were lysed in 1× PBS lysis buffer (1% Triton X-100, 2 mM EDTA). Cell lysates were then precleared by centrifugation at 14,000 × g for 15 min at 4°C. Protein samples were heated for 5 min at 70°C in Laemmli sample buffer. Following separation with SDS-PAGE, proteins were transferred onto nitrocellulose membranes (Hybond-ECL; Amersham) using a Transblot apparatus and revealed with specific antibodies. Following incubation with primary antibodies, membranes were incubated with the corresponding peroxidase-conjugated anti-species antibody. Proteins were then revealed by enhanced chemiluminescence (ECL) (Thermo Fisher) as recommended by the manufacturer.
Quantitative binding and virus internalization assays.
Virions bound to Huh-7 cells were determined by quantitative real-time reverse transcription-PCR (RT-PCR) assay as described previously (38). Internalization was measured as described previously (39).
Selection of a B5-resistant virus and identification of resistance mutations.
Supernatants of HCV-infected cells were serially passaged with increasing concentrations of B5. The genomic sequence of HCV from core to NS2 was amplified by RT-PCR and sequenced. Amino acid changes that arose during inhibitor selection were identified by analysis of the DNA sequence compared to the initial and control passages in the absence of drug. The identified mutations were reintroduced into the JFH1 plasmid by PCR mutagenesis, and the plasmids were sequenced.
Chimeric mice.
Fah−/− Rag2−/− IL2rγ−/− (FRG) mice with humanized liver (chimeric mice) were produced essentially as described before but with some modifications (40, 41). Briefly, at 24 h before transplantation, FRG mice were pretreated with 1.25 × 109 PFU of an adenoviral vector expressing urokinase plasminogen activator (uPA) (CuRx uPA Liver Tx Enhancer; Yecuris Corporation, Tualatin, OR, USA). At that moment, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)-containing drinking water (8 mg/ml) was replaced by regular drinking water. Mice were anesthetized with isoflurane and transplanted intrasplenically with approximately one million cryopreserved primary human hepatocytes (BD Biosciences, Erembodegem, Belgium). After transplantation, FRG mice were kept off NTBC for 7 days, followed by a 5-day NTBC treatment at 8 mg/ml. This regimen was repeated once and was followed by another 2 cycles consisting of 7 days of withdrawal and a shorter NTBC treatment of 4 days. This was followed by another 7 days of withdrawal and 3 days of NTBC treatment. The mice were then kept on a long-cycle NTBC protocol consisting of a withdrawal period of 21 days followed by 5 days of NTBC treatment. The long-cycle NTBC protocol was maintained until the mouse livers were sufficiently humanized. To evaluate successful engraftment and repopulation of the mouse liver, the amount of human albumin in mouse plasma was quantified by enzyme-linked immunosorbent assay (ELISA) (Bethyl Laboratories, Montgomery, TX, USA). All animals used in this study had human albumin plasma levels of between 2.5 and 11.5 mg/ml. B5 was administered in drinking water at a concentration of 0.25 mg/ml. The B5 treatment was started 4 days before the mice were challenged with 5 × 104 IU of virus and was continued until the end of the experiment. The animals were bled at different time points, and HCV RNA was quantified using the COBAS AmpliPrep/COBAS TaqMan HCV test (Roche Diagnostics, Vilvoorde, Belgium). Due to dilution of the plasma, this assay has a limit of detection (LOD) of 750 IU/ml. All animal experiments were approved by the Animal Ethics Committee of the Ghent University.
Graphs and statistics.
Prism v5.0c (GraphPad Software Inc., La Jolla, CA) software was used to prepare graphs, to calculate IC50s, and to determine statistical significance of differences between data sets.
RESULTS
B5 inhibits HCV infection.
We first tested 11 compounds among 4-aminoquinoline-based derivatives. As shown in Fig. 1, these derivatives belong to three different families: family A designed from CQ and linked with a bis-aminoalkyl piperazine moiety in order to improve its ability to accumulate into acidic cell compartments, family B from the best antimalarial compounds of family A bearing other aromatic cycles, and family C designed from amodiaquine, another prescribed aminoquinoline antimalarial. The compounds used in this work are presented in Fig. 2A. Compounds A1, B2, B3, B5, C1, and C3 showed efficient antimalarial activities against CQ-resistant strains (18–22) and were selected according to their chemical diversity. The others were chosen as structural analogs to allow structure-activity relationship studies. Compounds B1 and B4 are analogs of B2 and B3, A2 is an analog of A1, B6 is an analog of B5, and C2 is an analog of C1. All compounds showed only low toxicity in the MTS assay (Fig. 2B). To test the effects of these derivatives on the HCV life cycle, the compounds were added to Huh-7 target cells before as well as during infection with HCVcc. We identified six different molecules able to inhibit HCV infection (Fig. 2C) at 5 μM, a dose that did not show any cytotoxic effect for all compounds (Fig. 2B). Results obtained with families A and B showed that the 4-aminoquinoline nucleus can be substituted with a bis-aminopropylpiperazine chain and replaced by a tetrahydroacridine ring (A2), a benzimidazole ring (B5), or a benzylamino function (B2 and B3). In all cases, R1 and R2 substituents should be acyclic and apolar. Results obtained with family C showed the importance of one basic amino side chain as a substituent. Compound C1 was previously noted for its high efficiency upon CQ-resistant strain K1 and in vivo activity on Plasmodium berghei in a murine model (18). Nevertheless, inhibition activities of compounds A2, B1, B2, B3, and C1 were accompanied by noticeable morphological alteration on infected Huh-7 cells. We did not further investigate whether the antiviral activities of these compounds are linked to the observed morphological alterations. We rather focused on B5, which did not show any effect on cell morphology. Importantly, this compound succeeded all absorption, distribution, metabolism, and excretion (ADME) (42), toxicity, and pharmacodynamic measurements and is currently ending clinical phase I evaluation for neurodegenerative diseases (unpublished data).
FIG 1.

Chemical structures of compounds. Compounds from three different families were derived from 4-aminoquinoline or bis-aminoalkylpiperazine.
FIG 2.

B5 inhibits HCV infection. (A) Chemical structures of the compounds. (B) B5 toxicity was tested on Huh-7 cells. Cells were cultured in the presence of 5 μM B5, and their viability was monitored using an MTS-based viability assay at 48 h. (C) Anti-HCV activity of the compounds tested at 5 μM, a noncytotoxic concentration. (D to G) Anti-HCV activities of B5 (D and E) and CQ (F and G). Huh-7 cells were pretreated for 1 h with B5 (D and E) or CQ (F and G) before infection with JFH1 (multiplicity of infection [MOI] of 1), and infected cells were in contact with the drugs until the end of the experiment. At 48 h postinfection, infected cells were quantified by indirect immunofluorescence (C, D, and F), and virus released in the supernatant was titrated (E and G). For the infected cells, results are expressed as percentage of infection compared to the control infection in the presence of the solvent. Error bars indicate standard errors of the means (D and F) or standard deviations (SD) (E and G) from at least two independent experiments.
To characterize the anti-HCV activity of B5, we compared the effect of this compound in two different assays of HCV infection. The first assay measures the effect of the molecule on HCV infection by directly counting the number of cells expressing HCV proteins in the presence of the drug (Fig. 2D). The second assay measures the effect of the molecule on the whole HCV life cycle by titrating the virus released from infected Huh-7 cells treated with the compound (Fig. 2E). As shown in Fig. 2D, B5 exhibited a dose-dependent inhibition of the expression of HCV proteins, indicating that it specifically affects the HCV life cycle during virus entry and/or genome replication. The inhibitory effect was slightly more potent in the second assay, suggesting a slight additional effect of the compound on virus assembly and/or release. Indeed, the estimated IC50 was 1.36 (±0.69) μM in the first assay, whereas it was 0.91 (±0.64) μM in the second assay. Furthermore, the IC90 was 2.57 (±1.07) μM in the first assay and 2.27 (±0.46) μM in the second assay. The inhibitory effect was not due to cytotoxicity, since parallel experiments did not show any toxic effect of the drug at the concentrations tested (data not shown). B5 showed a 50% cytotoxic concentration (CC50) of 16.32 μM and a therapeutic index of 18. As shown in Fig. 2F and G, CQ was less effective against HCV. Indeed, the IC50 and IC90 values for CQ were 2.82 (±1.57) μM and 3.91 (± 1.04) μM, respectively. Furthermore, CQ had a CC50 of 18.75 μM and a therapeutic index of 6.65. To determine whether other members of the Flaviviridae family are also affected by B5, we tested this compound on yellow fever virus, another member of this viral family. However, B5 did not show any inhibitory effect on this virus (data not shown). Altogether, these results indicate that B5 has a specific antiviral activity against HCV.
Effect of B5 on the HCV life cycle.
To determine at which step B5 inhibits HCV, the compound was added or removed at different time points before, during, or after inoculation of Huh-7 cells with HCVcc (Fig. 3A). As shown in Fig. 3B and C, the greatest decrease in HCV infection was observed when B5 was present from the beginning of infection. However, some antiviral effect was still detected when B5 was added postinfection. As shown in Fig. 3D and E, similar results were obtained with CQ. Parallel control experiments performed with well-characterized HCV inhibitors affecting virus entry (anti-CD81 antibody JS-81) (43), replication (boceprevir) (44), or assembly (quinidine) (45) are shown in Fig. 4. These results suggest that B5 might potentially inhibit several steps of the HCV life cycle.
FIG 3.

B5 inhibits HCV entry. Huh-7 cells were infected with JFH1 (MOI of 1) and treated for different periods of time with B5 (B and C) or CQ (D and E) as indicated in panel A. At 48 h postinfection, infected cells were quantified by immunofluorescence. Results are expressed as percentage of infection relative to the control infection in the presence of solvent (Ctrl). Error bars indicate standard errors of the means from at least three experiments. The absence of cell toxicity was controlled in parallel experiments (data not shown).
FIG 4.

HCV inhibition with characterized HCV inhibitors added during infection or at different time postinfection. Huh-7 cells were infected with JFH1 and treated with MAb JS-81, boceprevir (BCP), or quinidine added during infection or at different times postinfection as indicated in Fig. 3A. At 48 h postinfection, infected cells were quantified by indirect immunofluorescence. Results are expressed as percentage of infection compared to the control infection in the absence of drug.
B5 inhibits HCV infection in primary hepatocytes.
Although human hepatocellular carcinoma-derived cell lines allow the study of the complete HCV replication cycle, these cells are phenotypically and functionally very different from hepatocytes in vivo. Since primary human hepatocytes more closely resemble the hepatocytes that are the main reservoir for HCV within the infected host, we assessed the effects of B5 in a previously described model of productive infection in these cells (25). As shown in Fig. 5A, B5 efficiently inhibited HCV infection of primary hepatocytes, with an IC50 of 4.04 (±1.85) μM. Moreover, no cytotoxicity was observed in primary human hepatocytes at the concentrations used in our experiments (Fig. 5B). These data indicate that B5 is able to block HCV infection of its natural host cells.
FIG 5.
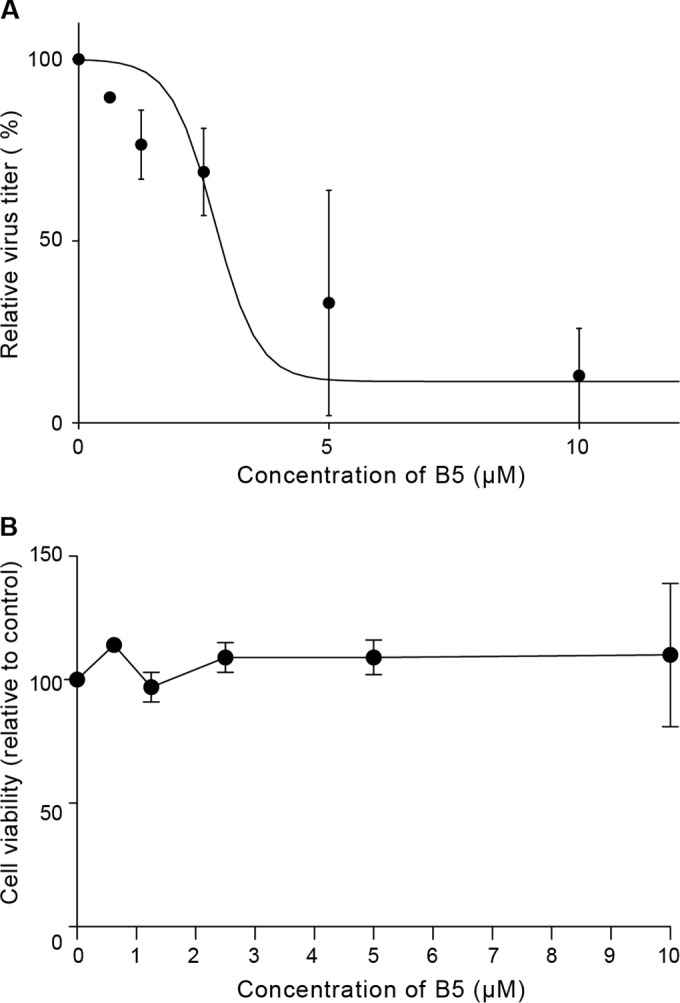
B5 inhibits HCV infection in primary human hepatocytes. (A) Dose-dependent decrease of infectious virus titers released from freshly isolated primary human hepatocytes infected with JFH1 (MOI of 2.5) and treated with increasing amounts of B5 or solvent for 3 days. Shown are infectivity titers (in FFU/ml) expressed as percentage of the solvent control value. Mean values ± SD (error bars) from two different experiments are presented. (B) Toxicity of B5 on primary human hepatocytes. Cells were cultured in the presence of different concentrations of B5, and their viability was monitored after 3 days using CytoTox 96 nonradioactive cytotoxicity assay-based measurement of lactate dehydrogenase leakage by determining optical density at 490 nm. Results are means ± SD (error bars) from at least two experiments.
Effect of B5 on HCV entry.
To investigate the effect of B5 on HCV entry, we used the HCVpp system, based on retroviral cores carrying HCV glycoproteins in their envelope. In this context, only the early steps of the viral life cycle, i.e., virus interaction with receptors, uptake, and fusion are specific to HCV, whereas all later steps are dependent on the retroviral nucleocapsid elements. Interestingly, B5 inhibited HCVpp entry in a dose-dependent manner with an IC50 of 1.80 (±0.78) μM (Fig. 6A). Furthermore, this effect was specific to HCV envelope glycoproteins, since B5 did not affect the entry of control pseudoparticles containing the envelope glycoprotein of the feline endogenous retrovirus RD114. These data indicate that B5 is an inhibitor of HCV entry. We then further investigated the entry step affected by this compound. To determine whether B5 directly impairs the binding of particles to the cell surface, we analyzed virus binding in the presence of B5. Cells were inoculated with purified HCVcc virions at 4°C in the presence or absence of B5, and the amount of bound virions was determined by quantifying HCV genomic RNA. Heparin was used as a control of inhibition of HCV binding. As expected, heparin strongly reduced HCV attachment to the cell surface (Fig. 6B). In contrast, in the presence of B5, no effect on virus binding was observed (Fig. 6B), indicating that B5 does not inhibit HCV entry by impairing virus binding to the cell surface. Thus, we also analyzed the effect of this molecule on the internalization of the viral particle. As shown in Fig. 6B, HCV internalization was not affected by B5 treatment, indicating that this molecule blocks a postinternalization step. To further analyze the mechanism by which B5 inhibits HCV entry, we finally assessed the surface expression of some known essential HCV entry factors (CD81, SR-B1, and claudin 1) in the presence of B5. However, we did not detect any effect of the compound on these receptors (data not shown).
FIG 6.
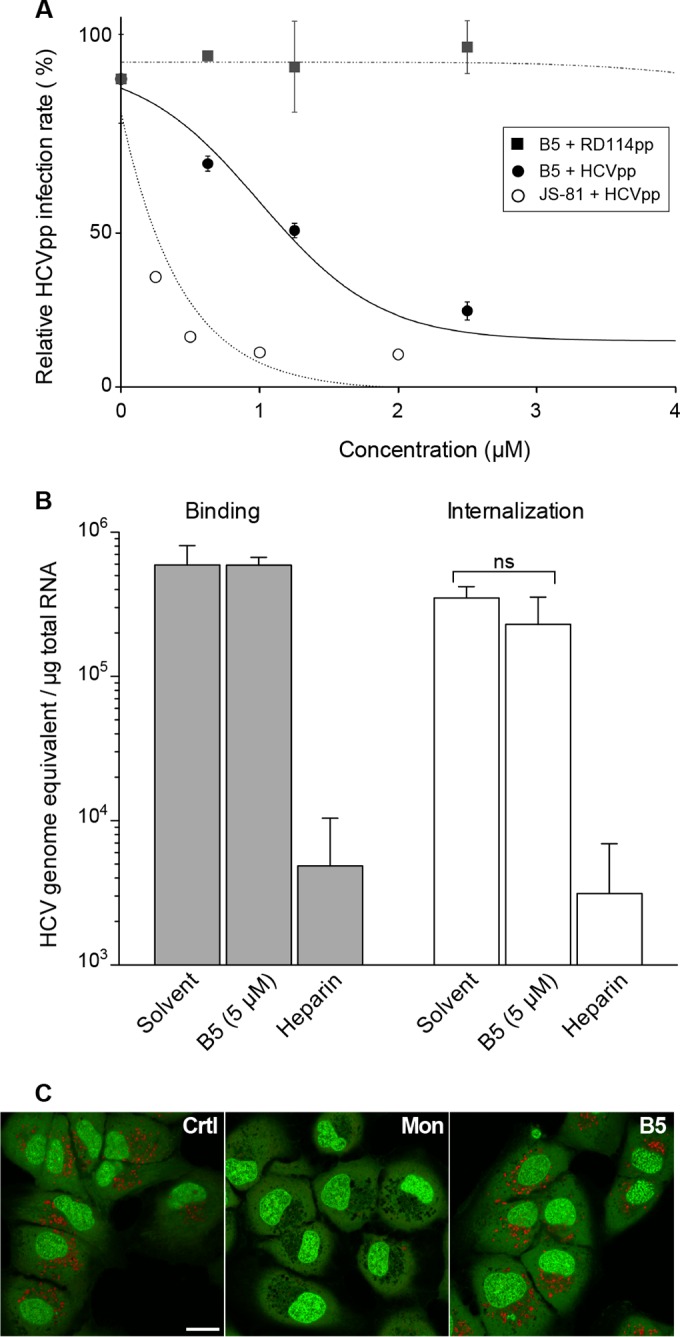
B5 inhibits a late step of HCV entry. (A) B5 inhibits HCVpp entry. Huh-7 cells were infected with pseudoparticles containing the HCV envelope glycoproteins of JFH1 (HCVpp) or with pseudoparticles containing the envelope glycoprotein of the feline endogenous retrovirus RD114 (RD114pp). Cells were treated with B5 for 1 h before infection and for the whole duration of the experiment. At 48 h postinfection, cells were lysed to quantify the luciferase activity. A control experiment with anti-CD81 MAb JS-81 was performed in parallel. HCVpp infectivity varied between 105 and 106 relative light units (RLUs). Results are expressed as percentage of infection compared to the control infection in the presence of solvent. Error bars indicate standard errors of the means from at least two independent experiments. (B) Effect of B5 on HCV binding and internalization. Huh-7 cells were inoculated for 1 h at 4°C with purified HCVcc at an MOI of 10 in the presence of solvent (PBS), B5, or 500 μg/ml of heparin. Cells were washed thrice with ice-cold PBS, and total RNA was extracted. For internalization, the binding step was followed by a 30-min incubation at 37°C, cells were then chilled on ice, and noninternalized virions were removed by trypsinization for 1 h at 4°C. Bound HCV virions or internalized viral particles were detected by quantification of HCV genomic RNA by quantitative RT-PCR. Mean values ± SD (error bars) from three different experiments are presented. Turkey's multiple-comparison test was used for statistical analysis. To verify the efficacy of trypsin treatment, virus was bound to cells at 4°C and then removed by trypsinization (data not shown). No significant difference (ns) was observed between treated and untreated cells. (C) B5 does not induce alkalization of intracellular organelles. Huh-7 cells were treated for 90 min with B5 (10 μM) or monensin (1 μM) or were left untreated (control). After incubation with acridine orange, cells were observed by confocal microscopy.
One possibility for the effect of B5 on HCV entry is that this compound could block acidification of endosomes. To test this hypothesis, we analyzed the effect of B5 on organelle acidification in Huh-7 cells. Acridine orange was used to test for acidic organelles. This compound is a weakly basic fluorescent probe that emits green fluorescence at low concentrations and gives a red fluorescence at high concentrations. It accumulates in acidic compartments, where it oligomerizes and fluoresces. In contrast, alkalization of the endocytic structure is accompanied by a change in acridine orange fluorescence (46). As shown in Fig. 6C, acridine orange produced a highly punctate red staining in untreated Huh-7 cells. As observed previously (47), the addition of monensin led to a drastic decrease of red acridine orange fluorescence (Fig. 6C). In contrast, no change in acridine orange color was observed after B5 treatment, strongly suggesting that the antiviral activity of B5 is not related to its effect on organelle acidification.
Together, these data indicate that B5 inhibits HCV entry by targeting a postbinding step without affecting the pH of intracellular organelles.
B5 resistance maps to E1 glycoprotein.
To further investigate the mechanism of action of B5, we selected a partially resistant mutant by propagation for several passages in the presence of increasing concentrations of the drug. After 14 passages, we obtained a resistant virus (data not shown). After sequencing, three mutations were identified in E1 (F291I, Y297H, and M356V), one in p7 (V787A), and one in NS2 (A880V). Since B5 inhibits HCV entry, we focused on the mutations present in E1, and, using reverse genetics, we reintroduced them into the JFH1 background, either alone or in combination. A mutant containing the three E1 mutations (FYM mutant) exhibited the same phenotype as the resistant virus, indicating that a mutation(s) conferring resistance is indeed located in E1 (Fig. 7 and data not shown). A double mutant (carrying F291I and Y297H) also showed a similar resistance phenotype (Fig. 7, FY mutant). The single Y297H mutation caused only a slight increase in resistance to B5 (Y mutant), whereas F291I alone caused a strong resistance to B5 (Fig. 7, F mutant). It is worth noting that phenylalanine 291 is located at the C terminus of a segment predicted to be a putative fusion peptide (48), suggesting that B5 could inhibit HCV entry by affecting membrane fusion.
FIG 7.

Mutation F291I in E1 confers resistance to B5 treatment. Huh-7 cells were pretreated for 1 h with B5 before infection with JFH1 or mutant viruses (MOI of 1), and infected cells were in contact with the drug until the end of the experiment. At 30 h postinfection, infected cells were quantified. Results are expressed as percentage of infection in the presence of the solvent. Error bars indicate standard errors of the means from at least three independent experiments. Mutant FYM contains E1 mutations F291I, Y297H, and M356V. Mutant FY contains E1 mutations F291I and Y297H. Mutants F and Y contain mutations F291I and Y297H, respectively.
B5 shares a mode of action with other 4-aminoquinoline-based molecules.
Since FQ and CQ, two other 4-aminoquinoline-based molecules, have been shown to inhibit HCV entry (13, 15), we wondered whether B5 might share a mechanism of action with these 2 compounds. For this purpose, we first analyzed the effect of CQ on the B5-resistant mutant. As shown on Fig. 8A, the B5-resistant mutant is approximately 3 times less sensitive to CQ treatment, suggesting that CQ and B5 may partially share some mechanism of action against HCV. We also analyzed the effect of FQ on the B5-resistant mutant. The B5-resistant mutant was also resistant to FQ treatment (Fig. 8B). To further compare B5 and FQ, we also analyzed the effect of B5 on an FQ-resistant mutant that has been previously reported (15). This mutant has a serine replaced by an alanine at position 327, which is located in a region of E1 different from that for the B5-resistant mutant. Interestingly, the FQ-resistant mutant was also resistant to B5 treatment (Fig. 8C), indicating that resistance to FQ treatment also primes HCV envelope glycoproteins to resist to B5 treatment. Together, these data suggest that B5 shares a mode of action with other 4-aminoquinoline-based molecules.
FIG 8.

Antiviral activities of other 4-aminoquinoline-based molecules against a B5-resistant mutant. Huh-7 cells were pretreated for 1 h with CQ (A), FQ (B), or B5 (C) before infection with JFH1 or mutant viruses (MOI of 1), and infected cells were in contact with the drug until the end of the experiment. At 30 h postinfection, infected cells were quantified. Results are expressed as percentage of infection in the presence of the solvent. Error bars indicate standard errors of the means from at least three independent experiments. Mutant FY contains E1 mutations F291I and Y297H selected in the presence of B5. Mutant SA contains mutation S327A selected in the presence of FQ (15).
Effect of B5 on other steps of the HCV life cycle.
The above data show that B5 has an effect on HCV entry. However, one cannot exclude some effect on other steps of the HCV life cycle. To analyze the effect of B5 on HCV genome replication, Huh-7 cells were electroporated with in vitro-transcribed JFH1 RNA to bypass the entry step and avoid any interference with late steps of the HCV life cycle. Boceprevir was used in parallel as a control of inhibition of viral replication (Fig. 9B). However, as shown in Fig. 9A, B5 did not inhibit HCV genome replication. We also analyzed the potential effect of B5 on late steps of the HCV life cycle. As shown in Fig. 9C and D, treatment of infected cells with B5 late during infection led to a decrease in core secretion, suggesting that B5 can also affect HCV egress and/or assembly. However, this effect was less drastic than what was observed with control inhibitors which inhibit HCV release (brefeldin A) or assembly (quinidine) (45, 49).
FIG 9.

Effect of B5 on other steps of the HCV life cycle. (A and B) Effect of B5 on HCV replication. JFH1 RNA was electroporated into Huh-7 cells, and at 4 h postelectroporation, cells were treated or not with B5 (A) or boceprevir (BCP) (B). HCV replication was measured at 30 h postelectroporation by counting the number of cells expressing E1 by immunofluorescence. (C and D) Effect of B5 on HCV virion production. Huh-7 cells were infected with JFH1 (MOI of 1) for 2 h at 37°C. At 40 h postinfection, the cells were treated with either B5 (5 μM), brefeldin A (1 μM), quinidine (100 μM), or their solvent for 8 h. Supernatants were harvested, and cells were lysed for core protein quantification.
B5 inhibits infection with HCVcc chimeras from other genotypes.
To further characterize the inhibition of HCV infection by B5, we tested its effect on other genotypes in the context of the HCVcc system, using JFH1-derived chimeras. As shown in Table 1, a similar antiviral activity was observed on the other HCV genotypes tested, indicating that B5 anti-HCV activity is not genotype specific.
TABLE 1.
B5 inhibits infection with different HCV genotypesa
| Genotype | IC50, μM (mean ± SD)b |
|---|---|
| gt1b | 0.77 ± 0.25 |
| gt2a | 0.91 ± 0.64 |
| gt3a | 2.93 ± 1.24 |
| gt4a | 1.81 ± 0.48 |
| gt5a | 1.15 ± 0.53 |
| gt6a | 1.87 ± 0.99 |
The antiviral effect of B5 was tested on JFH1 (gt2a) or chimeric viruses expressing the structural proteins of different genotypes: GT1b(Con1)/JFH1 (gt1b), GT3a(S52)/JFH1 (gt3a), GT4a(ED43)/JFH1 (gt4a), GT5a(SA13)/JFH1 (gt5a), or GT6a(HK6a)/JFH1 (gt6a).
IC50s were determined based on the titration of infectious virus released in the supernatant of treated cells.
B5 partially protects chimeric mice from HCV infection.
To study the efficacy of B5 in preventing in vivo HCV infections, we used the chimeric FRG mouse model (41). The livers of these chimeric mice are largely engrafted with functional primary human hepatocytes and can be infected with hepatotropic pathogens such as HCV (50–52). Three humanized chimeric FRG mice received B5 in drinking water at a concentration of 0.25 mg/ml. In addition, two untreated control animals were used. The B5 treatment was initiated 4 days before the animals were challenged with serum-derived HCV and was continued for up to 31 days after injection of the virus. A similar pretreatment approach is usually used to characterize the antiviral activity of entry inhibitors in vivo (53–57). As shown in Fig. 10, at 3 to 6 days after infection, HCV RNA could easily be detected in the two control animals, while HCV RNA was detected in only one out of three B5-treated animals. In the remaining two treated mice, the viral load was below the limit of detection (750 IU/ml). At day 10, viral RNA was detected in two out of three treated animals, albeit at much lower concentrations than in the control animals, which experienced full-blown viremia, indicating a delay in the kinetics of the infection in B5-treated mice. The third treated chimeric mouse remained completely HCV negative (plasma HCV RNA level of <750 IU/ml) throughout the 31-day observation period. The absence of viremia in this mouse was not due to a failure of engraftment, since this mouse had the highest human albumin level, a proof of successful engraftment. The E1E2 genes from the two partially protected mice and the control mouse were sequenced. However, we did not find the presence of resistance mutations in treated mice (data not shown). Together, these data indicate that B5 delays the kinetics of the viral infection in vivo. However, optimization of the experimental conditions will be needed to evaluate the full potential antiviral activity of B5.
FIG 10.

B5 partially protects chimeric mice from HCV infection. Three human liver chimeric mice (dashed lines) received a 35-day-long treatment starting 4 days before viral challenge. Two control chimeric mice (solid lines) were inoculated with the same virus but received placebo treatment. All animals were injected with 5 × 104 IU of HCV (isolate mP05), and HCV RNA levels were quantified in plasma at different time points after infection. The gray area represents the treatment window. The limit of detection (LOD) is represented by the dotted line.
DISCUSSION
HCV entry is a complex multistep process that remains poorly understood. Pharmacological inhibitors are useful tools to potentially decipher the early steps of the HCV life cycle. Here, we describe the characterization of B5, a new 2-aminobenzimidazole derivative with a potent anti-HCV activity. Our data show that B5 inhibits HCV infection at an IC50 close to 1 μM by blocking HCV entry. At higher concentrations it can also inhibit virus replication. Furthermore, we showed that this compound is active on different HCV genotypes and that it delays the kinetics of the viral infection in vivo.
B5 was first designed as an antimalarial compound derived from CQ, but the presence of the piperazine core provided a different mechanism of action against this parasite (58). Here we show that B5 is more potent than CQ. CQ is an aminoquinoline known since 1934 that has been developed as an antimalarial drug. Apart from its well-known antimalarial effects, the drug has also interesting antiviral effects (59). CQ can indeed inhibit the pH-dependent steps of the replication of several viruses, including HCV (13, 16). CQ is also an inhibitor of autophagy (60), and this effect can also contribute to its anti-HCV activity (17). Moreover, CQ has immunomodulatory effects, leading to its potential use against some infectious diseases. Indeed, in HIV-infected individuals, CQ was repeatedly reported to be effective in counteracting the deleterious immune activation associated with the disease (61). However, in clinical trials against influenza virus and chikungunya virus, CQ was rather disappointing (62, 63). This could be due to high viral loads in vivo, poor penetration in specific tissues, and narrow therapeutic indexes. The antiviral activity of B5 shows strong improvements compared to that of CQ. B5 has a much higher therapeutic index of 18 instead of 6.65 and is approximately 3 times more potent than CQ in our experimental conditions. Furthermore, the demonstration that B5 can also inhibit HCV infection in primary human hepatocytes indicates that this compound remains effective under more physiological conditions.
B5 is as potent as FQ. Recently, we reported that FQ, a ferrocenic analog of CQ, is a promising inhibitor of HCV (15). This bio-organometallic compound is currently one of the most promising new candidate drugs in the antimalarial pipeline, and it is also in phase II clinical trials as a treatment for uncomplicated malaria (62). With IC50s of 0.91 μM (this work) and 0.80 μM (15) for B5 and FQ, respectively, these two compounds indeed show similar potency. However, FQ has a therapeutic index of 6.7, the same as that of CQ, whereas B5 has a therapeutic index of 18, indicating that B5 has a wider therapeutic window for the potential treatment of HCV infection. Furthermore, mice with humanized liver treated with B5 showed a delay in the kinetics of the viral infection, suggesting some potential therapeutic applications for this compound in HCV-infected patients. However, further optimization of experimental conditions will be needed to determine the full antiviral potential of this molecule. Interestingly, B5 has curative properties against neurodegenerative diseases as shown in animal models of Alzheimer's disease (unpublished data), and it recently succeeded in a phase I clinical trial as a promising molecule against neurodegenerative diseases (unpublished data).
B5 inhibits a late step in HCV entry. Our data indicate that B5 inhibits a postattachment stage of the entry step. Furthermore, we identified a mutation in E1 that confers resistance to HCV. Indeed, the F291I mutation is able by itself to confer resistance to B5. Although the precise role of HCV envelope glycoproteins in virus entry remains poorly understood, recent crystallographic data suggest that E1, alone or in association with E2, is the fusion protein (10, 11). Furthermore, phenylalanine 291 is located at the C terminus of a segment predicted to be a putative fusion peptide (48), strongly suggesting that B5 affects the fusion step in HCV entry. It is worth noting that resistance mutations to other molecules affecting HCV fusion have been shown to map in E1 glycoprotein (15, 64), with one of them also being located in the putative fusion peptide (64).
B5 shares a mode of action with other 4-aminoquinoline-based molecules. Among 4-aminoquinolines, CQ is also an inhibitor of HCV entry (13, 16), and its mechanism of action involves impaired endosomally mediated virus entry, most likely through the prevention of endocytosis and/or endosomal acidification. The two compounds present structural similarities (presence of an aromatic core decorated with an amino side chain). The effect of B5 on a postinternalization step of HCV entry is indeed in favor of a mechanism involving impaired endosomally mediated virus entry. Due to the presence of the amino piperazine side chain in the structure of B5, the ability of this compound to accumulate into acidic compartments is improved compared to that of CQ. However, as shown in our experiments with acridine orange, the lack of effect of B5 on endosome acidification suggests a different mode of action. This hypothesis is reinforced by the observation that yellow fever virus is sensitive to CQ treatment (65) but not to B5 (data not shown). However, the decreased sensitivity of a B5-resistant mutant to CQ treatment suggests that CQ and B5 may partially share some mode of action against HCV. FQ, another 4-aminoquinoline, also affects a postinternalization step of HCV entry (15). The mode of action of this molecule has been suggested to be linked to its capacity to generate reactive oxygen species and induce lipid peroxidation (66, 67). The observations that a B5-resistant mutant is resistant to FQ treatment and that an FQ-resistant mutant is resistant to B5 treatment suggest that B5 and FQ might have a shared mode of action against HCV.
In conclusion, B5 is an interesting compound currently in development against Alzheimer's disease, which could be used as a major tool to further dissect the late steps of HCV entry into host cells, a viral life cycle step that is still not fully understood.
ACKNOWLEDGMENTS
We thank P.-E. Larchanché for the synthesis of the compounds. We are grateful to Gilles Duverlie and Véronique Descamps for core protein quantification. We thank Laure Saas for technical assistance. We are also grateful to R. Bartenschlager, C. Biot, J. Bukh, F.L. Cosset, P. Halfon, J. McKeating, and T. Wakita for providing essential reagents. The immunofluorescence analyses were performed with the help of the imaging core facility of the BioImaging Center Lille Nord-de-France.
This work was supported by the French National Agency for Research on AIDS and Viral Hepatitis (ANRS), the ANR through ERA-NET Infect-ERA program (ANR-13-IFEC-0002-01). Philip Meuleman was supported by Ghent University (Concerted Action Grant 01G01712) and the Belgian Science Policy Office (BELSPO; IUAP P7/47-HEPRO-2). Thibaut Vausselin and Matthieu Lemasson were supported by a fellowship from the ANRS. Sandrine Belouzard was supported by a Marie Curie International Reintegration Grant (PIRG-GA-2009-256300). Ahmed Atef Mesalam is the recipient of a Ph.D. fellowship provided by the Egyptian Government. The 300-MHz NMR facilities were funded by the Région Nord-Pas de Calais (France), the Ministère de la Jeunesse, de l'Education Nationale et de la Recherche (MJENR), and the Fonds Européens de Développement Régional (FEDER).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
T. Vausselin, K. Séron, M. Lavie, Y. Rouillé, L. Cocquerel, A. R. Rosenberg, P. Meuleman, and J. Dubuisson conceived and designed the experiments; T. Vausselin, K. Séron, M. Lavie, A. A. Mesalam, M. Lemasson, S. Belouzard, L. Fénéant, A. Danneels, L. Cocquerel, L. Foquet, and C. Wychowski performed the experiments; T. Vausselin, K. Séron, M. Lavie, A. A. Mesalam, A. R. Rosenberg, Y. Rouillé, L. Cocquerel, P. Meuleman, P. Melnyk, and J. Dubuisson analyzed the data; and T. Vausselin, K. Séron, M. Lavie, P. Melnyk, and J. Dubuisson wrote the paper.
REFERENCES
- 1.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, Barnes E. 2015. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 61:77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levrero M. 2006. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 25:3834–3847. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 3.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N Engl J Med 368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Chung RT, Baumert TF. 2014. Curing chronic hepatitis C—the arc of a medical triumph. N Engl J Med 370:1576–1578. doi: 10.1056/NEJMp1400986. [DOI] [PubMed] [Google Scholar]
- 6.Graham CS, Swan T. 2015. A path to eradication of hepatitis C in low- and middle-income countries. Antiviral Res 119:89–96. doi: 10.1016/j.antiviral.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Lindenbach BD, Thiel HJ, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152. In Knipe DM, Howley PM (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 9.Vieyres G, Dubuisson J, Pietschmann T. 2014. Incorporation of hepatitis C virus e1 and e2 glycoproteins: the keystones on a peculiar virion. Viruses 6:1149–1187. doi: 10.3390/v6031149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509:381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubuisson J, Cosset FL. 2014. Virology and cell biology of hepatitis C virus life cycle—an update. J Hepatol 61:S3–S13. doi: 10.1016/j.jhep.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 13.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coller KE, Berger KL, Heaton NS, Cooper JD, Yoon R, Randall G. 2009. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog 5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vausselin T, Calland N, Belouzard S, Descamps V, Douam F, Helle F, Francois C, Lavillette D, Duverlie G, Wahid A, Feneant L, Cocquerel L, Guerardel Y, Wychowski C, Biot C, Dubuisson J. 2013. The antimalarial ferroquine is an inhibitor of hepatitis C virus. Hepatology 58:86–97. doi: 10.1002/hep.26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. 2011. Lysosomotropic agents as HCV entry inhibitors. Virol J 8:163. doi: 10.1186/1743-422X-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, Ikejima K, Kitamura T, Enomoto N, Sakai T, Kominami E, Watanabe S. 2010. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J Gastroenterol 45:195–203. doi: 10.1007/s00535-009-0132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paunescu E, Susplugas S, Boll E, Varga RA, Mouray E, Grellier P, Melnyk P. 2008. Synthesis and antimalarial activity of new amino analogues of amodiaquine. Med Chem 4:407–425. doi: 10.2174/157340608785700153. [DOI] [PubMed] [Google Scholar]
- 19.Ryckebusch A, Deprez-Poulain R, Debreu-Fontaine MA, Vandaele R, Mouray E, Grellier P, Sergheraert C. 2003. Synthesis and antimalarial evaluation of new 1,4-bis(3-aminopropyl)piperazine derivatives. Bioorg Med Chem Lett 13:3783–3787. doi: 10.1016/j.bmcl.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Ryckebusch A, Deprez-Poulain R, Maes L, Debreu-Fontaine MA, Mouray E, Grellier P, Sergheraert C. 2003. Synthesis and in vitro and in vivo antimalarial activity of N1-(7-chloro-4-quinolyl)-1,4-bis(3-aminopropyl)piperazine derivatives. J Med Chem 46:542–557. doi: 10.1021/jm020960r. [DOI] [PubMed] [Google Scholar]
- 21.Melnyk P, Burlet S, Le Fur N, Delacourte A. November 2012. New heterocycle compounds and uses thereof for the prevention or treatment of diseases involving formation of amyloid plaques and/or where a dysfunction of the APP metabolism occurs. US patent application 2012/0283256 A1.
- 22.Melnyk P, LeFur N, Gay M, Sergeant N, Buée L. 2 May 2016. Novel 1,4-bis(3-aminopropyl)piperazine derivative and its use. European patent 15305384.8.
- 23.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 24.Wahid A, Dubuisson J. 2013. Virus-neutralizing antibodies to hepatitis C virus. J Viral Hepat 20:369–376. doi: 10.1111/jvh.12094. [DOI] [PubMed] [Google Scholar]
- 25.Podevin P, Carpentier A, Pene V, Aoudjehane L, Carriere M, Zaidi S, Hernandez C, Calle V, Meritet JF, Scatton O, Dreux M, Cosset FL, Wakita T, Bartenschlager R, Demignot S, Conti F, Rosenberg AR, Calmus Y. 2010. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 139:1355–1364. doi: 10.1053/j.gastro.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 26.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol 73:6235–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouille Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 31.Rocha-Perugini V, Montpellier C, Delgrange D, Wychowski C, Helle F, Pillez A, Drobecq H, Le Naour F, Charrin S, Levy S, Rubinstein E, Dubuisson J, Cocquerel L. 2008. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 3:e1866. doi: 10.1371/journal.pone.0001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottwein JM, Scheel TK, Hoegh AM, Lademann JB, Eugen-Olsen J, Lisby G, Bukh J. 2007. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133:1614–1626. doi: 10.1053/j.gastro.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 34.Jensen TB, Gottwein JM, Scheel TK, Hoegh AM, Eugen-Olsen J, Bukh J. 2008. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J Infect Dis 198:1756–1765. doi: 10.1086/593021. [DOI] [PubMed] [Google Scholar]
- 35.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A 105:997–1002. doi: 10.1073/pnas.0711044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C pseudo-particles containing functional E1E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouille Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, Belouzard S, McKeating J, Patel AH, Maertens G, Wakita T, Wychowski C, Dubuisson J. 2006. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol 80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castelain S, Descamps V, Thibault V, Francois C, Bonte D, Morel V, Izopet J, Capron D, Zawadzki P, Duverlie G. 2004. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 39.Albecka A, Belouzard S, de Beeck AO, Descamps V, Goueslain L, Bertrand-Michel J, Terce F, Duverlie G, Rouille Y, Dubuisson J. 2012. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55:998–1007. doi: 10.1002/hep.25501. [DOI] [PubMed] [Google Scholar]
- 40.Bissig KD, Wieland SF, Tran P, Isogawa M, Le TT, Chisari FV, Verma IM. 2010. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest 120:924–930. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. 2007. Robust expansion of human hepatocytes in Fah−/− Rag2−/− Il2rg−/− mice. Nat Biotechnol 25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melnyk P, Vingtdeux V, Burlet S, Eddarkaoui S, Grosjean ME, Larchanche PE, Hochart G, Sergheraert C, Estrella C, Barrier M, Poix V, Plancq P, Lannoo C, Hamdane M, Delacourte A, Verwaerde P, Buee L, Sergeant N. 2015. Chloroquine and chloroquinoline derivatives as models for the design of modulators of amyloid Peptide precursor metabolism. ACS Chem Neurosci 6:559–569. doi: 10.1021/cn5003013. [DOI] [PubMed] [Google Scholar]
- 43.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong X, Chase R, Skelton A, Chen T, Wright-Minogue J, Malcolm BA. 2006. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res 70:28–38. doi: 10.1016/j.antiviral.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Chockalingam K, Simeon RL, Rice CM, Chen Z. 2010. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc Natl Acad Sci U S A 107:3764–3769. doi: 10.1073/pnas.0915117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266:17707–17712. [PubMed] [Google Scholar]
- 47.Feneant L, Potel J, Francois C, Sane F, Douam F, Belouzard S, Calland N, Vausselin T, Rouille Y, Descamps V, Baumert TF, Duverlie G, Lavillette D, Hober D, Dubuisson J, Wychowski C, Cocquerel L. 2015. New insights into the understanding of hepatitis C virus entry and cell-to-cell transmission by using the ionophore monensin A. J Virol 89:8346–8364. doi: 10.1128/JVI.00192-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flint M, Thomas JM, Maidens CM, Shotton C, Levy S, Barclay WS, McKeating JA. 1999. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J Virol 73:6782–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vercauteren K, de Jong YP, Meuleman P. 2015. Animal models for the study of HCV. Curr Opin Virol 13:67–74. doi: 10.1016/j.coviro.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vercauteren K, de Jong YP, Meuleman P. 2014. HCV animal models and liver disease. J Hepatol 61:S26–33. doi: 10.1016/j.jhep.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 52.de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, Gerges S, Vega K, Labitt RN, Donovan BM, Giang E, Krishnan A, Chiriboga L, Charlton MR, Burton DR, Baltimore D, Law M, Rice CM, Ploss A. 2014. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med 6:254ra129. doi: 10.1126/scitranslmed.3009512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desombere I, Fafi-Kremer S, Van Houtte F, Pessaux P, Farhoudi A, Heydmann L, Verhoye L, Cole S, McKeating JA, Leroux-Roels G, Baumert TF, Patel AH, Meuleman P. 2016. Monoclonal anti-envelope antibody AP33 protects humanized mice against a patient-derived hepatitis C virus challenge. Hepatology 63:1120–1134. doi: 10.1002/hep.28428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 55.Meuleman P, Albecka A, Belouzard S, Vercauteren K, Verhoye L, Wychowski C, Leroux-Roels G, Palmer KE, Dubuisson J. 2011. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob Agents Chemother 55:5159–5167. doi: 10.1128/AAC.00633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meuleman P, Bukh J, Verhoye L, Farhoudi A, Vanwolleghem T, Wang RY, Desombere I, Alter H, Purcell RH, Leroux-Roels G. 2011. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 53:755–762. doi: 10.1002/hep.24171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meuleman P, Catanese MT, Verhoye L, Desombere I, Farhoudi A, Jones CT, Sheahan T, Grzyb K, Cortese R, Rice CM, Leroux-Roels G, Nicosia A. 2012. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 55:364–372. doi: 10.1002/hep.24692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ryckebusch A, Deprez-Poulain R, Debreu-Fontaine MA, Vandaele R, Mouray E, Grellier P, Sergheraert C. 2002. Parallel synthesis and anti-malarial activity of a sulfonamide library. Bioorg Med Chem Lett 12:2595–2598. doi: 10.1016/S0960-894X(02)00475-4. [DOI] [PubMed] [Google Scholar]
- 59.Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. 2003. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect Dis 3:722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janku F, McConkey DJ, Hong DS, Kurzrock R. 2011. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 8:528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 61.Savarino A. 2011. Use of chloroquine in viral diseases. Lancet Infect Dis 11:653–654. doi: 10.1016/S1473-3099(11)70092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biot C, Nosten F, Fraisse L, Ter-Minassian D, Khalife J, Dive D. 2011. The antimalarial ferroquine: from bench to clinic. Parasite 18:207–214. doi: 10.1051/parasite/2011183207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaur P, Chu JJ. 2013. Chikungunya virus: an update on antiviral development and challenges. Drug Discov Today 18:969–983. doi: 10.1016/j.drudis.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perin PM, Haid S, Brown RJ, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, Baktash YM, Vondran FW, Speerstra S, Awadh A, Mukhtarov F, Schang LM, Kirschning A, Muller R, Guzman CA, Kaderali L, Randall G, Meuleman P, Ploss A, Pietschmann T. 2016. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63:49–62. doi: 10.1002/hep.28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brandriss MW, Schlesinger JJ. 1984. Antibody-mediated infection of P388D1 cells with 17D yellow fever virus: effects of chloroquine and cytochalasin B. J Gen Virol 65:791–794. doi: 10.1099/0022-1317-65-4-791. [DOI] [PubMed] [Google Scholar]
- 66.Chavain N, Vezin H, Dive D, Touati N, Paul JF, Buisine E, Biot C. 2008. Investigation of the redox behavior of ferroquine, a new antimalarial. Mol Pharm 5:710–716. doi: 10.1021/mp800007x. [DOI] [PubMed] [Google Scholar]
- 67.Dubar F, Egan TJ, Pradines B, Kuter D, Ncokazi KK, Forge D, Paul JF, Pierrot C, Kalamou H, Khalife J, Buisine E, Rogier C, Vezin H, Forfar I, Slomianny C, Trivelli X, Kapishnikov S, Leiserowitz L, Dive D, Biot C. 2011. The antimalarial ferroquine: role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem Biol 6:275–287. doi: 10.1021/cb100322v. [DOI] [PubMed] [Google Scholar]