

ABSTRACT
Varicella-zoster virus (VZV) is an alphaherpesvirus that causes varicella upon primary infection and zoster upon reactivation from latency in sensory ganglion neurons. The replication of herpesviruses requires manipulation of cell signaling pathways. Notably, CREB, a factor involved in the regulation of several cellular processes, is activated upon infection of T cells with VZV. Here, we report that VZV infection also induced CREB phosphorylation in fibroblasts and that XX-650-23, a newly identified inhibitor of the phosphorylated-CREB (pCREB) interaction with p300/CBP, restricted cell-cell spread of VZV in vitro. CREB phosphorylation did not require the viral open reading frame 47 (ORF47) and ORF66 kinases encoded by VZV. Evaluating the biological relevance of these observations during VZV infection of human skin xenografts in the SCID mouse model of VZV pathogenesis showed both that pCREB was upregulated in infected skin and that treatment with XX-650-23 reduced infectious-virus production and limited lesion formation compared to treatment with a vehicle control. Thus, processes of CREB activation and p300/CBP binding are important for VZV skin infection and may be targeted for antiviral drug development.
IMPORTANCE Varicella-zoster virus (VZV) is a common pathogen that causes chicken pox and shingles. As with all herpesviruses, the infection is acquired for life, and the virus can periodically reactivate from latency. Although VZV infection is usually benign with few or no deleterious consequences, infection can be life threatening in immunocompromised patients. Otherwise healthy elderly individuals who develop zoster as a consequence of viral reactivation are at risk for postherpetic neuralgia (PHN), a painful and long-lasting complication. Current vaccines use a live attenuated virus that is usually safe but cannot be given to many immunodeficient patients and retains the capacity to establish latency and reactivate, causing zoster. Antiviral drugs are effective against severe VZV infections but have little impact on PHN. A better understanding of virus-host cell interactions is relevant for developing improved therapies to safely interfere with cellular processes that are crucial for VZV pathogenesis.
INTRODUCTION
Varicella-zoster virus (VZV) is the causative agent of varicella (chicken pox), resulting from primary infection, and zoster (shingles), due to viral reactivation from latency in neurons of the sensory ganglia (1). The current model of primary infection is initiation by upper respiratory tract inoculation of the virus and replication in mucosal epithelial cells, followed by transfer to T cells in the tonsils or other regional lymph nodes. This leads to a primary viremia allowing the delivery of the virus to the skin, where it causes the typical rash associated with varicella, and to neurons in the sensory ganglia, where latency is established (2). Virions also gain access to sensory nerve cells in ganglia by retrograde axonal transport from the skin (3). Zoster is characterized by a vesicular rash that usually appears in a dermatomal distribution and follows the delivery of virions to the skin by anterograde axonal transport from neurons where VZV has reactivated. In both varicella and zoster, the vesicular lesions contain high titers of infectious virions (1), allowing the virus to complete its epidemiological cycle by transmission to susceptible individuals.
VZV is a large DNA virus belonging to the family Herpesviridae and the subfamily Alphaherpesvirinae. The VZV genome encodes at least 71 predicted proteins involved in various processes, including viral replication to maintain its epidemiological cycle and manipulating the host cell in order to delay recognition by the immune system and clearance from the host (1, 4). VZV is highly host restricted for human tissue, although it can also infect guinea pigs (5). For many years, this species specificity of VZV limited laboratory studies of the virus primarily to in vitro analyses that do not represent the tissue environment encountered in the human host. To overcome this challenge, our laboratory has developed a model of VZV pathogenesis using human tissues engrafted in mice with severe combined immunodeficiency (SCID) (6). The absence of any adaptive immune system in SCID mice allows both the engraftment of human tissue and the analysis of VZV pathogenesis solely in the context of the presence of innate but not adaptive immune responses. This model has been successfully used to unravel the importance and functionality of various VZV proteins and protein subdomains during infection of skin, dorsal root ganglia (DRG), or T cells by VZV (7–10). The contributions of several host transcription factors to the regulation of VZV gene promoters and their tissue-specific functions have also been demonstrated in the SCID mouse model (8, 11, 12).
The cyclic AMP (cAMP) response element binding protein (CREB) is a transcription factor that is crucial for regulating several host genes that mediate important cellular processes. CREB is phosphorylated by various host cell kinases, including PKA; Akt; and CamK I, II, and IV (13–16). Phosphorylated CREB (pCREB) translocates to the nucleus, where it dimerizes, forming homodimers or heterodimers, and recruits p300/CREB binding protein (CBP) to initiate transcription of its numerous target genes (17). pCREB is an important regulator of apoptosis (18) and is also involved in modulating several immune processes (19) and neuronal plasticity (20). p300/CBP is a histone acetyltransferase and a transcriptional cofactor containing several domains, allowing its interaction with various partners. Importantly, the p300/CBP KIX domain interacts with the kinase-inducible domain (KID) found in CREB. The p300/CBP KIX domain also interacts with the KID domain in ATF-1 and CREM (21), two other member of the same family of proteins (22), and binds other important transcriptional regulators, including c-Myb and c-Jun (23–25). Recently, we used analysis of VZV-infected T cells by single-cell mass cytometry to identify cellular factors altered by VZV replication (26) and demonstrated that VZV induced CREB phosphorylation, among other changes. Similar effects were observed in VZV-infected melanoma cells (27). These data raise the possibility that CREB may be involved in the pathogenesis of VZV.
The activation of CREB and its molecular interactions with its required coactivators have been well characterized (22, 23, 25, 28). Biophysical and biochemical screenings have facilitated the development of small-molecule inhibitors of the CREB-CBP/p300 interaction. These small molecules serve as a useful tool in assessing the effects of disrupting CREB-driven transcription.
Here, we investigated the role of CREB activation in VZV infection in vitro and in the pathogenesis of VZV infection of human skin xenografts in vivo. We found that CREB phosphorylation is increased upon VZV infection of human fibroblasts but that this increase in phosphorylation was not dependent on the functions of the two VZV ORF47 and ORF66 serine-threonine kinases. The importance of pCREB activation and the p300 interaction through its KIX domain for VZV infection and skin xenografts was demonstrated using a novel small-molecule inhibitor of CREB transcription, XX-650-23, to disrupt this event in VZV-infected cells. Blocking the interaction using XX-650-23 not only inhibited VZV replication in cultured cells, but also had significant antiviral activity against VZV infection of human skin in vivo.
MATERIALS AND METHODS
Cells and viruses.
Melanoma cells were propagated in culture medium (minimal essential Eagle medium [MEM] supplemented with 10% fetal bovine serum [Gemini Bio-Products, Woodland, CA], nonessential amino acids [100 μM; Omega Scientific, Inc., Tarzana, CA], penicillin G [100 units/ml; Omega Scientific, Inc., Tarzana, CA], streptomycin [100 units/ml; Omega Scientific, Inc., Tarzana, CA], and amphotericin [0.5 mg/ml; Omega Scientific, Inc., Tarzana, CA]). Human embryonic lung fibroblasts (HELFs) were propagated in the same culture medium without nonessential amino acids. The viruses that were used included recombinant VZV parent Oka (pOka) (29); rOka-ORF10-GFP, which expresses green fluorescent protein (GFP) as a fusion protein with the ORF10 protein (30); two VZV kinase mutants, pOkaORF66G102A (31) and VZV-ORF47D-N (8), which have point mutations that block kinase function; and VZV-chimera D (VZV-ORF47D-N was generated from this strain) (32). The viruses were propagated in melanoma cells and HELFs. Inactivated virus preparations were obtained by treating infected fibroblasts for 20 min with UV light at a distance of ∼10 cm from the lamp. The titers of untreated control wells were determined to find the amount of virus in the preparation.
XX-650-23.
The inhibitory molecule XX-650-23 was prepared in the laboratory of Xiangshu Xiao. The specificity and binding characteristics of this compound have been previously described (33) The compound was solubilized in dimethyl sulfoxide (DMSO) to a concentration of 20 mg/ml for use in the in vivo studies and to 50 mM for in vitro studies; stocks were stored at −20°C. For the in vivo studies, the stock was diluted 10-fold in sterile normal saline (0.9% NaCl in water) immediately before intraperitoneal (i.p.) injection. For the in vitro experiments, the stock was diluted in tissue culture medium to a concentration of 1 μM. The carrier control was prepared by replacing the solubilized drug with an equal amount of pure DMSO.
Infection of skin xenografts in SCID mice.
Skin xenografts were prepared in male homozygous C.B.-17 scid-scid mice (Taconic Biosciences, Oxnard, CA) as previously described (23), using human fetal tissue provided by Advanced Bioscience Resource (Alameda, CA) and obtained with informed consent according to federal and state regulations. Four weeks after engraftment, the xenografts were inoculated with HELFs infected with rOka-R6362FL (34). Infectious-virus titers were determined for the inoculum at the time of the injection. Xenografts were harvested at 22 days postinfection (dpi); one half of each tissue was homogenized and resuspended in phosphate-buffered saline (PBS) for viral titration or in DNAzol for DNA extraction to measure genome copies, and the other half was fixed in paraformaldehyde (4% in PBS) for histology.
In vivo imaging.
Mice with skin xenografts infected with rOka-R6362FL were injected i.p. with either DMSO or XX-650-23 every day from 0 to 14 dpi. Mice with mock-infected xenografts were also included as controls. For imaging, the mice were anesthetized (isoflurane), injected with d-luciferin (3 mg/mouse) i.p., and imaged after 10 min using a Xenogen IVIS 100 instrument. A standardized region of interest (ROI) was identified and used to determine the radiance value (photons per second per square centimeter per steradian) for each xenograft at each time point (Living Image 4.3.1). A threshold that defined the bioluminescence signal as positive was determined as the mean bioluminescence of the same ROI on animals with xenografts that were not inoculated with VZV. Animal procedures were conducted in compliance with the Animal Welfare Act and were approved by the Stanford University Administrative Panel on Laboratory Animal Care.
Plaque analysis, viral titrations, and genome copies.
For plaque size analysis, HELF monolayers were infected with rOka-ORF10-GFP, fixed after 24 or 48 h with paraformaldehyde (4% in PBS), blocked for 1 h with PBS-fetal calf serum (10%), and sequentially incubated with a mixture of monoclonal mouse anti-VZV antibodies (Meridian Lifescience, Memphis TN; catalog no. C05108MA), biotinylated anti-mouse IgG antibody (Vector Laboratories, Burlingame, CA; catalog no. BA-9200), and alkaline phosphatase-conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA; catalog no. 016-050-084); the signal was developed using Fast Red substrate (Sigma). Student's t test was used to compare the numbers of plaques in treated and mock-treated wells.
Viral titers in skin xenografts were assessed in suspensions of harvested skin xenograft tissues made in PBS, serially diluted 10-fold, and added to melanoma cell monolayers prepared in 24-well plates 24 h before use (100 μl per well; triplicate wells for each dilution). The cells were fixed after 4 days and stained as described above.
DNA was extracted from skin xenograft tissue homogenates with DNAzol as recommended by the manufacturer and tested by qualitative PCR to detect VZV genomes using 1.25 units of Taq polymerase (NEB, Ipswich, MA; catalog no. M0273), 0.2 μM each primer, and 0.2 μM deoxynucleoside triphosphates (dNTPs). The primers that were used hybridize to the promoter region of ORF59 were 5′-GAGCTCCGGTATCAAACCTCAGCGAG-3′ and 5′-CCCGGGTTTCCGGGATATTTGTGGAA-3′. The PCR cycle was as follows: 95°C for 5 min, followed by 25 cycles of 30 s at 95°C, 30 s at 58°C, and 30 s at 68°C and a final extension time of 10 min at 68°C. The PCR products were analyzed on agarose gels to confirm the presence of a fragment corresponding to the expected size. A positive control (a cosmid fragment containing the ORF59 promoter) was included, and uninfected samples and water were used as negative controls.
Western blot analysis.
HELFs were infected with VZV and treated with either DMSO or XX-650-23 (1 μM) at the time of infection. Cytoplasmic and nuclear fractions were prepared after 24 h of infection using a nuclear extract kit (Active Motif, Carlsbad, CA; catalog no. 40010) following the manufacturer's instructions. The lysates were diluted in Laemmli sample buffer (Bio-Rad, Hercules, CA), boiled for 5 min, and loaded on a precast 4 to 20% SDS-PAGE gel (Bio-Rad). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. For protein detection, the membrane was blocked in Tris-buffered saline (TBS)–Tween–5% milk and then hybridized with antibodies against IE62 (purified rabbit serum), pCREB (Cell Signaling, Danvers, MA; catalog no. 9198), alpha tubulin (Sigma, St. Louis, MO; catalog no. T5268), or lamin A/C (Cell Signaling; catalog no. 4777).
Flow cytometry.
HELFs were collected in cold PBS-EDTA, fixed with 1.5% paraformaldehyde, and permeabilized with cold methanol before staining. Detection of VZV-infected cells was done either by infecting them with rOka-ORF10-GFP or using mouse antibodies to VZV glycoprotein E (gE). Antibody binding was detected with anti-mouse Alexa-488 (Life Technologies, Carlsbad, CA; catalog no. A11001). pCREB was detected using pCREB(pS133)/ATF-1(pS63) conjugated to Alexa-647 (BD Biosciences, San Jose CA; catalog no. 558434). The data were analyzed with FlowJo software (TreeStar, Ashland, OR). Quantification of living cells was done with LIVE/DEAD fixable violet dead staining (Life Technologies; catalog no. L3595) following the manufacturer's instructions. As a control for this experiment, cells were heat treated (1 h at 50°C) and mixed with untreated cells at each time point to validate the staining and to determine the baseline for positive staining.
Immunofluorescence.
HELFs were seeded on glass coverslips 24 h prior to infection, infected with rOka-ORF10-GFP virus for 24 h, and treated with XX-650-23 for 24 h before fixation with 2% paraformaldehyde. The cells were treated with PBS-0.3% Triton X100-5% donkey serum for 1 h prior to probing with rabbit anti-pCREB (Cell Signaling) or rabbit anti-CREB (Cell Signaling) antibody overnight at 4°C. The antibody binding was detected using donkey anti-rabbit antibody conjugated to Alexa 647 fluorophore (Life Technologies), and nuclei were stained with Hoechst.
Immunohistochemistry and immunofluorescence analysis of skin sections.
Skin xenograft tissues fixed in paraformaldehyde (4% in PBS) were embedded in paraffin before sectioning (10-μm/section). The sections were deparaffinized, rehydrated, and treated for antigen retrieval (antigen-unmasking solution; H-3300; Vector Laboratories). The sections were stained for VZV with mouse monoclonal anti-VZV glycoprotein E antibody (monoclonal antibody [MAb] 81612; Millipore), biotinylated secondary anti-rabbit and anti-mouse IgG (Millipore; 20775), and horseradish peroxidase (HRP)-conjugated streptavidin (Millipore; 20774). VZV protein expression was detected with a peroxidase-HRP kit (Vector Laboratories; SK-4100), and sections were counterstained with hematoxylin. For immunofluorescence, sections were stained with the mouse anti-VZV glycoprotein E antibody (MAb 81612; Millipore) and rabbit anti-pCREB antibody (Cell Signaling). Antibody binding was detected using donkey anti-rabbit antibody conjugated to Alexa 647 and donkey anti-mouse antibody conjugated to Alexa 488.
RESULTS
VZV infection induces CREB phosphorylation in fibroblasts in vitro.
Since human fibroblasts are permissive for VZV replication in vitro, we analyzed these cells for CREB phosphorylation levels. When rOka-ORF10-GFP-infected fibroblasts were stained for pCREB and analyzed by flow cytometry, the level of pCREB was shown to be increased (Fig. 1A). To further investigate the increase in pCREB induced by VZV infection, the localization of CREB and pCREB in fibroblasts was analyzed by confocal microscopy. CREB and pCREB both showed a nuclear pattern in infected and uninfected cells (Fig. 1B and C). The intensities of nuclear expression of CREB and pCREB were quantified from images of uninfected and infected samples, distinguishing three populations of cells: uninfected, bystanders (uninfected cells in an infected sample), and infected cells, identified by GFP expression using Volocity software. The quantification showed that CREB levels were comparable in the three populations (Fig. 1D). However, pCREB was increased significantly in infected cells compared to either bystanders or uninfected cells (Fig. 1E), and pCREB localized primarily to the nuclei of infected cells.
FIG 1.

CREB and pCREB expression in VZV-infected fibroblasts in vitro. (A) Fibroblasts were infected for 24 h with rOka-ORF10-GFP virus and then fixed and probed for pCREB before analysis by flow cytometry. The levels of pCREB in control cells (shaded), bystanders (dotted line), and infected cells (solid line) are shown; the x axis shows the intensity of fluorescence, and the y axis shows the number of cells. (B to E) Fibroblasts were infected for 24 h with rOka-ORF10-GFP virus and then fixed and probed with antibodies to CREB (B) or pCREB (C). Binding was detected using Alexa-647 (red), and nuclei were stained with Hoechst. The intensity of red fluorescence indicating the level of CREB (D) or pCREB (E) was quantified in the nuclei after distinguishing infected, bystander, and uninfected populations using Velocity 6.3 software (PerkinElmer). Quantification was done on 4 to 6 fields of view, including at least 200 nuclei for each condition. The results from the different fields of view were plotted and analyzed using a Mann-Whitney test. **, P < 0,005. The error bars indicate standard errors.
We also compared the levels of pCREB in infected and uninfected fibroblasts by Western blotting of cytoplasmic and nuclear protein fractions collected 24 h postinfection. pCREB was largely present in the nuclear fraction and was induced about 2-fold in the nuclei of VZV-infected cells compared to uninfected fibroblasts (Fig. 2A). VZV replication was needed to induce CREB phosphorylation in infected cells, as shown by an absence of increased pCREB in cells infected with UV-inactivated VZV compared to VZV-infected cells (Fig. 2B). When 1 μM XX-650-23 was added to HELFs, pCREB was slightly decreased in uninfected fibroblasts. However, an increase in CREB activation following VZV infection was maintained in the presence of the drug.
FIG 2.

Analysis of pCREB expression in VZV-infected fibroblasts and effects of inhibition of pCREB transcriptional activity. (A) HELFs were infected with VZV and treated with either 0.1% DMSO or 1 μM XX-650-23 at the time of infection. Twenty-four hours postinfection, cytoplasmic and nuclear fractions were prepared. The lysates were analyzed by Western blotting with antibodies against VZV IE62 (control for infection), pCREB, alpha-tubulin (cytoplasmic protein control), and lamin A/C (nuclear protein control). Whole-cell lysates from forskolin-treated SK-H-MC human neural cells (Sigma) were included as a control for the detection of pCREB. Quantification was done using ImageJ software; the intensities of the bands detected with anti-pCREB antibody were collected and normalized to the intensities of the bands detected with anti-alpha-tubulin for cytoplasmic extracts and with anti-lamin A/C for nuclear extracts. (B) Fibroblasts were left uninfected or inoculated with VZV or UV-inactivated VZV. Extracts were prepared 24 h postinfection and analyzed by Western blotting to detect IE62, pCREB, and alpha-tubulin as a control. (C) Lysates prepared at 24 h were analyzed by Western blotting for cyclin D1 and cyclin D2. Alpha-tubulin was used as a control for cell proteins and IE62 as a control for infection. UI, uninfected; I, infected; ctrl, control. Numbers to the left of each blot are molecular masses (in kilodaltons).
Since XX-650-23 is known to affect the cell cycle by inducing arrest at the G1/S phase (33), we investigated whether VZV infection altered the expression of cyclin D1 and cyclin D2 by using Western blots comparing infected and uninfected cells. Indeed, we hypothesized that the drug might alter the cell cycle in a way that counteracted the alteration led by VZV infection. Cyclin D1 was decreased in infected cells compared to uninfected cells, and cyclin D2 might have been slightly decreased in infected cells (Fig. 2C). Investigating the levels of the two proteins in VZV-infected cells in the context of the drug treatment would have been of interest, but the inhibition of infection in the presence of the drug was so extensive that we were unable to retrieve enough infected cells treated with the drug to evaluate the combined action of the two factors on cyclin levels.
VZV kinases are not required for CREB phosphorylation.
The VZV genes ORF47 and ORF66 both encode viral serine/threonine kinases. The transcriptional activity of CREB requires phosphorylation of serine residue S133 prior to binding CBP (35, 36). To determine if the viral kinases were responsible for CREB phosphorylation, we infected HELFs with mutant loss-of-function viruses. VZV ORF47D-N contains a point mutation in the DYS region of the kinase domain (8), and pOkaORF66G102A contains a point mutation causing the replacement of the guanine at position 102 by an alanine; these mutations abolish kinase function (31). Cells were infected with parental or mutant viruses or left uninfected, collected after 24 h, stained for VZV gE and pCREB, and analyzed by flow cytometry. pCREB was increased to equivalent levels in HELFs infected with each of the viruses (Fig. 3). These results indicated that the increased phosphorylation of CREB in infected cells is related to cellular mechanisms triggered by VZV infection rather than being caused directly by VZV-encoded kinases.
FIG 3.

Analysis of involvement of VZV kinases in CREB phosphorylation upon infection. Fibroblasts were infected with VZV-pOka, VZV-ORF66G102A, VZV-chimera D, or VZV-ORF47-DN 24 h prior to fixation and staining with mouse anti-gE and rabbit anti-pCREB antibodies and analysis by flow cytometry. Shown are the levels of pCREB in uninfected control cells (shaded), bystanders (dotted lines), and infected cells (solid lines) for the indicated viruses.
Inhibiting the interaction of pCREB with p300/CBP by XX-650-23 limits VZV spread in vitro.
Although the absence of toxicity of XX-650-23 has been proven for various normal human bone marrows (33), no data were available regarding fibroblasts. Consequently, before testing whether disrupting pCREB-CBP interactions in VZV-infected cells affected viral growth, we evaluated the effects of the CREB inhibitor on uninfected HELFs. Cells were treated with either DMSO or XX-650-23 (1 μM) for 24 and 48 h before counting the cells for viability or staining them with a LIVE/DEAD fixable fluorophore. The LIVE/DEAD staining reagent binds to amine groups present at the surface of the cell and inside permeabilized cells. Dead cells are more permeable, and thus, the fluorescent molecule can enter the cytoplasm, while living cells are resistant to intracellular staining. In the presence of the drug, we observed a slight increase in fluorescence (Fig. 4A), indicating a modification of the amine groups at the surfaces of the cells or an increase in membrane permeability, allowing some of the dye to enter the cells. However, the fluorescence signal detected was lower than that detected in dead cells from the control samples prepared at the same time points. Although cell growth was also slightly slower, cell counts increased over the 48-h time period in the presence of the drug, indicating that cellular division occurred (Fig. 4B). These data showing that cell toxicity was limited established that the drug was suitable for investigating effects on VZV replication when pCREB-p300/CBP binding was inhibited.
FIG 4.

Inhibition of pCREB interaction with the p300/CBP KIX domain does not significantly affect cell survival or growth but limits VZV spread in vitro. (A) Fibroblasts were seeded 24 h prior to treatment with DMSO (carrier) or XX-650-23 (1 μM) for 24 or 48 h. The cells were stained with a violet LIVE/DEAD stain to evaluate cell death. A heat-treated control aliquot was tested at each time point. Curves with shading are DMSO treated; open curves are cells treated with two different batches of XX-650-23. (B) Cell growth was assessed by counting cells that were DMSO or XX-650-23 treated for 24 or 48 h. The results are representative of two different lots of XX-650-23. (C and D) Cells were infected with rOka-ORF10-GFP and treated with DMSO or XX-650-23 (1 μM) for 24 h or 48 h. Cells were fixed at each time point and stained for VZV antigens with a mixture of monoclonal antibodies. (C) The images on the right show magnifications of specific areas in the wells represented on the left. (D) Plaque sizes were determined from photographs by using ImageJ software (at least 35 plaques per condition). Shown are means ± standard deviations (SD); statistical significance was determined using unpaired Student t tests to compare the groups at each time point. ****, P < 0.0001.
To determine the effect of XX-650-23 on VZV spread in vitro, fibroblasts were treated with XX-650-23 at a concentration of 1 μM or with DMSO (carrier) at the time of the infection and fixed at 24 or 48 h postinfection (hpi). At both 24 and 48 hpi, the size of the plaques was decreased significantly in the presence of the drug (Fig. 4C and D). These results indicate that VZV spread in cultured cells is defective when the binding of pCREB to p300/CBP via the KIX domain is prevented.
Inhibiting the interaction of pCREB with p300/CBP limits VZV infection in skin xenografts.
To determine the biological relevance of blocking pCREB function by interfering with its interaction with p300/CBP via the KIX domain, we investigated the effects of treatment with XX-650-23 on VZV infection of skin xenografts in SCID mice in vivo. Two xenografts per mouse in two groups of five mice each were inoculated with rOka-R6362L expressing the firefly luciferase. The mice were treated daily from the day of inoculation through 14 dpi with either DMSO (carrier) or XX-650-23 (20 mg/kg of body weight per day) by i.p. injection. Uninfected control mice were included to determine a baseline of bioluminescence and to test for drug effects on the skin xenografts in the absence of VZV infection. The mice were imaged every 2 or 3 days between 7 and 14 dpi (Fig. 5). The daily treatment dose was given after imaging to avoid interference with the imaging process.
FIG 5.

Inhibition of pCREB interaction with p300/CBP interferes with VZV infection of skin xenografts as shown by in vivo imaging. Two xenografts per mouse were infected with VZV expressing firefly luciferase (rOka-R6362FL; 5.7 × 105 PFU/ml) 4 weeks after engraftment and were treated with either DMSO (left) or XX-650-23 (right) daily from 0 to 14 dpi. Mice were imaged from day 7 to day 21 postinfection at the indicated time points. Radiance units (photons per second per square centimeter per steradian) were used to compare the images. The results shown are from one experiment representative of two independent experiments.
At day 7 postinfection, 8/10 skin xenografts in the DMSO-treated group had a bioluminescence signal above the baseline versus 4/10 in the XX-650-23-treated group (P = 0.894; comparison of bioluminescence intensities) (Fig. 6A). By day 9, 10/10 xenografts were positive in the DMSO-treated group compared to 6/10 in the XX-650-23-treated group (P = 0.748). By day 21, 8/10 xenografts in the XX-650-23-treated animals were positive while 10/10 xenografts in those given DMSO remained positive (P = 0.196). When the signal intensities were compared, bioluminescence was significantly lower (P = 0.003) only at 14 dpi for the xenografts in the XX-650-23-treated group.
FIG 6.
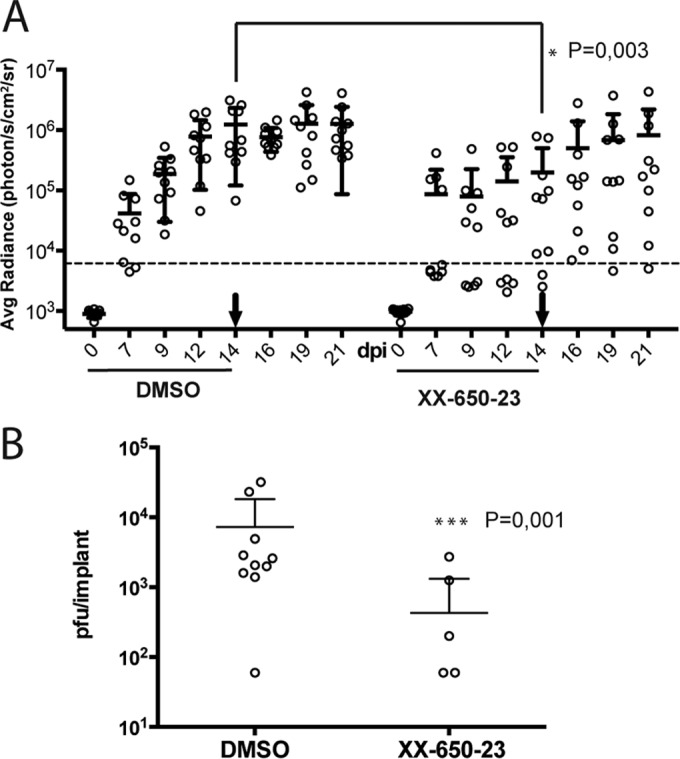
Inhibition of pCREB interaction with p300/CBP interferes with VZV replication in skin xenografts. (A) The bioluminescence signal was quantified by measuring the average radiance inside a region of interest standardized for size over each skin xenograft. The average radiance was plotted against the day postinfection. Treatments received by the mice are indicated on the x axis, and the arrows indicate the last day of treatment. The values obtained at each day were analyzed with unpaired Student t tests, and significantly different values are indicated with asterisks. Shown are means ± SD. (B) Xenografts were harvested 22 days postinfection, and titers were determined by plaque assay. Shown are means ± SD; P = 0.001 (Mann-Whitney, unpaired, nonparametric test).
In addition to the delay in the detection of a positive signal in the treated animals, the antiviral activity of XX-650-23 was further demonstrated by the increase of the bioluminescence signal in skin xenografts after discontinuing the treatment (day 14 and later) in the XX-650-23 group, indicating an increase in viral replication. In contrast, the bioluminescence signal was relatively constant between 14 and 21 days postinfection in xenografts of the DMSO-treated group, indicating a relatively constant level of infection. This seems to indicate that the treatment inhibits the replication of VZV without clearing the viral infection for at least 14 days in this experimental setting.
By 22 days postinfection, infectious VZV was recovered and VZV genomic DNA was detected (data not shown) from 10/10 xenografts in DMSO-treated animals compared to only 5/10 xenografts from animals treated with XX-650-23. The VZV titers in the skin xenografts from animals treated with the inhibitor that yielded virus were also significantly lower than those in skin xenografts from DMSO-treated animals (Fig. 6B).
CREB phosphorylation is increased in VZV skin lesions, and inhibiting pCREB interaction with p300/CBP impairs lesion formation.
Skin sections prepared from xenografts recovered 22 dpi were stained by immunochemistry to detect VZV gE. The skin xenografts recovered from DMSO-treated mice showed the expected typical VZV lesions, whereas those recovered from XX-650-23-treated mice showed smaller, more localized lesions (Fig. 7A). The effects of either the inhibitor alone or DMSO treatment on skin morphology was examined by hematoxylin-eosin staining of skin sections (22 dpi) and showed no differences between the groups (Fig. 7B).
FIG 7.

The progression of VZV skin lesion formation is limited by inhibition of pCREB interaction with p300/CBP. Human skin xenografts were harvested at 22 days postinfection, and sections were stained with anti-VZV-gE antibodies to show the foci of VZV infection (A) or with hematoxylin-eosin (B). (A) Dark-brown staining indicates DAB-positive areas, revealing VZV lesions in skin tissue. Given the size of lesions in DMSO-treated samples, images are a composite of several images (Adobe Photoshop; automated photomerge). Two different lesions are shown for each experimental condition. (B) Skin sections stained with hematoxylin-eosin show the integrity of the xenograft tissue in the absence of treatment, when treated with DMSO, or when treated with XX-650-23.
To determine if the increase in pCREB in HELFs infected with VZV in vitro was also observed in vivo, immunofluorescence dual staining was done on skin sections from infected and uninfected xenografts. pCREB was readily detected in sections of infected skin xenografts primarily in cells within the lesion area and was not detectable, or only at very low levels, in the noninfected areas of the skin tissue (Fig. 8A and B). No or very little pCREB was detected in uninfected skin (Fig. 8C). pCREB was identified predominantly in cells at the margins of the skin lesions, where gE expression was lower. Since gE is expressed most abundantly at late times, cells at the centers of the lesions are likely to be in the end stage of lytic infection, when nuclear membranes have been disrupted or the cells are nonviable. Lack of pCREB detection by immunofluorescence does not exclude the presence of some low levels of pCREB in uninfected cells surrounding the lesions or in more distant, unaffected areas, but VZV infection increased levels to where they were readily detected in cells within skin lesions.
FIG 8.

Detection of pCREB in lesions in VZV-infected skin xenografts. Human skin xenografts were harvested 22 days postinfection, and sections were stained with mouse anti-VZV-gE antibody (Alexa-488; green) and rabbit anti-pCREB antibodies (Alexa-647; red). Nuclei were detected using Hoechst staining. (A) Skin section from a mouse treated with DMSO for 14 days. (B) Skin section from a mouse treated with XX-650-23 for 14 days. (C) Skin section from a mouse that uninfected, left untreated. (D) Skin section stained with secondary antibodies and Hoechst only.
DISCUSSION
The purpose of this study was to investigate the consequences of disrupting pCREB transcriptional activity on VZV infection, using fibroblasts in vitro and human tissue xenografts in SCID mice as an in vivo system to study requirements for VZV skin pathogenesis. As part of its natural infectious cycle, VZV must infect several different cell types (6), and although similar modifications of the cellular environment are expected to occur in these different cell types, some of the remodeling of the cells is also expected to be cell type specific. In addition, pCREB has target genes involved in many cellular processes, and the consequences of an increase in pCREB could be cell type specific. Here, we determined that CREB phosphorylation is upregulated in fibroblasts, as well as in T cells (31), and together, this suggests that CREB phosphorylation is a common mechanism designed to modulate the cellular transcriptional program of VZV-infected cells. VZV encodes two viral serine/threonine kinases (ORF47 and ORF66) (37, 38). As CREB activation is mostly regulated by phosphorylation on the serine 133 residue (13, 35) and pCREB is increased upon infection, we hypothesized that one or both viral kinases could be responsible for this modification. However, measurement of the levels of CREB phosphorylation in cells infected by wild-type VZV and by mutant viruses lacking the active kinases (8, 31) showed that the viral kinases are not responsible for the increase in CREB phosphorylation. Since no other viral kinases have been identified in VZV, these observations indicate that VZV infection triggers one of the cellular pathways involved in CREB phosphorylation. CREB activation can be triggered by several signals, leading to the activation of various signaling cascades and ultimately several CREB kinases, including calmodulin kinase, PKA, and Akt, all of which are in turn involved in several regulatory pathways (39–41).
Since the phosphorylation of CREB increases following VZV infection, we hypothesized that CREB participates in a transcriptional program important in establishing VZV infection. To test this hypothesis, we used XX-650-23, which disrupts the binding of CREB to CBP, its required coactivator. This approach facilitated a direct study of the importance of CREB function regardless of which kinase cascades participate in its activation. Previous data had shown that this molecule has minimal toxicity to nonmalignant cells, and indeed, our data also show that this small molecule similarly had little effect on cell viability and fibroblast growth. We observed a decrease of pCREB when the drug was added to fibroblasts as opposed to what is observed when the drug is added to acute myeloid leukemia (AML) cells (33); however, AML cells are cancer cells, and the biology and the regulation seen in this type of cell is often not relevant to normal cells. Mice treated with up to 60 mg/kg intraperitoneally demonstrated no adverse effects (33), and in this study, there was no measurable dermal toxicity.
CREB inhibition dramatically decreased VZV propagation in vitro. Although the observation of a small-plaque phenotype in tissue culture usually predicts the outcome of VZV infection in differentiated human tissues in vivo, we have observed exceptions. Therefore, we confirmed the biological relevance of in vitro observations in the SCID mouse model of VZV pathogenesis in vivo (42). The effects on VZV infection of human skin associated with XX-650-23 treatment of the mice confirmed the phenotype observed in vitro. VZV pathogenicity was reduced significantly, as documented by reduced lesion size and lower VZV titers in xenografts when binding of p300/CBP to its partners through the KIX domain was prevented. Moreover, the results obtained by in vivo imaging were confirmed by viral titration, PCR, and immunohistochemistry, all of which showed a significant drug effect.
While these results show that VZV infection induces CREB phosphorylation and that disrupting the interaction of pCREB with its cofactor, CBP, leads to a dramatic decrease in VZV pathogenicity, further investigation will be required in order to unravel the precise mechanisms and cellular pathways involved in the process. Several hypotheses, not mutually exclusive, can be suggested. First, the pCREB/CBP complex could be required to ensure the necessary level of transcriptional activity of one or several crucial viral and/or cellular promoters. Indeed, the pCREB interaction with CBP has been demonstrated to be an important event during infection by several viral pathogens. Notably, the function of the human T-cell leukemia virus protein Tax as an activator of transcription is strictly dependent on the presence of pCREB and its association with p300/CBP (43). Along the VZV genome, CREB binding elements have been found in at least 11 viral promoters. Cellular proteins that are necessary for VZV pathogenesis may also be transcribed under the control of pCREB. Second, CREB is involved in several crucial cellular processes, such as apoptosis, cell growth and differentiation, and immune response. We showed that cyclin D1 and D2 were decreased in VZV-infected cells. It is possible that the drug used here induced opposite effects and consequently limited VZV spread. Also, CREB regulates the transcription of the antiapoptotic proteins, including Bcl-2, in various cell types, including T cells (18, 44). Although it has been shown that Bcl-2 transcription is not increased in human fibroblasts upon VZV infection (12) and that no variation in the levels of Bad and Bax, two other important apoptosis regulators, is observed (data not shown), the possibility that this mechanism could be relevant for T cells is not excluded. Also, other downstream targets involved in apoptosis inhibition, like Bcl-XL, should be investigated in the different cell types considered.
Immune evasion mechanisms are a hallmark of herpesviruses. Indeed, as persistent pathogens establishing lifelong latent infection and potentially sporadically reactivating, herpesviruses have evolved numerous immunoevasion strategies, and VZV is certainly no exception (45). In this context, it can be hypothesized that the requirement for an intact p300/CBP-pCREB interaction for the formation of skin lesions is linked to the disruption of the normal immune innate response. Indeed, the Toll-like receptor (TLR)-triggered proinflammatory response involves at least elements of the NF-κB pathway that recruits p300/CBP and interacts in the same domain as pCREB. Consequently, it has been suggested that the presence of high levels of pCREB could inhibit the proinflammatory response by competing for the recruitment of p300/CBP (19, 46, 47). Without excluding other mechanisms, CREB phosphorylation could therefore support VZV infection by its involvement in immunoevasion strategies. It is most likely that the increase in pCREB upon infection has several molecular consequences. Identifying the targets of pCREB that are important for VZV infection will be of interest in the future.
It has recently been suggested that p300/CBP could have a role in modulating the reactivation from latency of herpes simplex virus 1 (HSV-1) (48), and pCREB is also well known to be important for neuron survival (21). As VZV, like HSV, establishes latency in neurons, it will be interesting to evaluate the role of pCREB in VZV infection of neurons, both latent and lytic, which can be done in our SCIDhu mouse model of human DRG xenograft (49), to determine the effects of interfering with p300/CBP-pCREB at different stages of the in vivo infection.
Finally, current antiherpesviral therapies, like acyclovir and valacyclovir, are designed to interfere with the viral thymidine kinase and DNA polymerase (50–52), preventing viral replication. This study shows that an alternative approach may be to interfere with cell signaling pathways that are required for viral pathogenesis. The expected downside of such “cell-directed” antiviral therapies is the risk of dysregulating and potentially killing uninfected cells, as occurs during cancer chemotherapy. However, here, we showed that such a candidate drug has limited toxicity while still limiting VZV pathogenesis. Also, it can be expected that therapies directed toward cellular components required for viral infection might be less at risk for viral adaptation and resistance development. Identifying the upstream molecule(s) involved in pCREB increase will be a challenge, as several of the pathways are intricate. However, determining the critical pathway(s) involving CREB that is essential to VZV pathogenesis will be of interest for further understanding of host-pathogen interactions and also to develop new antiviral strategies.
ACKNOWLEDGMENTS
We are grateful to Leigh Zerboni, Marvin Sommer, and Phil Sung for their technical assistance in this study.
REFERENCES
- 1.Arvin A, Gliden D. 2013. Varicella-zoster virus, p 2015–2057. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Ku CC, Zerboni L, Ito H, Graham BS, Wallace M, Arvin AM. 2004. Varicella-zoster virus transfer to skin by T cells and modulation of viral replication by epidermal cell interferon-alpha. J Exp Med 200:917–925. doi: 10.1084/jem.20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilden DH, Vafai A, Shtram Y, Becker Y, Devlin M, Wellish M. 1983. Varicella-zoster virus DNA in human sensory ganglia. Nature 306:478–480. doi: 10.1038/306478a0. [DOI] [PubMed] [Google Scholar]
- 4.Cohen JI. 2010. The varicella-zoster virus genome. Curr Top Microbiol Immunol 342:1–14. doi: 10.1007/82_2010_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers MG, Duer HL, Hausler CK. 1980. Experimental infection of guinea pigs with varicella-zoster virus. J Infect Dis 142:414–420. doi: 10.1093/infdis/142.3.414. [DOI] [PubMed] [Google Scholar]
- 6.Zerboni L, Sen N, Oliver SL, Arvin AM. 2014. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol 12:197–210. doi: 10.1038/nrmicro3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arvin AM. 2006. Investigations of the pathogenesis of varicella zoster virus infection in the SCIDhu mouse model. Herpes 13:75–80. [PubMed] [Google Scholar]
- 8.Besser J, Sommer MH, Zerboni L, Bagowski CP, Ito H, Moffat J, Ku CC, Arvin AM. 2003. Differentiation of varicella-zoster virus ORF47 protein kinase and IE62 protein binding domains and their contributions to replication in human skin xenografts in the SCID-hu mouse. J Virol 77:5964–5974. doi: 10.1128/JVI.77.10.5964-5974.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moffat JF, Zerboni L, Sommer MH, Heineman TC, Cohen JI, Kaneshima H, Arvin AM. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc Natl Acad Sci U S A 95:11969–11974. doi: 10.1073/pnas.95.20.11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zerboni L, Reichelt M, Arvin A. 2010. Varicella-zoster virus neurotropism in SCID mouse-human dorsal root ganglia xenografts. Curr Top Microbiol Immunol 342:255–276. doi: 10.1007/82_2009_8. [DOI] [PubMed] [Google Scholar]
- 11.Sato B, Ito H, Hinchliffe S, Sommer MH, Zerboni L, Arvin AM. 2003. Mutational analysis of open reading frames 62 and 71, encoding the varicella-zoster virus immediate-early transactivating protein, IE62, and effects on replication in vitro and in skin xenografts in the SCID-hu mouse in vivo. J Virol 77:5607–5620. doi: 10.1128/JVI.77.10.5607-5620.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. 2012. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci U S A. 109:600–605. doi: 10.1073/pnas.1114232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez GA, Montminy MR. 1989. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 14.Shaywitz AJ, Greenberg ME. 1999. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 15.Sun P, Maurer RA. 1995. An inactivating point mutation demonstrates that interaction of cAMP response element binding protein (CREB) with the CREB binding protein is not sufficient for transcriptional activation. J Biol Chem 270:7041–7044. doi: 10.1074/jbc.270.13.7041. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, McMurray CT. 2001. Calmodulin kinase II attenuation of gene transcription by preventing cAMP response element-binding protein (CREB) dimerization and binding of the CREB-binding protein. J Biol Chem 276:1735–1741. doi: 10.1074/jbc.M006727200. [DOI] [PubMed] [Google Scholar]
- 17.Carlezon WA Jr, Duman RS, Nestler EJ. 2005. The many faces of CREB. Trends Neurosci 28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Wilson BE, Mochon E, Boxer LM. 1996. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol 16:5546–5556. doi: 10.1128/MCB.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen AY, Sakamoto KM, Miller LS. 2010. The role of the transcription factor CREB in immune function. J Immunol 185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lonze BE, Ginty DD. 2002. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623. doi: 10.1016/S0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi M, Shimomura A, Hagiwara M, Kawakami K. 1997. Phosphorylation of ATF-1 enhances its DNA binding and transcription of the Na, K-ATPase alpha 1 subunit gene promoter. Nucleic Acids Res 25:877–882. doi: 10.1093/nar/25.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brindle PK, Montminy MR. 1992. The CREB family of transcription activators. Curr Opin Genet Dev 2:199–204. doi: 10.1016/S0959-437X(05)80274-6. [DOI] [PubMed] [Google Scholar]
- 23.Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. 1995. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene 11:2509–2514. [PubMed] [Google Scholar]
- 24.Dai P, Akimaru H, Tanaka Y, Hou DX, Yasukawa T, Kanei-Ishii C, Takahashi T, Ishii S. 1996. CBP as a transcriptional coactivator of c-Myb. Genes Dev 10:528–540. doi: 10.1101/gad.10.5.528. [DOI] [PubMed] [Google Scholar]
- 25.Goodman RH, Smolik S. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev 14:1553–1577. [PubMed] [Google Scholar]
- 26.Sen N, Mukherjee G, Sen A, Bendall SC, Sung P, Nolan GP, Arvin AM. 2014. Single-cell mass cytometry analysis of human tonsil T cell remodeling by varicella zoster virus. Cell Rep 8:633–645. doi: 10.1016/j.celrep.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desloges N, Rahaus M, Wolff MH. 2008. The phosphorylation profile of protein kinase A substrates is modulated during varicella-zoster virus infection. Med Microbiol Immunol 197:353–360. doi: 10.1007/s00430-007-0068-8. [DOI] [PubMed] [Google Scholar]
- 28.Bannister AJ, Kouzarides T. 1995. CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J 14:4758–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niizuma T, Zerboni L, Sommer MH, Ito H, Hinchliffe S, Arvin AM. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J Virol 77:6062–6065. doi: 10.1128/JVI.77.10.6062-6065.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nour AM, Reichelt M, Ku CC, Ho MY, Heineman TC, Arvin AM. 2011. Varicella-zoster virus infection triggers formation of an interleukin-1beta (IL-1beta)-processing inflammasome complex. J Biol Chem 286:17921–17933. doi: 10.1074/jbc.M110.210575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaap-Nutt A, Sommer M, Che X, Zerboni L, Arvin AM. 2006. ORF66 protein kinase function is required for T-cell tropism of varicella-zoster virus in vivo. J Virol 80:11806–11816. doi: 10.1128/JVI.00466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zerboni L, Hinchliffe S, Sommer MH, Ito H, Besser J, Stamatis S, Cheng J, Distefano D, Kraiouchkine N, Shaw A, Arvin AM. 2005. Analysis of varicella zoster virus attenuation by evaluation of chimeric parent Oka/vaccine Oka recombinant viruses in skin xenografts in the SCIDhu mouse model. Virology 332:337–346. doi: 10.1016/j.virol.2004.10.047. [DOI] [PubMed] [Google Scholar]
- 33.Mitton B, Chae H-D, Hsu K, Dutta R, Aldana-Masangkay G, Ferrari R, Davis K, Tiu BC, Kaul A, Lacayo N, Dahl G, Xie F, Li BX, Breese MR, Landaw EM, Nolan G, Pellegrini M, Romanov S, Xiao X, Sakamoto KM. 23 May 2016. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia, in press. doi: 10.1038/leu.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones JO, Sommer M, Stamatis S, Arvin AM. 2006. Mutational analysis of the varicella-zoster virus ORF62/63 intergenic region. J Virol 80:3116–3121. doi: 10.1128/JVI.80.6.3116-3121.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez GA, Yamamoto KK, Fischer WH, Karr D, Menzel P, Biggs W III, Vale WW, Montminy MR. 1989. A cluster of phosphorylation sites on the cyclic AMP-regulated nuclear factor CREB predicted by its sequence. Nature 337:749–752. doi: 10.1038/337749a0. [DOI] [PubMed] [Google Scholar]
- 36.Papavassiliou AG. 1994. The CREB/ATF family of transcription factors: modulation by reversible phosphorylation. Anticancer Res 14:1801–1805. [PubMed] [Google Scholar]
- 37.Heineman TC, Seidel K, Cohen JI. 1996. The varicella-zoster virus ORF66 protein induces kinase activity and is dispensable for viral replication. J Virol 70:7312–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kenyon T, Grose C. 2010. VZV ORF47 serine protein kinase and its viral substrates. Curr Top Microbiol Immunol 342:99–111. doi: 10.1007/82_2009_5. [DOI] [PubMed] [Google Scholar]
- 39.Bir SC, Xiong Y, Kevil CG, Luo J. 2012. Emerging role of PKA/eNOS pathway in therapeutic angiogenesis for ischaemic tissue diseases. Cardiovasc Res 95:7–18. doi: 10.1093/cvr/cvs143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carnero A, Paramio JM. 2014. The PTEN/PI3K/AKT pathway in vivo, cancer mouse models. Front Oncol 4:252. doi: 10.3389/fonc.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu B, Ragazzon B, Rizk-Rabin M, Bertherat J. 2012. Protein kinase A alterations in endocrine tumors. Horm Metab Res 44:741–748. doi: 10.1055/s-0032-1316292. [DOI] [PubMed] [Google Scholar]
- 42.Moffat JF, Stein MD, Kaneshima H, Arvin AM. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol 69:5236–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geiger TR, Sharma N, Kim YM, Nyborg JK. 2008. The human T-cell leukemia virus type 1 tax protein confers CBP/p300 recruitment and transcriptional activation properties to phosphorylated CREB. Mol Cell Biol 28:1383–1392. doi: 10.1128/MCB.01657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang P, Yan H, Li J-C. 2007. CREB-mediated Bcl-2 expression in trichosanthin-induced Hela cell apoptosis. Biochem Biophys Res Commun 363:101–105. doi: 10.1016/j.bbrc.2007.08.141. [DOI] [PubMed] [Google Scholar]
- 45.Abendroth A, Arvin AM. 2001. Immune evasion as a pathogenic mechanism of varicella zoster virus. Semin Immunol 13:27–39. doi: 10.1006/smim.2001.0293. [DOI] [PubMed] [Google Scholar]
- 46.Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. 1996. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem 271:20828–20835. doi: 10.1074/jbc.271.34.20828. [DOI] [PubMed] [Google Scholar]
- 47.Parry GC, Mackman N. 1997. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol 159:5450–5456. [PubMed] [Google Scholar]
- 48.Du T, Zhou G, Roizman B. 2013. Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc Natl Acad Sci U S A 110:E2621–E2628. doi: 10.1073/pnas.1309906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zerboni L, Ku CC, Jones CD, Zehnder JL, Arvin AM. 2005. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc Natl Acad Sci U S A 102:6490–6495. doi: 10.1073/pnas.0501045102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elion GB. 1982. Mechanism of action and selectivity of acyclovir. Am J Med 73:7–13. doi: 10.1016/0002-9343(82)90055-9. [DOI] [PubMed] [Google Scholar]
- 51.Gnann JW Jr, Barton NH, Whitley RJ. 1983. Acyclovir: mechanism of action, pharmacokinetics, safety and clinical applications. Pharmacotherapy 3:275–283. doi: 10.1002/j.1875-9114.1983.tb03274.x. [DOI] [PubMed] [Google Scholar]
- 52.Matthews T, Boehme R. 1988. Antiviral activity and mechanism of action of ganciclovir. Rev Infect Dis 10(Suppl 3):S490–S494. doi: 10.1093/clinids/10.Supplement_3.S490. [DOI] [PubMed] [Google Scholar]