

ABSTRACT
Human noroviruses (HuNoVs), named after the prototype strain Norwalk virus (NV), are a leading cause of acute gastroenteritis outbreaks worldwide. Studies on the related murine norovirus (MNV) have demonstrated the importance of an interferon (IFN) response in host control of virus replication, but this remains unclear for HuNoVs. Despite the lack of an efficient cell culture infection system, transfection of stool-isolated NV RNA into mammalian cells leads to viral RNA replication and virus production. Using this system, we show here that NV RNA replication is sensitive to type I (α/β) and III (interleukin-29 [IL-29]) IFN treatment. However, in cells capable of a strong IFN response to Sendai virus (SeV) and poly(I·C), NV RNA replicates efficiently and generates double-stranded RNA without inducing a detectable IFN response. Replication of HuNoV genogroup GII.3 strain U201 RNA, generated from a reverse genetics system, also does not induce an IFN response. Consistent with a lack of IFN induction, NV RNA replication is enhanced neither by neutralization of type I/III IFNs through neutralizing antibodies or the soluble IFN decoy receptor B18R nor by short hairpin RNA (shRNA) knockdown of mitochondrial antiviral signaling protein (MAVS) or interferon regulatory factor 3 (IRF3) in the IFN induction pathways. In contrast to other positive-strand RNA viruses that block IFN induction by targeting MAVS for degradation, MAVS is not degraded in NV RNA-replicating cells, and an SeV-induced IFN response is not blocked. Together, these results indicate that HuNoV RNA replication in mammalian cells does not induce an IFN response, suggesting that the epithelial IFN response may play a limited role in host restriction of HuNoV replication.
IMPORTANCE Human noroviruses (HuNoVs) are a leading cause of epidemic gastroenteritis worldwide. Due to lack of an efficient cell culture system and robust small-animal model, little is known about the innate host defense to these viruses. Studies on murine norovirus (MNV) have shown the importance of an interferon (IFN) response in host control of MNV replication, but this remains unclear for HuNoVs. Here, we investigated the IFN response to HuNoV RNA replication in mammalian cells using Norwalk virus stool RNA transfection, a reverse genetics system, IFN neutralization reagents, and shRNA knockdown methods. Our results show that HuNoV RNA replication in mammalian epithelial cells does not induce an IFN response, nor can it be enhanced by blocking the IFN response. These results suggest a limited role of the epithelial IFN response in host control of HuNoV RNA replication, providing important insights into our understanding of the host defense to HuNoVs that differs from that to MNV.
INTRODUCTION
Noroviruses (NoVs) are a group of positive-strand RNA viruses classified into the Norovirus genus in the Caliciviridae family. They are genetically divided into at least six genogroups associated with specific hosts: GI (human), GII (human), GIII (bovine), GIV (human and feline), GV (murine), and GVI (canine), which can be further divided into different genotypes. The prototype strain Norwalk virus (NV) represents genogroup I, genotype 1 (GI.1). NoVs that infect humans belong to genogroups GI, GII, and GIV, together referred to as human noroviruses (HuNoVs). HuNoVs are the leading cause of epidemic gastroenteritis worldwide, and illness can be particularly severe in infants, young children, and the elderly (1–4). Among HuNoVs, GII.4 noroviruses account for the majority of epidemic outbreaks of viral gastroenteritis, and new GII.4 variants emerge every 2 to 3 years replacing the previously dominant variants (5). Recent examples include the 2012-2013 winter outbreak of gastroenteritis caused by an emergent GII.4 variant, Sydney/2012 (6), and the rapid emergence of a fast-evolving GII.17 variant in late 2014 (7, 8).
Despite the disease burden of HuNoVs that documents the need for effective prevention and therapy strategies, currently there are no vaccines or antiviral drugs available to counter these viruses. This is largely due to the inability to efficiently propagate HuNoVs in cell culture and the lack of a simple small-animal infection model. Experimental infection studies in volunteers are currently the main strategy used to study antibody and serological responses to virus infection with NV and other HuNoVs (9–11). Studies using gnotobiotic pigs and calves inoculated with a GII.4 strain of HuNoV have shown that the infected animals develop diarrhea and virus shedding, similar to infections in humans, with histopathological changes in the intestinal epithelium and the presence of viral capsid protein in intestinal epithelial cells (12, 13), but these costly animal models are not widely used. The discovery that murine norovirus (MNV) can be grown in cultured macrophages and dendritic cells has provided a new model to investigate norovirus biology and pathogenesis (14, 15). However, since HuNoVs and MNV infect different cell types (15, 16) (also see Discussion), it remains unclear whether MNV is a model that recapitulates all the biological characteristics of HuNoVs. Recent studies have reported that GII.4 HuNoV can infect B cells in vitro (17) and macrophage-like cells in immune-deficient mice in vivo (18), representing some progress toward an in vitro cultivation system and a small-animal model for HuNoV. However, considering the immune cell tropism in these systems and new evidence detecting HuNoV antigen in intestinal biopsy specimens of chronically infected transplant patients (19), in vitro or in vivo models in which HuNoV can infect intestinal epithelial cells are still needed.
The HuNoV RNA genome is a single positive-strand RNA typically 7.5 to 7.7 kb in length, covalently linked to a small virus-encoded protein, VPg (viral protein genome-linked), at the 5′ end and polyadenylated at the 3′ end (20). The genomic RNA contains three open reading frames (ORFs). ORF1 encodes a nonstructural polyprotein that is cleaved by its own protease (Pro) to generate at least six distinct proteins: p48-p41-p22-VPg-Pro-Pol (where Pol is polymerase), while ORF2 encodes the major capsid protein VP1, and ORF3 encodes the minor capsid protein VP2 (Fig. 1A) (21–23). Upon infection of cells and uncoating of the virus particle, the released viral genomic RNA is believed to function as mRNA to direct the translation of the ORF1 polyprotein, which is auto-processed into individual nonstructural proteins. The nonstructural proteins assemble into a replication complex to produce both new genomic RNA and a 3′ coterminal subgenomic RNA expressing structural proteins VP1 (ORF2) and VP2 (ORF3). The structural proteins and newly synthesized genomic RNAs assemble into new virus particles that are subsequently released from the cells (24). Since expression of VP1 is predominantly dependent on the production of a subgenomic RNA, it is widely considered a hallmark of HuNoV RNA replication (24).
FIG 1.

NV RNA replicates in 293FT cells. (A) Schematic drawing of NV genome RNA showing viral proteins encoded by each of the three ORFs. (B) Western blotting detection of VPg precursors and VP1 in NV RNA-transfected 293FT cells. Cells were transfected with carrier RNA (−) or NV RNA (+) and incubated for 48 h. Note that mature VPg (20 kDa) was not detectable by Western blotting. Actin and a nonspecific protein band served as equal loading controls. (C) NV VP1 expression is RNA replication dependent. 293FT cells were transfected with in vitro-transcribed NV genomic RNA (NV IVT RNA) or stool-isolated NV RNA and incubated for 48 h. VPg precursors and VP1 in cell lysates were detected by Western blotting. VP1 was concentrated by immunoprecipitation (IP) prior to Western blotting. (D and E) Kinetics of expression of VP1 and NV RNA during NV RNA replication. 293FT cells were transfected with carrier RNA or NV RNA and incubated for 8, 24, 48, and 72 h. At each time point, cells were either lysed for IP and Western blotting of VP1 (D) or extracted for cellular RNA to quantify NV RNA by RT-qPCR (E). WB, Western blotting.
Due to the short course of HuNoV disease, which typically lasts for 1 to 3 days in healthy adults, it has been hypothesized that innate immune responses may play an important role in the control of HuNoV infection. Innate immune responses, including the synthesis and secretion of interferons (IFNs), form the first line of defense against virus infections. The RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation-associated gene 5) cellular helicases and Toll-like receptor 3 (TLR3) are pattern recognition receptors (PRRs) that sense virus-derived RNA within infected cells and initiate signal transduction cascades through their adaptor proteins, mitochondrial antiviral signaling protein (MAVS) and TRIF (TIR domain-containing adaptor inducing IFN-β), respectively. These adaptors engage downstream kinases to activate two crucial transcription factors, NF-κB (nuclear factor kappa B) and interferon regulatory factor 3 (IRF3), resulting in the induction of IFNs. Secreted IFNs can function through autocrine and paracrine signaling by binding to IFN receptors (IFNRs) and activating the JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathway to induce IFN-stimulated genes (ISGs) that ultimately establish an antiviral state (25, 26). Among the three types of IFNs, type I IFNs, including mainly IFN-α and IFN-β, and type III IFNs, including interleukin-29 ([IL-29] IFN-λ1), IL-28A (IFN-λ2), and IL-28B (IFN-λ3), share similar expression patterns and antiviral functions, while the sole type II IFN, IFN-γ, is produced by T cells and natural killer cells and is involved in immune and inflammatory responses (27, 28).
Studies using MNV have provided strong support for the role of the IFN responses in control of norovirus replication in vivo and in vitro. In particular, these studies have shown that MNV infection strongly induces type I IFNs in cultured macrophages and dendritic cells (29) and that MNV replicates to higher titers in cultured macrophages or dendritic cells derived from STAT1−/−, MDA5−/−, IFNAR1−/−, or IRF3−/− mice (15, 29, 30). Recent studies also showed that type III IFNs control persistent enteric MNV infection and that type III IFN treatment cures persistent MNV infection in mice (31, 32). However, without a robust in vitro infection system for HuNoVs, it remains unclear whether the IFN response also plays a key role in control of HuNoV infection.
To overcome the lack of a cell culture infection system for HuNoVs, our laboratory has reported that transfection of human hepatoma Huh-7 cells with NV RNA isolated from stool samples of infected volunteers leads to a single cycle of RNA replication and release of viral particles into the culture medium (33). Recently, our laboratory also reported a plasmid-based reverse genetics system for the GII.3 HuNoV strain U201 that allows a low level of RNA replication and virus production (34). The goal of the studies reported here was to use these two systems to investigate the cellular IFN response to HuNoV RNA replication. We found that replication of NV and U201 RNA in cultured mammalian cells does not induce an IFN response and that NV RNA replication is not enhanced by neutralization of type I/III IFNs or by knockdown of MAVS or IRF3 in the IFN induction pathways. Furthermore, we show that NV RNA replication does not block IFN induction elicited by an exogenous virus. These results suggest an unexpected, limited role of IFN response in control of HuNoV RNA replication in cultured mammalian epithelial cells.
MATERIALS AND METHODS
Cells and viruses.
The 293FT cell line, derived from human embryonic kidney HEK293 cells transformed with the simian virus 40 (SV40) large T antigen, was purchased from Invitrogen. The interferon-stimulated response element (ISRE)-luciferase (Luc) reporter cell line, 293FT-ISRE-Luc, was generated by transduction of 293FT cells with ISRE-Luc reporter lentiviral particles (CLS-008L; SABioscinces) and selected with 30 μg/ml puromycin (Sigma-Aldrich). Stable short hairpin RNA (shRNA) knockdown 293FT cell lines 293FT-shMAVS and 293FT-shIRF3 were generated by transduction of 293FT cells with shRNA lentiviral particles of MAVS (kindly provided by Zongdi Feng, Nationwide Children's Hospital) and IRF3 (sc-35710-V; Santa Cruz Biotechnology), respectively, and selected with 30 μg/ml puromycin. The human hepatoma cell line Huh-7 was kindly provided by Shinji Makino (University of Texas Medical Branch). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 8% fetal bovine serum (FBS), with the addition of 30 μg/ml puromycin for puromycin-selected cells.
NV stool specimen lot 51899 was obtained from a human volunteer experimentally infected with NV and processed as previously reported for lot 42399 (35, 36). The stool sample was diluted 1:10 (wt/vol) with phosphate-buffered saline (PBS), clarified by centrifugation, and filtered through a 0.45-μm-pore-size filter. Human volunteer studies were approved by the Institutional Review Board of Baylor College of Medicine, and written consent was obtained from all volunteers. The cell culture-adapted hepatitis A virus (HAV) strain HM175/18f was kindly provided by Stanley Lemon (University of North Carolina at Chapel Hill) and propagated in Huh-7 cells. Sendai virus (SeV) Cantell strain (Charles River) was kindly provided by Shinji Makino (University of Texas Medical Branch).
Plasmids, antibodies, and proteins.
The following expression plasmids were originally constructed (34) and kindly provided by Kazuhiko Katayama (National Institute of Infectious Diseases [NIID], Japan): empty vector plasmid pKS435; the plasmid containing human norovirus GII.3 strain U201 full-length genome cDNA, pHuNoV-U201F; the mutant full-length plasmid expressing U201-Δ4607, pHuNoV-U201FΔ4607, in which a deletion of a guanine (G) nucleotide at position 4607 (Δ4607) of the ORF1 region causes a frameshift and premature termination of the viral polymerase (Pol); the full-length U201-RLuc reporter genome plasmid, pHuNoV-U201F-ORF2RLuc, in which a Renilla luciferase (RLuc) gene was inserted in frame into the VP1 (ORF2) region; and the mutant U201-Δ4607-RLuc reporter genome plasmid, pHuNoV-U201FΔ4607-ORF2RLuc, which contains the same Δ4607 mutation as described above. Details of these plasmids have been previously published (34). The plasmid pT7-NV, which contains the complete NV cDNA under the T7 promoter, has been previously described (37). The IFN-β promoter-luciferase (IFN-β-Luc) reporter plasmid was kindly provided by Hiroki Kato (Osaka University, Japan). The TK-RLuc reporter plasmid pGL4.74, which constitutively expresses RLuc under the human thymidine kinase (TK) promoter, was purchased from Promega.
A rabbit polyclonal antiserum to NV VPg was generated by immunizing rabbits with recombinant NV VPg that was expressed and purified from Escherichia coli as previously reported (11). Rabbit and guinea pig polyclonal antisera to NV VP1 were generated by immunizing animals with virus-like particles (VLPs) formed by NV VP1 and VP2 that were expressed and purified from a baculovirus expression system (38). A rabbit polyclonal antiserum to NV p48 was previously generated in our laboratory by immunizing rabbits with a synthetic peptide of NV p48 amino acids (aa) 70 to 83 (39). Rabbit polyclonal antisera to U201 Pol, p35 (N terminus), and p41, mouse monoclonal antibody to U201 VPg, and guinea pig polyclonal antiserum to U201 VP1 were kindly provided by Kazuhiko Katayama (NIID, Japan). Other antibodies used in Western blotting and immunofluorescence (IF) staining included the following: rabbit monoclonal anti-IRF3 D6I4C (11904S; Cell Signaling Technology), rabbit anti-ISG56 (p56) (PA3-848; Thermo Fisher Scientific), mouse monoclonal anti-double-stranded RNA (dsRNA) J2 (Scicons, Hungary), mouse monoclonal anti-HAV K3-4C8 (Commonwealth Serum Laboratories, Victoria, Australia), rabbit anti-MAVS (A300-782A; Bethyl Laboratories), and mouse monoclonal anti-actin (ab6276; Abcam).
IFN neutralizing antibodies and their dilutions included the following: sheep anti-IFN-α (31100-1; PBL Assay Science) at 1:4,000, sheep anti-IFN-β (31400-1; PBL Assay Science) at 1:4,000, rabbit anti-IFN-β (31410-1; PBL Assay Science) at 1:4,000, mouse monoclonal anti-IFN-α/β receptor chain 2 (IFNAR2) (21385-1; PBL Assay Science) at 1:1,000, a cocktail of the above four antibodies at the same dilutions for neutralization of type I IFNs, goat anti-IL-29 (IFN-λ1) with cross-reactivity to IL-28A (IFN-λ2) and IL-28B (IFN-λ3) (AF1598; R&D Systems) at 1:100, goat anti-IL-10 receptor β (IL10R2) (AF874; R&D Systems) at 1:100, sheep anti-IL-28 receptor α (IL28R1) (AF5260; R&D Systems,) at 1:100, and a cocktail of the above three antibodies at the same dilutions for neutralization of type III IFNs.
IFN-α (PHP108Z; AbD Serotec) and IFN-β (407318; CalBiochem) were used at 1,000 U/ml unless otherwise indicated. IL-29 (IFN-λ1) (1598-IL; R&D Systems) was used at 50 ng/ml or as indicated in the dose-dependence experiment. The vaccinia virus B18R recombinant protein (14-8185-62; eBioscience), supplied in 1% bovine serum albumin (BSA) carrier, was used at 125 ng/ml. Equally diluted BSA was used as a negative control.
In addition to NV VLPs (38), VLPs formed by VP1 and VP2 capsid proteins of HuNoV GII.3 strain TCH-104 were expressed and purified from a baculovirus expression system as previously described (40).
NV stool RNA isolation and transfection.
Purification of NV particles from NV stool filtrate using rabbit anti-NV VP1 antibody-coated magnetic beads (Bio-Rad) and isolation of NV RNA from purified virus particles by a modified QIAamp viral RNA minikit (Qiagen) protocol have been described previously (41). The yield of NV stool RNA from 20-ml stool filtrate was typically 60 μl of RNA at a concentration of 60 to 70 ng of RNA/μl (mostly carrier RNA) containing 3 × 107 to 4 × 107 NV genome copies/μl. For RNA transfection, 293FT cells were seeded into 48-well plates with 250 μl of medium/well or 96-well plates with 100 μl of medium/well and cultured to 60 to 70% confluence. In some experiments the cells were pretreated with IFNs (24 h), IFN neutralizing antibodies (6 h), or B18R protein (1 h). NV stool RNA or carrier RNA as a negative control was transfected into the cells using TransIT-mRNA transfection reagent (Mirus Bio) according to the manufacturer's instructions with the following modifications. For a 48-well plate (for Western blotting) or eight-chamber slide (for IF) format, 200 ng of NV stool RNA (typically containing 1 × 108 NV genome copies) was mixed sequentially with 25 μl of Opti-MEM (Invitrogen) containing 0.4 U/μl Optizyme RNase inhibitor (Fisher Scientific), 0.5 μl of mRNA boost reagent (Mirus Bio), and 0.5 μl of TransIT-mRNA reagent (Mirus Bio); the mixture was then incubated for 1 min at room temperature and added to the cell culture. Following the same procedure, for a 96-well plate format (for 293FT-ISRE-Luc assay), 240 ng of NV stool RNA was mixed sequentially with 30 μl of Opti-MEM, 0.6 μl of mRNA boost reagent, and 0.6 μl of TransIT-mRNA reagent; the mixture was incubated for 1 min and distributed at 10 μl/well in triplicate to the cell culture. In most experiments, cells were lysed or fixed at 48 h post-RNA transfection unless otherwise indicated.
For an experiment that compared transfection of NV stool RNA to that of in vitro-transcribed NV genomic RNA (NV IVT RNA), the plasmid pT7-NV (37) was linearized with SalI as the template to synthesize NV genomic RNA by in vitro transcription using a mMESSAGE mMACHINE T7 transcription kit (Thermo Fisher Scientific). Transfection of NV IVT RNA was performed in essentially the same manner as described above for NV stool RNA.
RT-qPCR of NV RNA.
To quantify NV RNA replication in transfected cells, a reverse transcription-quantitative PCR (RT-qPCR) assay was performed as previously described (33, 36) with the following modifications. Standard NV RNA transcripts were produced by in vitro transcription from the plasmid pT7-NV (37) as described above. Five 10-fold dilutions of the NV RNA transcripts (500 to 5,000,000 genome copies), each in triplicate, were used to generate a standard curve. At 8, 24, 48, and 72 h following NV stool RNA transfection in a 48-well plate format, cellular RNAs were extracted using an RNeasy minikit (Qiagen), and 1/10 (5 μl) of the total RNA (50 μl/well) was used in the RT-qPCR assay. All samples were quantified using RNAs from triplicate wells. The primers for the RT-qPCR assay were antisense primer p165 (complementary to NV nucleotides [nt] 4689 to 4715) and sense primer p166 (NV nt 4641 to 4658); the probe was p167 (NV nt 4660 to 4685; 5′ 6-carboxyfluorescein [FAM], 3′ 6-carboxytetramethylrhodamine [TAMRA]) (36). Reactions were performed using a qScript XLT One-Step RT-qPCR ToughMix reagent with 6-carboxy-X-rhodamine (ROX) (Quanta Biosciences) on a StepOnePlus real-time PCR system (Thermo Fisher Scientific) with StepOne Software (Thermo Fisher Scientific). The levels of NV RNA in transfected cells are presented as genome copies/well.
293FT-ISRE-Luc reporter assay.
For experiments that measured the IFN response to NV RNA replication, the 293FT-ISRE-Luc reporter cells were transfected with NV stool RNA or with carrier RNA as a negative control and incubated for 48 h or for 8, 24, 48, and 72 h in a time course experiment. For an experiment that measured the IFN response to VLPs, the 293FT-ISRE-Luc reporter cells were treated with VLPs of NV (38) or GII.3 HuNoV strain TCH-104 (40) at 12- and 36-μg/ml concentrations in the medium for 6 h. As a positive control for both experiments, cells were treated with 100 μg/ml poly(I·C) (Invivogen) in the medium for 6 h. For experiments that evaluated efficacies and stabilities of IFN neutralizing antibodies, at the end of the IFN neutralization/NV RNA transfection experiments, the spent medium containing the antibody was collected before the cells were lysed for Western blotting. The spent medium was added at 50 μl/well to 293FT-ISRE-Luc reporter cells that were seeded into 96-well plates with 100 μl of medium/well and incubated for 1 h. IFN-α, IFN-β, or IL-29 was then added at 50 μl/well (total volume, 200 μl/well) to a final concentration of 500 U/ml (IFN-α/β) or 50 ng/ml (IL-29) and incubated for 18 h. At the end of the incubation, cells were lysed with 100 μl/well cell culture lysis buffer (Promega). For the luciferase assay, 2 μl of the cell lysate was mixed with 50 μl of luciferase assay reagent (Promega) in a 3.5-ml polypropylene tube (Sarstedt) and immediately measured for luminescence on a Sirius Luminometer (Berthold) programmed with 2 s of delay and 10 s of signal integration. Statistically significant differences between samples were calculated by Student's t test. The results presented are representative of experiments performed at least twice with similar results.
Plasmid transfection and dual-luciferase assay.
For U201 reverse genetics experiments, 293FT cells seeded into a 48-well plate (for Western blotting) or eight-chamber slide (for IF) with 250 μl of medium/well and cultured to 70 to 80% confluence were transfected with 200 ng of vector, U201, or U201-Δ4607 full-length genome plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 24 h posttransfection (hpt), cells were either lysed with 100 μl/well cell culture lysis buffer (Promega) for Western blotting or fixed with methanol-acetone (1:1) for IF microscopy.
For an IFN-β-Luc reporter assay, 293FT cells seeded into a 48-well plate were transfected with a combination of 100 ng of IFN-β-Luc reporter plasmid, 10 ng of TK-RLuc reporter plasmid (as an internal control for transfection efficiency), and 100 ng of vector, U201, or U201-Δ4607 full-length genome plasmid. For combined IFN-β-Luc and U201-RLuc reporter assays, cells were transfected with a combination of 100 ng of IFN-β-Luc reporter plasmid and 100 ng of vector, U201-RLuc, or U201-Δ4607-RLuc reporter genome plasmid. As a positive control for both assays, at 7 hpt, the vector-transfected cells were infected with SeV at 40 hemagglutination units (HA)/ml. At 24 hpt (17 h postinfection [hpi] for SeV), cells were lysed with 100 μl of passive lysis buffer (Promega). The Luc (firefly luciferase) and RLuc (Renilla luciferase) activities in 10 μl of cell lysate were measured using dual-luciferase assay reagents (Promega), according to the manufacturer's instructions, on a Sirius Luminometer (Berthold) as described above.
Western blotting and IF microscopy.
For Western blotting of protein expression, the cell monolayer in a 48-well plate was lysed in 100 μl/well NP-40 lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 10% glycerol, 1 mM dithiothreitol [DTT], 1× protease inhibitor cocktail [CalBiochem]). In some experiments, cell lysates from luciferase assays, when supplemented with 1× protease inhibitor cocktail (CalBiochem), were also used for Western blotting. Each cell lysate was clarified by centrifugation, and 10 μl of lysate was subjected to SDS-PAGE and Western blotting using antibodies against viral or cellular proteins. In some experiments, NV VP1 in the cell lysates was first concentrated by immunoprecipitation with rabbit anti-NV VP1 antibody-coated magnetic beads and then analyzed by Western blotting using guinea pig anti-NV VP1 antibody.
For IF microscopy, cells cultured in eight-chamber slides (for confocal IF) or 48-well plates (for low-magnification IF) were fixed with methanol-acetone (1:1) and blocked with 1% BSA. The cell monolayer was incubated with primary antibodies against viral antigens, dsRNA, or cellular proteins, followed by corresponding fluorophore-conjugated secondary antibodies. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). Low-magnification (×4) images were collected using an Olympus IX70 inverted fluorescence microscope. Confocal images were collected using a Nikon A1 confocal laser scanning microscope with Nikon Elements software (version 3.5; Melville, NY) at the Integrated Microscopy Core of Baylor College of Medicine.
RESULTS
Replication of NV RNA in 293FT cells.
To investigate the IFN response to NV RNA replication in cultured mammalian cells, we first sought a cell line that is both permissive for efficient NV RNA replication and capable of an IFN response. Although our laboratory previously reported that NV RNA can replicate in Huh-7 hepatoma cells following transfection (33), these cells are defective in the TLR3 signaling pathway of IFN induction (42, 43), which we confirmed by extracellular poly(I·C)-stimulated IFN-β-Luc and ISRE-Luc reporter assays (data not shown). Through screening of different cell lines, 293FT cells, which were derived from SV40 large T antigen-transformed HEK293 cells, were found to support NV RNA replication. Transfection of 293FT cells with NV RNA isolated from stool samples of an NV-challenged volunteer resulted in expression of ORF1-encoded nonstructural proteins such as VPg precursors and ORF2-encoded structural protein VP1, both readily detected by Western blotting (Fig. 1B). The appearance of multiple VPg precursors, among which p22-VPg-Pro-Pol seemed to be a major precursor, confirmed the synthesis and auto-processing of the ORF1 polyprotein (Fig. 1B). Consistent with previous cell culture expression of ORF1 (41), full-length ORF1 or precursors larger than p22-VPg-Pro-Pol were not detected due to rapid cotranslational processing of p48 and p41 (Fig. 1B). Interestingly, the smallest VPg precursor detected by Western blotting was p22-VPg (Fig. 1B), believed to be a membrane-associated form of VPg (via the membrane association domain in p22) that is important for initiation of RNA replication (44). The mature VPg, a 20-kDa protein believed to be covalently linked to the 5′ end of viral genome RNA, was not detected by Western blotting (Fig. 1B). In our previous study, mature VPg was detected by Western blotting in cell culture expression of the ORF1 polyprotein processing precursors (41). Whether the mature VPg is unstable or its level is very low in the context of RNA replication remains to be further investigated.
The detection of VP1 (Fig. 1B) confirmed RNA replication since this protein is not present in the input RNA and not translated from the input RNA following transfection (due to the frameshift between ORF1 and ORF2), but it is produced from subgenomic RNA as result of RNA replication (24). It has been reported that in a bovine GIII NoV, VP1 can be produced by translation termination reinitiation (TTR) between ORF1 and ORF2 (VP1) without the need for subgenomic RNA (45). To confirm that NV VP1 expression is due to RNA replication, not to the TTR mechanism, we compared transfection of stool-isolated NV RNA to that of NV genomic RNA synthesized from in vitro transcription (IVT). Instead of a 5′ VPg that is essential for NV RNA replication (33), the NV IVT RNA contains a 5′ cap analog that is incorporated during IVT and, therefore, is capable of translation but unable to replicate. Indeed, translation and auto-processing of the ORF1 polyprotein were observed in both NV RNA- and NV IVT RNA-transfected cells, as evidenced by the Western blot detection of VPg precursors (Fig. 1C, VPg panel). However, the NV IVT RNA, unlike NV RNA, failed to produce VP1 (Fig. 1C, VP1 panel), indicating that NV VP1 expression is strictly dependent on RNA replication.
To further confirm the correlation between VP1 expression and NV RNA replication, we compared the kinetics of expression of VP1 and NV RNA in transfected cells in a time course experiment. Immunoprecipitation (IP) and Western blotting of VP1 in NV RNA-transfected cells showed a typical time-dependent expression pattern in which VP1 was undetectable at 8 h, appeared at 24 h, peaked at 48 h, and declined at 72 h (Fig. 1D). Similar replication kinetics of NV RNA in transfected cells were shown by RT-qPCR assay (Fig. 1E). These results, together with the NV IVT RNA data, confirmed that VP1 expression depends on and closely correlates with NV RNA replication. We thus used VP1 as a hallmark of NV RNA replication in this study.
NV RNA replication is sensitive to type I and III IFN treatment.
Having established that NV RNA can replicate in 293FT cells, we next tested whether NV RNA replication is sensitive to the antiviral effects of type I (α/β) or III (IL-29) IFN. Pretreatment of 293FT cells with increasing concentrations (100 and 1,000 U/ml) of IFN-α or IFN-β for 24 h, followed by NV RNA transfection, showed a dose-dependent inhibitory effect on NV RNA replication, as measured by VP1 expression (Fig. 2A). In a similar experiment, IL-29 also showed a dose-dependent inhibitory effect on NV RNA replication although it was less effective at 10 and 100 ng/ml than IFN-α or IFN-β at 100 and 1,000 U/ml (equivalent to 0.37 and 3.7 ng/ml IFN-β), respectively (Fig. 2A). When 1,000 U/ml of IFN-β was added to the cell culture at 12 h after NV RNA transfection, which allowed NV RNA to establish initial replication, an inhibitory effect was also observed and, as expected, was less effective than pretreatment (Fig. 2B). By IF staining of VP1 in NV RNA-transfected cells, we also observed markedly reduced numbers of VP1-positive cells in IFN-β-pretreated cells compared to the number in untreated cells (Fig. 2C). A similarly reduced number of VP1-positive cells was observed in IFN-α-pretreated cells (data not shown). These results collectively showed that NV RNA replication is sensitive to the antiviral effects of exogenously added type I and III IFNs.
FIG 2.
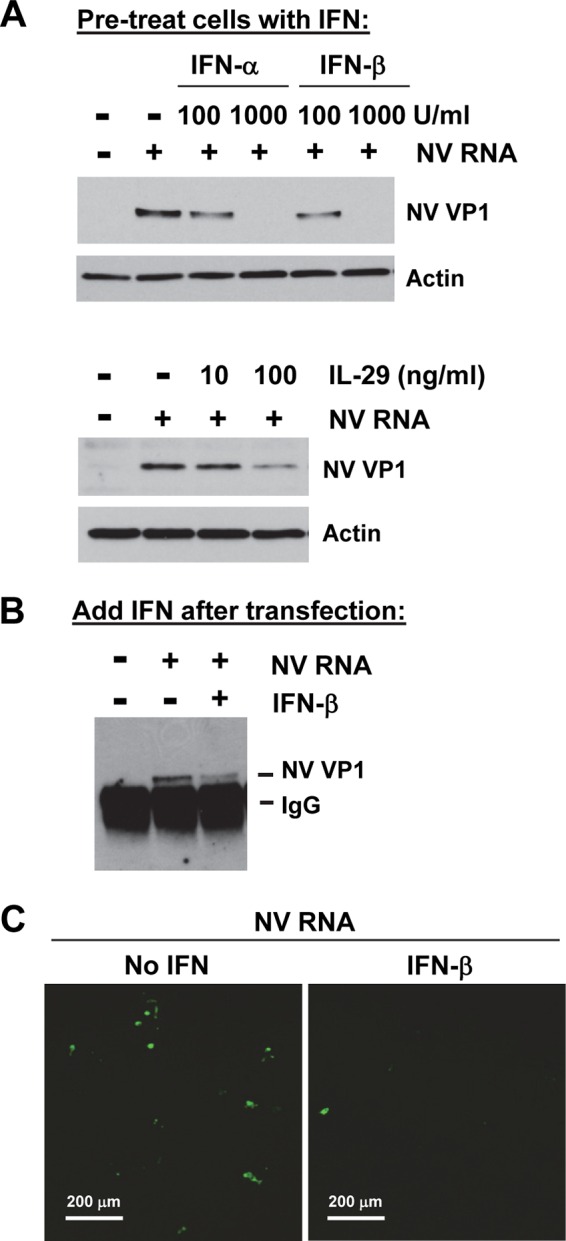
NV RNA replication is sensitive to type I and III IFN treatment. (A) Effect of type I and III IFN pretreatment on NV RNA replication. 293FT cells were pretreated with increasing doses of type I (100 and 1,000 U/ml IFN-α or IFN-β) or III (10 and 100 ng/ml IL-29) IFN for 24 h and then transfected with carrier RNA (−) or NV RNA (+) and incubated for 48 h. NV VP1 in cell lysate was detected by Western blotting. Actin served as an equal loading control. (B) Effect of posttransfection IFN treatment on NV RNA replication. 293FT cells were transfected with NV RNA and treated with 1,000 U/ml IFN-β at 12 h post-RNA transfection. Cells were lysed at 48 h post-RNA transfection, and NV VP1 in cell lysate was detected by IP and Western blotting. (C) Immunofluorescence staining of NV VP1 in NV RNA-transfected 293FT cells either untreated (no IFN) or pretreated with 1,000 U/ml IFN-β. Cells were fixed at 48 h post-RNA transfection and stained with guinea pig antibody to NV VP1. Scale bars, 200 μm.
NV RNA replication does not induce an IFN response.
To test whether NV RNA replication induces an IFN response in 293FT cells, we first confirmed that these cells are capable of an IFN response to a variety of stimuli. Using the 293FT-ISRE-Luc reporter cells that were derived from 293FT cells, we observed strong responses to SeV and the double-stranded RNA (dsRNA) mimic poly(I·C), as well as to type I (α and β) and III (IL-29) IFNs (Fig. 3A), confirming that 293FT cells have functional RIG-I/MDA5, TLR3 (both for IFN induction), and JAK-STAT (for IFN signaling) pathways. ISRE, which is widely found in the promoter regions of many ISGs (e.g., ISG56), is activated by both type I and III IFNs through the JAK-STAT pathway but also can be activated by IRF3 in an IFN-independent manner (46–48). Indeed, the vaccinia virus B18R protein, a soluble type I IFN decoy receptor (49), was able to completely block IFN-α- and IFN-β-induced ISRE-Luc activities but only partially blocked SeV- and extracellular poly(I·C)-induced ISRE-Luc activities (Fig. 3A). These results showed that in the cases of SeV and poly(I·C), ISRE-Luc detected both IFN induction (resistant to B18R) and signaling (sensitive to B18R). As expected, the B18R protein had no effect on ISRE-Luc activity induced by IL-29, a type III IFN (Fig. 3A). Using specific IFN neutralizing antibodies, we further confirmed that SeV- and poly(I·C)-induced IFNs from 293FT cells were mainly IFN-β, excluding the possibility that the B18R-resistant portions of ISRE-Luc activities induced by SeV and poly(I·C) might be due to type III IFN signaling (data not shown). We therefore chose the 293FT-ISRE-Luc reporter cell line to detect both induction and signaling of type I and III IFNs for most of the experiments in this paper unless otherwise indicated.
FIG 3.
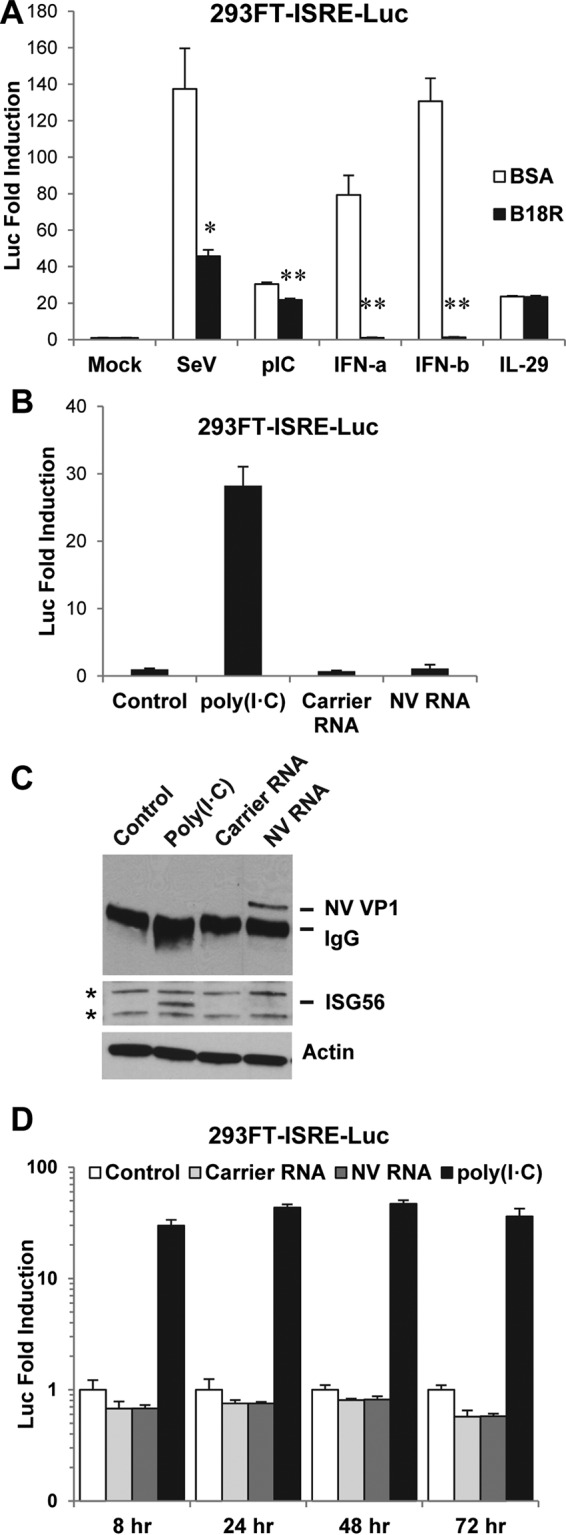
NV RNA replication does not induce an IFN response. (A) 293FT-ISRE-Luc reporter cells have strong IFN responses to various stimuli and detect both IFN induction and signaling. Cells were pretreated with B18R protein (125 ng/ml) or carrier protein BSA (control) for 1 h and stimulated with the indicated reagents for 18 h. Doses of stimuli were as follows: SeV, 40 HA/ml; poly(I·C), 100 μg/ml; IFN-α and IFN-β, 1,000 U/ml; IL-29, 100 ng/ml. Luciferase activity was normalized to that of BSA-pretreated and unstimulated (mock) cells and is presented as fold induction. *, P < 0.05; **, P < 0.01, compared to results with BSA pretreatment. (B) 293FT-ISRE-Luc reporter cells were untransfected (control) or transfected with carrier RNA or NV RNA and incubated for 48 h. As a positive control, 100 μg/ml poly(I·C) was added to the medium of untransfected cells for 6 h. Luciferase activity was normalized to that of untransfected cells (control) and is presented as fold induction. (C) Western blotting of NV VP1 and ISG56 in the same cell lysates from the experiment shown in panel B. NV VP1 was concentrated by IP prior to Western blotting. Actin and two nonspecific protein bands (marked by asterisks) served as equal loading controls. (D) Time course of 293FT-ISRE-Luc reporter assay. 293FT-ISRE-Luc reporter cells were untransfected (control) or transfected with carrier RNA or NV RNA and incubated for 8, 24, 48, and 72 h. As a positive control, 100 μg/ml poly(I·C) was added to the medium of untransfected cells for 6 h before each time point. Luciferase activity was normalized to that of untransfected cells (control) and is presented as fold induction. Note that the vertical axis is in logarithmic scale. pIC, poly(I·C).
Using the 293FT-ISRE-Luc reporter cells, we next examined whether NV RNA replication induces an IFN response. In cells transfected with NV RNA, we observed a basal level of ISRE-Luc activity similar to that of both untransfected (control) and carrier RNA-transfected (another negative control) cells at 48 h post-RNA transfection (Fig. 3B). This result was further confirmed by RT-qPCR, which showed no increase of IFN-α or IFN-β mRNA by NV RNA transfection (data not shown). As a positive control, poly(I·C), when added to the medium for 6 h, induced a strong ISRE-Luc activity (Fig. 3B). NV RNA replication in transfected cells was confirmed by IP and Western blotting of VP1 (Fig. 3C). Consistent with ISRE-Luc reporter assay results, the induction of ISG56, an ISG, was observed only in poly(I·C)-treated cells and not in control or carrier RNA- or NV RNA-transfected cells (Fig. 3C). To address a possible early or delayed IFN response to NV RNA replication, we also performed a time course experiment in which the IFN response was examined at 8, 24, 48, and 72 h following NV RNA transfection of 293FT-ISRE-Luc cells. At each time point, the IFN response was not detected by the ISRE-Luc assay in NV RNA-transfected cells, similar to the result in untransfected (control) or carrier RNA-transfected cells (Fig. 3D). As a positive control, poly(I·C), when added to the medium for 6 h, induced strong ISRE-Luc activity at each time point (Fig. 3D). The lack of IFN response in NV RNA-transfected cells at each time point, contrasted with the dynamic changes of VP1 (Fig. 1D) and NV RNA (Fig. 1E) during the same period, demonstrated that a measurable IFN response was not induced throughout the course of NV RNA replication.
Replication of GII.3 HuNoV RNA does not induce an IFN response.
In addition to NV, a GI.1 HuNoV strain, we also examined the IFN response to RNA replication of the GII.3 HuNoV strain U201 in 293FT cells. To this end, we used a recently developed plasmid-based reverse genetics system, in which the entire U201 genome is cloned downstream a mammalian EF-1α promoter in a plasmid containing the SV40 origin of replication (34). Cells transfected with the plasmid express the ORF1 polyprotein and initiate RNA replication at low frequency, resulting in expression of VP1 and VP2 capsid proteins and production of progeny viruses (34). As a negative control, we used a replication-defective mutant U201 genome that contains a guanine (G) nucleotide deletion at position 4607 (Δ4607) in the region encoding the viral polymerase (Pol), which causes a frameshift and premature termination of Pol (Fig. 4A). To facilitate the detection of U201 RNA replication, a pair of wild-type and mutant reporter genome plasmids was used in which an RLuc reporter gene was inserted in frame into the ORF2 (VP1) region of both U201 and U201-Δ4607, creating a U201-RLuc reporter genome and its isogenic mutant U201-Δ4607-RLuc, respectively (34) (Fig. 4A). Following plasmid transfection of 293FT cells, Western blotting of nonstructural proteins p35 (N-terminal protein [Nterm], corresponding to NV p48), p41, and Pol showed that ORF1 polyprotein expression and auto-processing occurred for both U201 and the mutant U201-Δ4607, but Pol was produced only by U201, not by the mutant U201-Δ4607 (Fig. 4B). A truncated form of Pol was not detected by Western blotting (data not shown), suggesting that the prematurely terminated Pol may be unstable and rapidly degraded. The expression levels of p35 (Nterm) and p41 were elevated in mutant U201Δ4607 compared to the levels in U201 (Fig. 4B), probably due to reduced size and enhanced expression of ORF1 polyprotein as a result of premature termination of Pol. Consistent with the Western blotting results, IF staining of VPg and VP1 in plasmid-transfected cells showed that although both U201 and mutant U201-Δ4607 expressed ORF1 polyprotein (represented by VPg), only U201 was capable of RNA replication, as evidenced by expression of VP1 in some of the VPg-positive cells, whereas no VP1 was produced by the replication-defective U201-Δ4607 mutant (Fig. 4C).
FIG 4.

Replication of plasmid-derived U201 RNA does not induce an IFN response. (A) Schematic drawing of U201 genome transcript RNAs expressed from plasmids. Note that the Δ4607 mutation causes a frameshift and premature termination of Pol, creating an untranslated region between ORF1 and ORF2. (B) Western blotting of U201 polymerase (Pol), p35 (Nterm), and p41 in 293FT cells transfected with U201 or U201-Δ4607 plasmid for 24 h. A nonspecific protein band served as an equal loading control. (C) Laser scanning confocal microscopy images of 293FT cells transfected with U201 or U201-Δ4607 plasmid for 24 h. Cells were labeled with mouse monoclonal antibody to U201 VPg (red) and guinea pig antibody to U201 VP1 (green). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. The merged images show that both plasmids express ORF1 polyproteins (VPg-positive), but RNA replication (VP1-positive) occurs in only one of the U201-transfected cells, not in U201-Δ4607-transfected cells. (D) IFN-β-Luc reporter assay. 293FT cells were transfected with IFN-β-Luc reporter in combination with vector, U201, or U201-Δ4607 plasmid for 24 h. As a positive control, vector-transfected cells were infected with SeV (40 HA/ml) for 18 h. Luciferase activity was first normalized to that of an internal RLuc transfection control and then normalized to that of vector-transfected cells and is presented as fold induction. (E and F) RLuc and IFN-β-Luc dual reporter assay. 293FT cells were transfected with IFN-β-Luc reporter in combination with vector, U201-RLuc, or U201-Δ4607-RLuc plasmid for 24 h. As a positive control, vector-transfected cells were infected with SeV (40 HA/ml) for 18 h. RLuc activity is presented as original relative light units/second (RLU/s). Luciferase activity was normalized to that of vector-transfected cells and is presented as fold induction. (G) Western blotting of ISG56 in the same cell lysates as used in the experiments shown in panels E and F. Actin served as an equal loading control.
We next used this system to examine the IFN response to U201 RNA replication. Following cotransfection of U201 or U201-Δ4607 plasmid with an IFN-β-Luc reporter plasmid into 293FT cells, at 24 hpt, which was the peak of U201 RNA replication (34), no activation of the IFN-β promoter was detected for either U201 or U201-Δ4607 compared to the level with vector (negative control) transfection (Fig. 4D). As a positive control, SeV induced strong activation of the IFN-β promoter (Fig. 4D). We also performed a dual-luciferase assay by cotransfection of an IFN-β-Luc (firefly luciferase) reporter plasmid with the U201-RLuc or U201-Δ4607-RLuc (Renilla luciferase) reporter genome plasmid into 293FT cells. As an indication of viral RNA replication, U201-RLuc produced strong RLuc activity at 24 hpt, whereas only a basal level of RLuc activity was detected for the replication-defective mutant U201-Δ4607-RLuc (Fig. 4E). The RLuc levels of U201-RLuc versus U201-Δ4607-RLuc were consistent with those previously reported (34). However, despite their different replication abilities, no activation of IFN-β promoter was observed for either U201-RLuc or U201-Δ4607-RLuc compared to the levels with the vector negative control and the SeV positive control (Fig. 4F). Western blotting of ISG56, another indicator of IFN response, in the same cell lysates also confirmed that it was induced only by the SeV positive control, not by vector, U201-RLuc, or U201-Δ4607-RLuc (Fig. 4G). These results collectively showed that replication of U201 RNA derived from a reverse genetics system did not induce an IFN response.
As previously reported (34), a basal level of RLuc activity was observed for the mutant U201-Δ4607-RLuc (Fig. 4E). This could be due to a basal level of internal translation initiation but not likely to residual RNA replication or translation termination reinitiation (TTR) between ORF1 and ORF2, as reported for a bovine GIII NoV (45). In the same report, TTR was not observed for the GII.4 strain Lordsdale (45), which shares an identical ORF1/ORF2 junction sequence with U201 (data not shown). In the U201-Δ4607 mutant, the Δ4607 mutation creates an untranslated region between ORF1 and ORF2 (Fig. 4A), thus also preventing TTR between the two ORFs. The actual cause of the basal RLuc activity remains to be further investigated.
NV and U201 RNA replication generates dsRNA.
The lack of a detectable IFN response to NV and U201 RNA replication in 293FT cells prompted us to evaluate whether dsRNA is generated during NV and U201 RNA replication. As an intermediate in viral RNA replication, dsRNA is a pathogen-associated molecular pattern (PAMP) that induces IFN responses (25, 26) and has been detected in cells infected by other positive-strand RNA viruses using a dsRNA-specific antibody (50). We found that dsRNA was readily detected by IF in NV RNA-transfected (VP1-positive) cells (Fig. 5A). Different from VP1, which displayed both diffuse and punctate patterns in the cytoplasm, dsRNA appeared in highly concentrated, punctate, and limited (less than 10 per cell) foci in the cytoplasm at the peak (48 h post-RNA transfection) of NV RNA replication (Fig. 5A). Furthermore, dsRNA colocalized with the punctate, but not the diffuse, form of VP1 (Fig. 5A). In contrast to VP1, p48, a membrane-associated viral nonstructural protein involved in RNA replication (51, 52), shared the same punctate focus staining patterns with dsRNA and strongly colocalized with dsRNA (Fig. 5A). These results not only confirm that dsRNA is generated during NV RNA replication but also suggest that it is a component of the membrane-associated RNA replication complex.
FIG 5.

NV and U201 RNA replication generates dsRNA. (A and B) Laser scanning confocal microscopy images of 293FT cells transfected with NV RNA for 48 h or transfected with U201 or U201-Δ4607 plasmid for 24 h, as indicated. Cells were labeled with antibodies to dsRNA (red) and NV VP1 or p48 (green) (A) or with antibodies to dsRNA (red), U201 VP1 (green), and U201 VPg (magenta) (B). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm.
We next determined whether dsRNA can be detected by IF during U201 RNA replication using a plasmid-based reverse genetics system (Fig. 5B). In U201 plasmid-transfected cells (VPg-positive) that underwent RNA replication (VP1-positive), dsRNA was detected at 24 hpt by IF in both punctate and diffuse patterns within the same cells (Fig. 5B, U201). Neither VP1 nor dsRNA was detected in cells transfected with the U201-Δ4607 mutant plasmid, which is capable of ORF1 expression (VPg-positive) but unable to replicate (Fig. 5B, U201-Δ4607). Interestingly, both VP1 and the diffuse form of dsRNA were located in the cytoplasm, whereas the punctate form of dsRNA was located in the nucleus where VP1 was absent (Fig. 5B). The nuclear localization of dsRNA appeared to be associated with RNA replication since dsRNA was not observed in the U201-Δ4607 mutant plasmid-transfected cells (Fig. 5B), but the exact mechanism remains to be investigated. Overall, the results showed that dsRNA is generated in detectable amounts during NV and U201 RNA replication, thus excluding the possibility that the lack of IFN response is due to the absence of dsRNA.
NV and GII.3 VLPs do not induce an IFN response.
Since both NV stool RNA transfection and the U201 reverse genetics system bypass viral entry, these methods cannot address whether viral entry may induce an IFN response. Due to the lack of an efficient cell culture infection system to properly study viral entry, as an alternative approach we tested whether VLPs, which are morphologically and antigenically similar to virus particles (38), can induce an IFN response upon binding to the cell surface. Since U201 VLPs were not available for this study, we used VLPs of another GII.3 HuNoV strain, TCH-104 (40), which shares 98% similarity (96% identity [data not shown]) to the VP1 sequence of U201. Treatment of 293FT-ISRE-Luc reporter cells with two concentrations (12 and 36 μg/ml) of NV or GII.3 VLPs in the medium for 6 h showed no IFN response compared to results in untreated (negative control) cells (Fig. 6). As a positive control, treatment with poly(I·C) in the medium for 6 h induced strong ISRE-Luc activity (Fig. 6). These results suggested that binding of NV or GII.3 VLPs to the cell surface does not induce an IFN response.
FIG 6.
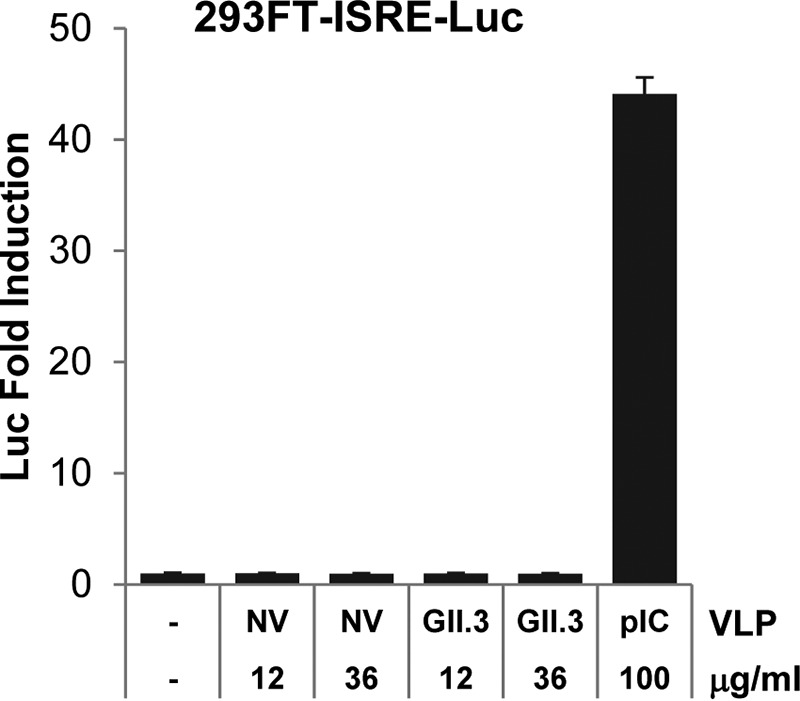
NV and GII.3 VLPs do not induce an IFN response. 293FT-ISRE-Luc reporter cells were treated with increasing doses (12 and 36 μg/ml) of NV or GII.3 (strain TCH-104) VLPs or with 100 μg/ml poly(I·C) (as a positive control) in the medium for 6 h. Luciferase activity was normalized to that of untreated cells and is presented as fold induction. pIC, poly(I·C).
NV RNA replication is not enhanced by neutralization of type I/III IFNs.
Although no IFN response to NV or U201 RNA replication was detected by 293FT-ISRE-Luc or IFN-β-Luc reporter assay or by Western blotting of ISG56, to exclude the possibility that these methods might be not sensitive enough to detect a trace level of IFN induction, we further performed IFN neutralization experiments using high concentrations of IFN neutralizing reagents. These included neutralizing antibodies to IFN-α, IFN-β, IFN-α/β receptor 2 (IFNAR2), IL-29 (IFN-λ1), and IFN-λ receptors IL10R2 and IL28R1, as well as the vaccinia virus B18R protein (49). We determined the dilutions/concentrations of these reagents that were enough to completely or efficiently block 1,000 IU/ml IFN-α/β or 100 ng/ml IL-29. Since such amounts of type I/III IFNs induced ISRE-Luc activities in the range of 20- to 120-fold increases (Fig. 3A) in contrast to no fold increase with NV RNA replication (Fig. 3B and D), the neutralizing reagents used in these experiments were at sufficient doses to block any trace levels of IFNs that could possibly be induced by NV RNA replication. When type I IFN neutralizing antibodies were added to 293FT cell culture, either individually or as a cocktail, followed by NV RNA transfection, NV RNA replication as measured by VP1 expression was not enhanced by any of them (Fig. 7A). Similarly, the B18R protein, which was able to completely block type I IFNs (Fig. 3A), also failed to enhance NV RNA replication as measured by VP1 and VPg expression (Fig. 7B and C). To exclude the possibility that the lack of enhancement might be due to instability/degradation of antibodies in the cell culture medium and also to validate antibody efficacies, the spent medium (containing the antibodies) was collected at the end of the NV RNA transfection experiment and added to the 293FT-ISRE-Luc reporter cells, followed by IFN-α or IFN-β stimulation. The results showed that individual antibodies in the spent medium effectively blocked their corresponding IFNs, albeit at different efficiencies, while a cocktail of the four antibodies completely blocked both IFN-α and IFN-β (Fig. 7D), confirming that each antibody was effective and stable in cell culture.
FIG 7.

NV RNA replication is not enhanced by neutralization of type I IFNs. (A) Western blotting of NV VP1 in 293FT cells pretreated with type I IFN neutralizing antibodies for 6 h and transfected with carrier RNA or NV RNA for 48 h. See Materials and Methods for the dilutions of the antibodies. Sh, sheep; Rb, rabbit; Ab, antibody. (B and C) Western blotting of NV VP1 and VPg in 293FT cells pretreated with BSA (−) or with 125 ng/ml B18R (+) for 1 h and transfected with carrier RNA or NV RNA for 48 h. Actin served as an equal loading control. (D) 293FT-ISRE-Luc reporter assay of type I IFN neutralizing antibodies. Spent medium (containing IFN neutralizing antibody) from the experiment shown in panel A was collected before lysis of cells and added to 293FT-ISRE-Luc reporter cells for 1 h, followed by stimulation with 500 U/ml IFN-α or IFN-β for 18 h. Luciferase activity (presented as fold induction) was normalized to that of the reporter cells that received medium from untreated (no antibody and no IFN) carrier RNA-transfected 293FT cells. * and ^, P < 0.05, compared to results with carrier RNA-transfected 293FT cells with IFN-α and IFN-β stimulation, respectively, and no antibody treatment.
In a similar experiment, NV RNA replication, as measured by both VP1 and VPg expression, was also not enhanced by type III IFN neutralizing antibodies either applied individually or as a cocktail (Fig. 8A). Similar to type I IFN neutralizing antibodies, each type III IFN neutralizing antibody was stable and effective against IL-29 with different efficiencies and worked efficiently as a cocktail (Fig. 8B). Together, these results showed that despite the effectiveness of the IFN neutralizing reagents, NV RNA replication was not enhanced by neutralization of type I or III IFNs.
FIG 8.
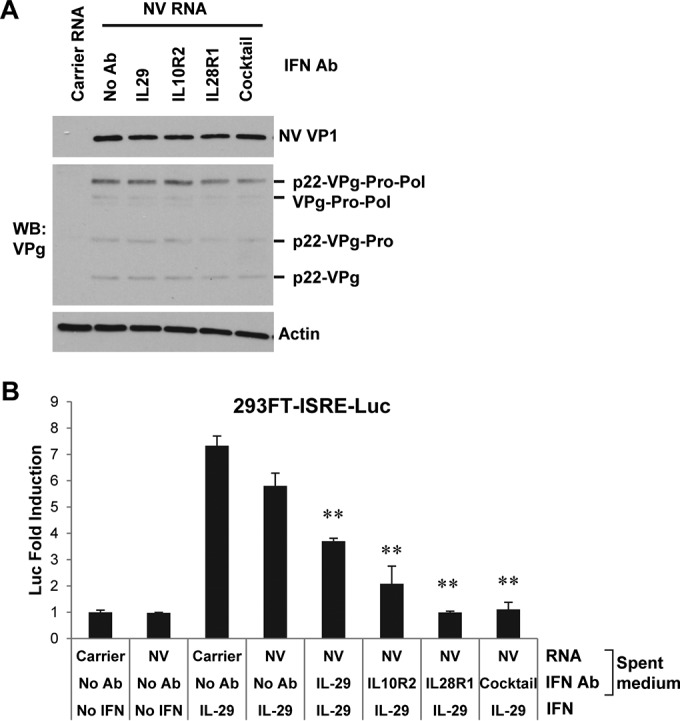
NV RNA replication is not enhanced by neutralization of type III IFNs. (A) Western blotting of NV VP1 and VPg in 293FT cells pretreated with type III IFN neutralizing antibodies for 6 h and transfected with carrier RNA or NV RNA for 48 h. See Materials and Methods for dilutions of the antibodies. Actin served as an equal loading control. (B) 293FT-ISRE-Luc reporter assay of type III IFN neutralizing antibodies. The experimental procedure was essentially the same as that described in the legend of Fig. 5D, except that type III IFN neutralizing antibodies were used, and reporter cells were stimulated with 50 ng/ml IL-29. **, P < 0.01, compared to results in carrier RNA-transfected 293FT cells with IL-29 stimulation and no antibody treatment.
NV RNA replication is not enhanced by knockdown of MAVS or IRF3.
Considering that some ISGs can be induced by activation of IFN induction pathways in an IFN-independent manner (46–48), as shown in Fig. 3A, and therefore may not be affected by neutralization of IFNs, we further tested whether NV RNA replication can be enhanced by short hairpin RNA (shRNA) knockdown of essential factors in the RIG-I/MDA5 and TLR3 pathways of IFN induction. For this purpose, stable knockdown cell lines were generated by transduction of 293FT cells with shRNA lentiviral particles. Knockdown of adaptor protein MAVS in the RIG-I/MDA5 signaling pathway, as confirmed by Western blotting (Fig. 9A), severely reduced an SeV-induced IFN response in 293FT cells (Fig. 9B) but failed to enhance NV RNA replication, as measured by VP1 expression (Fig. 9C). Similarly, knockdown of IRF3 (Fig. 9D), an essential transcription factor for both RIG-I/MDA5 and TLR3 pathways, significantly reduced a poly(I·C)-induced IFN response in 293FT cells (Fig. 9E) but did not enhance NV RNA replication as measured by either VP1 Western blotting (Fig. 9F) or the number of NV RNA replicating (VP1-positive) cells (Fig. 9G). These results, together with the IFN neutralization data, collectively show that NV RNA replication is not enhanced by inhibition of either IFN induction or the signaling pathway.
FIG 9.

NV RNA replication is not enhanced by knockdown of MAVS or IRF3. (A) Western blotting to confirm knockdown of MAVS. (B) ISRE-Luc assay to functionally confirm knockdown of MAVS. 293FT and 293FT-shMAVS cells were transduced with ISRE-Luc lentiviral particles for 24 h and then mock or SeV infected (40 HA/ml) for 18 h. Luciferase activity was normalized to that of mock-infected 293FT cells and is presented as fold induction. (C) Western blotting of NV VP1 and MAVS in 293FT and 293FT-shMAVS cells transfected with carrier RNA (−) or NV RNA (+) for 48 h. (D) Western blotting to confirm knockdown of IRF3. (E) ISRE-Luc assay to functionally confirm knockdown of IRF3. 293FT and 293FT-shIRF3 cells were transduced with ISRE-Luc lentiviral particles for 24 h and then mock or poly(I·C) treated (100 μg/ml in the medium) for 6 h. Luciferase activity was normalized to that of mock-treated 293FT cells and is presented as fold induction. (F) Western blotting of NV VP1 and IRF3 in 293FT and 293FT-shIRF3 cells transfected with carrier RNA (−) or NV RNA (+) for 48 h. (G) Effect of IRF3 knockdown on the number of NV RNA replicating cells. 293FT and 293FT-shIRF3 cells seeded in 48-well plates were transfected with NV RNA for 48 h. Cells were fixed for IF staining with antibody to NV VP1. The number of VP1-positive cells per well was counted and is presented as mean values from multiple wells. *, P < 0.05; **, P < 0.01; ns, not statistically significant, compared to results in 293FT cells. Actin (A, C, D, and F) and a nonspecific protein band (marked by a circle in C and F) served as equal loading controls.
Knockdown experiments of the IFN-α/β receptor 1 (IFNAR1) and IFN-λ receptors IL10R2 and IL-28R1 were also performed (data not shown) but were found to be less effective than the B18R protein or IFN neutralizing antibodies in blocking type I/III IFNs and therefore were not further tested on NV RNA replication.
NV RNA replication does not block exogenous-virus-induced IFN response.
Despite the detection of dsRNA (Fig. 5), the lack of IFN response to NV and U201 RNA replication (Fig. 3 and 4) suggests either that viral RNA replication is not sensed by cellular PRRs such as RIG-I/MDA5 and TLR3 that trigger the IFN response or that the virus has evolved a mechanism to antagonize the activation of the IFN response. The latter has been demonstrated by detailed studies on many other positive-strand RNA viruses. For example, hepatitis A virus (HAV) in the Picornaviridae family, which shares many common features with the Caliciviridae family, is known to block the RIG-I/MDA5 signaling pathway of IFN induction through cleavage of the adaptor protein MAVS by virus-encoded protease precursor 3ABC (53). We thus first examined MAVS in NV RNA-transfected versus HAV-infected 293FT cells. Western blotting showed that the abundance of MAVS was markedly reduced in HAV-infected 293FT cells due to cleavage by the viral 3ABC protease precursor (Fig. 10A). In contrast, the level of MAVS remained unchanged in NV RNA-transfected cells (Fig. 10A). Both NV RNA transfection and HAV infection were confirmed by Western blotting of NV VP1 and HAV VP1 and precursors, respectively (Fig. 10A). Considering that the efficiency of NV RNA transfection was lower than that of HAV infection, we also examined MAVS at the single-cell level by IF staining. Consistent with Western blotting results, in HAV-infected 293FT cells that were stained positive for HAV capsid proteins, MAVS was severely reduced compared to the level in neighboring uninfected cells (Fig. 10B, HAV). In contrast, both the abundance and staining pattern of MAVS in NV RNA replicating (VP1-positive) cells remained intact compared to those in neighboring untransfected cells (Fig. 10B, NV RNA), suggesting that, unlike HAV, NV RNA replication does not result in MAVS degradation.
FIG 10.
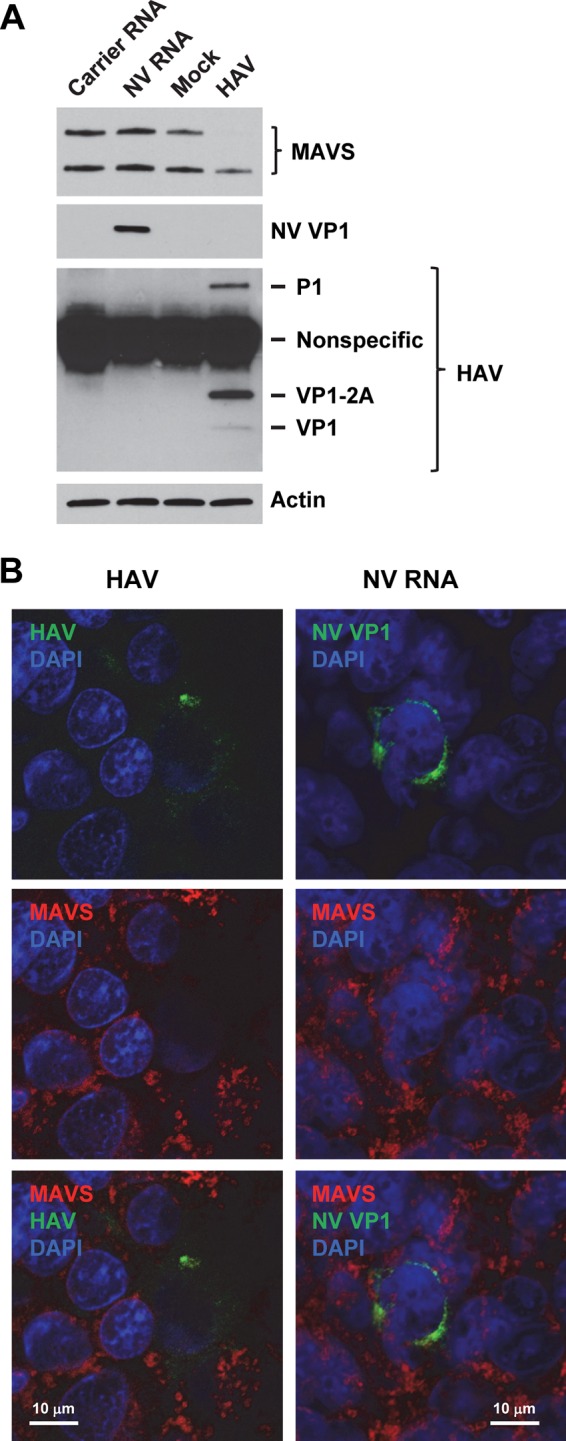
MAVS is not degraded in NV RNA replicating cells. (A) Western blotting of MAVS in 293FT cells transfected with carrier RNA or NV RNA or mock or HAV infected (multiplicity of infection of 1) for 48 h. NV RNA replication and HAV infection were confirmed by Western blotting of NV VP1 and HAV VP1/precursors, respectively. Actin served as an equal loading control. (B) Laser scanning confocal microscopy images of 293FT cells infected with HAV (multiplicity of infection of 0.1) or transfected with NV RNA for 48 h. Cells were labeled with antibodies to MAVS (red) and HAV VP1 or NV VP1 (green). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm.
We next performed an IF experiment to address whether NV RNA replication can actively block the activation of the IFN response induced by an exogenous virus, such as SeV. Due to an unknown reason, nuclear translocation of IRF3, a common indicator of an IFN response, was not observed in SeV-infected 293FT cells (data not shown) despite a strong IFN response as detected by ISRE-Luc and IFN-β-Luc assays (Fig. 3A and 4D and F) and Western blotting of ISG56 (Fig. 4G). Phosphorylation of IRF3 was observed in SeV-infected 293FT cells by Western blotting (data not shown). Although the exact mechanism remains to be further investigated, we speculate that SeV infection of 293FT cells induces phosphorylation and nuclear translocation of a subset of IRF3 molecules at a level sufficient for a strong IFN response, whereas the majority of IRF3 remains in the cytoplasm, resulting in an overall appearance of a lack of nuclear translocation. As an alternative approach, we selected ISG56 as an indicator of an IFN response. 293FT cells were first transfected with NV RNA for 30 h, subsequently infected with SeV for 18 h, and finally fixed for IF staining of both NV VP1 and ISG56. Consistent with the Western blotting result (Fig. 4G), the IF signal of ISG56 was strongly upregulated in SeV-infected cells, and this was observed in both NV RNA replicating cells (VP1-positive) and untransfected cells (Fig. 11), indicating that NV RNA replication does not block an SeV-induced IFN response.
FIG 11.
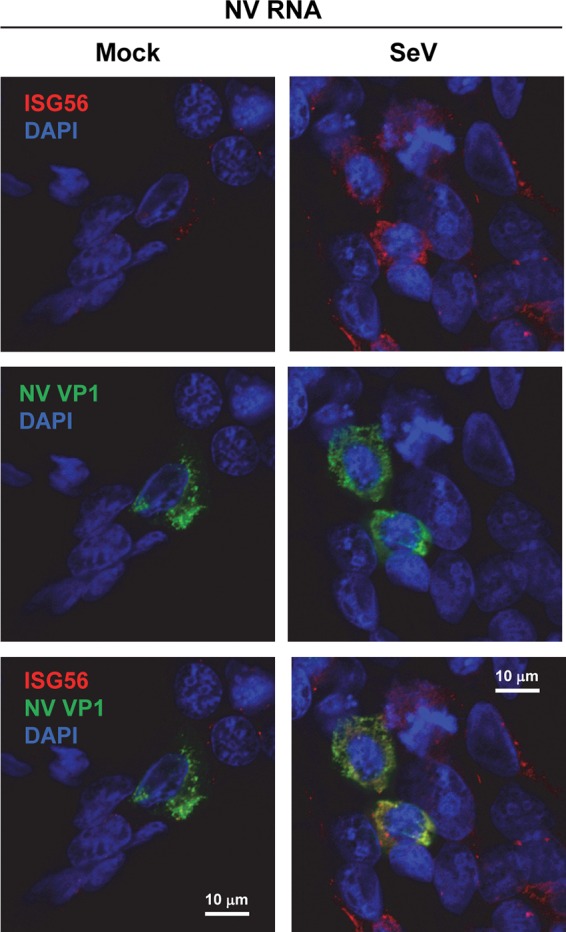
NV RNA replication does not block an SeV-induced IFN response. 293FT cells were transfected with NV RNA. At 30 h post-RNA transfection (hpt), cells were mock or SeV infected (4 HA/ml). At 48 hpt (18 hpi for SeV), cells were fixed and labeled with antibodies to ISG56 (red) and NV VP1 (green). Nuclei were counterstained with DAPI (blue). The laser scanning confocal microscopy images show that SeV-induced upregulation of ISG56 occurs in both NV VP1-positive and -negative cells. Scale bars, 10 μm.
DISCUSSION
Although IFN responses have been speculated to play a role in host control of HuNoV infection, we show here that RNA replication of two HuNoVs, NV (GI.1) and U201 (GII.3), in 293FT mammalian epithelial cells does not induce an IFN response. Consistent with the lack of an IFN response, NV RNA replication was not enhanced by IFN neutralization or knockdown of key factors in the IFN induction pathways. We further show that NV RNA replication does not block the activation of an IFN response induced by an exogenous virus. These results collectively suggest that HuNoV RNA replication in cultured mammalian epithelial cells does not trigger, and therefore is unlikely to be strongly controlled by, the host epithelial IFN response. It should be noted that the RNA replication systems used in this study—stool RNA transfection for NV and a reverse genetics system for U201—are close to, but do not fully represent, a complete virus infection cycle since both systems bypass viral entry. For some viruses, viral entry has been shown to induce innate immune responses. For example, membrane fusion during entry of enveloped viruses, such as SeV, Semliki Forest virus (SFV), and human cytomegalovirus (HCMV), has been shown to induce IFN-α/β or a subset of ISGs (54, 55). Although we showed that NV and GII.3 VLPs do not induce an IFN response when they are added to the medium of 293FT cell culture (Fig. 6), these results should be considered with the caveats that VLPs do not fully represent virus particles and that 293FT cells may lack the necessary receptor/coreceptor for viral entry. Until an efficient cell culture infection system is established for HuNoVs, the possibility of an IFN response triggered by viral entry remains an open question. Despite the absence of viral entry, the NV stool RNA transfection system and the U201 reverse genetics system recapitulate other major steps of the HuNoV life cycle, including translation of genomic RNA, auto-processing of polyprotein, assembly of replication complex and RNA replication, capsid protein expression from subgenomic RNA, virus particle assembly, and release of virus progenies (33, 34). The lack of an IFN response indicates that none of these steps triggers IFN induction.
In two previous studies (33, 41) and this report, the viruses produced from RNA replication did not spread in cell culture to initiate new infections, which was reflected by the decline of both VP1 and NV RNA after RNA replication peaked at 48 h (Fig. 1D and E). The replication of GII.3 strain U201 RNA generated from a reverse genetics system has also been shown to decline after it peaks at 24 hpt (34). Our attempts to passage whole-culture lysate of NV RNA-transfected cells (by repeated freeze-thaw) onto naive cells or to passage the transfected cells themselves were not successful (data not shown). The failure of reinfection has been speculated to be caused by the lack of a necessary cell surface receptor/coreceptor or by a virus-induced IFN response that is sensed by the neighboring cells (33). Our current results show that HuNoV RNA replication in cultured epithelial cells does not induce an IFN response and is not enhanced by neutralization of IFNs or knockdown of factors in the IFN induction pathways. In particular, the number of NV RNA replicating (VP1-positive) cells was not affected by IRF3 knockdown (Fig. 9G) or IFN neutralization (data not shown), suggesting that blocking the IFN response does not lower the threshold for NV RNA replication, nor does it allow progeny virus produced from the transfected cells to spread to untransfected cells. Therefore, the IFN response is unlikely to play a major role in restricting NV replication; other factors, such as the lack of receptor/coreceptor, must account for the block of virus spread. Our results should help refocus efforts to identify such factors in order to achieve an efficient cell culture infection system for HuNoVs.
The lack of an IFN response to HuNoV RNA replication in 293FT cells may have important implications for understanding the innate immune response to these viruses in epithelial cells. Studies using gnotobiotic pigs and calves inoculated with a GII.4 strain of HuNoV have shown that intestinal epithelial cells, especially enterocytes lining the small intestine, are the primary sites of HuNoV infection in vivo (12, 13). We showed here that NV RNA efficiently replicates in cultured 293FT kidney epithelial cells, indicating that these cells also have a favorable intracellular environment for HuNoV RNA replication. The commonly used Caco-2 intestinal epithelial cells have a strong IFN response to SeV infection (56) but were not used in this study due to low NV RNA transfection efficiency (data not shown). Similar to Caco-2 cells, 293FT cells mount robust IFN responses to a variety of stimuli including SeV (Fig. 3A), confirming that both cell types have functional IFN induction and signaling pathways. Therefore, the lack of IFN response to HuNoV RNA replication in 293FT cells is likely due to an intrinsic feature of HuNoV RNA replication rather than to cell type difference although the latter cannot be completely ruled out at this point.
Our results showing that NV RNA replication is inhibited by exogenous type I or III IFN treatment are consistent with previous studies on NV replicon cells (57, 58) and MNV infection of cultured macrophages and dendritic cells (59). However, our finding that NV RNA replication does not induce an IFN response in cultured epithelial cells was unexpected and currently cannot be conclusively compared to previous studies due to the lack of data specifically from intestinal epithelial cells. In human volunteers inoculated with Snow Mountain virus (SMV), a GII.2 HuNoV, local intestinal innate cytokine responses were not assessed (60). In gnotobiotic pigs inoculated with a GII.4 HuNoV, an elevated level of IFN-α was detected in the intestinal contents during both the early and late phases of infection, but it was unclear whether IFN-α was produced by infected intestinal epithelial cells or by immune cells in the lamina propria (12). MNV infection has been shown to strongly induce type I IFNs in cultured macrophages and dendritic cells (29), which are professional type I IFN-producing immune cells. Evidence of MNV infection of intestinal epithelial cells is lacking, and results from recent studies suggest the opposite. One report showed that MNV is transported across an in vitro-polarized mouse intestinal epithelial monolayer in the absence of viral replication by cells characteristic of microfold (M) cells (61). Further studies showed that MNV replication in mouse intestine requires M cells in Peyer's patches and that oral MNV infection is blocked in mice lacking Peyer's patches and mature M cells (62, 63). These results support a model in which M cells are the primary route for MNV to cross the intestinal epithelial barrier and infect the underlying immune cells, suggesting that, unlike the case of HuNoVs, intestinal epithelial cells may not be the primary infection sites for MNV. Overall, based on the existing data, it remains unclear if HuNoVs induce an IFN response in intestinal epithelial cells. Our results from cultured kidney epithelial cells thus add another piece to the puzzle from a viral RNA replication point of view. More studies and new models of HuNoV-permissive intestinal epithelial cells are needed to address this interesting question.
Despite their differences in tropism and disease symptoms, HuNoV and MNV share many common biological features, including a similar mechanism of RNA replication that could expose both viruses to the antiviral effects of IFNs. It is therefore unexpected that NV RNA replication in cultured cells is not enhanced by blocking the IFN response, which is known to enhance MNV replication in vivo and in vitro (15, 29, 30). Previous studies have shown that MNV replicates to higher titers in cultured macrophages or dendritic cells derived from STAT1−/−, MDA5−/−, IFNAR1−/−, or IRF3−/− mice (15, 29, 30). In our study, the use of type I or III IFN neutralizing antibodies (Fig. 7 and 8) and of the soluble type I IFN decoy receptor B18R protein (Fig. 3A) was functionally equivalent to IFNAR1 or STAT1 knockout, while shRNA knockdown of MAVS and IRF3 also efficiently inhibited RIG-I/MDA5 and TLR3 signaling pathways of IFN induction, as tested by SeV and poly(I·C), respectively (Fig. 9B and E). Thus, the discrepancy between our data on NV and previous studies on MNV is unlikely to be due to the difference in the approaches used. It is possible that since MNV strongly induces type I IFNs in cultured macrophages and dendritic cells, its replication can be greatly enhanced in the aforementioned knockout cells, whereas the lack of enhancement of NV RNA replication in 293FT cells by various methods to block an IFN response is consistent with the lack of IFN induction by viral RNA replication. Unlike MNV, NV does not replicate in human macrophages or dendritic cells (16). In order to assess the effect of blocking an IFN response on NV replication in a way similar to MNV, a special cell type is required in which NV can both replicate and induce a substantial IFN response. Such a cell type remains to be identified.
The lack of an IFN response to virus infection has been observed for some other positive-strand RNA viruses and can be attributed to diverse mechanisms evolved by the viruses to antagonize IFN induction or signaling pathways. For example, the hepatitis C virus (HCV) protease NS3/4A and HAV protease precursor 3ABC cleave the adaptor protein MAVS to block the RIG-I/MDA5 signaling pathway of IFN induction (53, 64). In both cases, the viruses are able to block an IFN response induced by an exogenous virus such as SeV (53, 65), similar to the approach used in this study. However, our results show that, unlike HCV or HAV, NV RNA replication does not cause degradation of MAVS (Fig. 10) and cannot block an SeV-induced IFN response (Fig. 11). A previous study also showed that the SeV-induced IFN response is not blocked in NV replicon cells (58). These observations are not without precedent. For example, murine hepatitis virus (MHV) and severe acute respiratory syndrome-coronavirus (SARS-CoV), both belonging to the Coronaviridae family, do not induce an IFN response but also do not prevent IFN induction by SeV or poly(I·C) (66–68). One possible explanation is that instead of blocking IFN induction, NV might have evolved a yet to be identified mechanism to protect viral RNA from detection by the host PRRs. This could partially explain why we detected dsRNA during NV and U201 RNA replication (Fig. 5) but could not detect an IFN response (Fig. 3 and 4). Our finding that dsRNA colocalizes with the membrane-associated nonstructural protein p48 in NV RNA-transfected cells (Fig. 5A) suggests that dsRNA is part of the membrane-associated replication complex. Recent studies have shown that some other positive-strand RNA viruses, such as dengue virus (DEN) and HCV, induce membrane rearrangement that conceals viral RNA from recognition by the PRRs (69, 70). It should be interesting to investigate whether a similar strategy is used by HuNoVs to evade an IFN response.
Alternatively, NV might have evolved a mechanism(s) to antagonize the antiviral activity of IFN. An example can be found again in the coronavirus MHV, of which the accessory protein 5a is a major antagonist of the antiviral action of IFN and is responsible for reduced IFN sensitivity of the MHV-A59 strain (71). A similar mechanism is possible for NV and does not directly contradict our observation that NV RNA replication is sensitive to type I and III IFN treatment (Fig. 2), as high concentrations of IFNs were used in these experiments. The same concentration (1,000 U/ml) of IFN-α has been shown to be inhibitory even for the less IFN-sensitive strain of MHV (71). In previous reports (57, 58) and this study, only NV, a GI.1 HuNoV, has been tested for sensitivity to IFN. It should be interesting to assess IFN sensitivities of HuNoVs in other genogroups and genotypes. Overall, the inability of NV RNA replication to block IFN induction raises an interesting question of how NV and other HuNoVs cope with the host IFN response and certainly requires more investigation.
Several results from this study, including that NV RNA replication is sensitive to type I IFN treatment and that NV RNA replication does not block an SeV-induced IFN response, are consistent with previous studies based on an NV replicon system (57, 58). The NV replicon is a valuable cell culture system representing NV RNA replication in the Huh-7 hepatoma cell line (58) but is associated with several limitations that affect the interpretation of results. For example, since most of ORF2 in the NV replicon is replaced with a neomycin phosphotransferase (neo) gene to facilitate antibiotic selection, structural proteins VP1 (ORF2) and VP2 (ORF3, which is translated by a TTR mechanism after ORF2) are not expressed (58). As a result of the selection process, the NV replicon contains adaptive mutations (58) that could reduce virulence or an innate immune response associated with viral RNA replication. Due to the mutual selection between replicon and the cells, the cells harboring NV replicon could also carry mutations that benefit NV RNA replication, similar to (but not necessarily the same as) the loss-of-function mutation in the RIG-I gene in the Huh7.5 cells supporting HCV replicon (72). In contrast, the NV stool RNA transfection system used in this study is an authentic RNA replication system in which the NV stool-isolated RNA, without cloning and also possibly containing natural quasi-species, is directly transfected into 293FT cells. Unlike the NV replicon which expresses only the nonstructural proteins, NV stool RNA transfection leads to expression of both nonstructural and structural proteins (Fig. 1B) that results in not only RNA replication but also virion assembly and release of progeny virus, providing a system closer to a complete virus life cycle than the NV replicon. However, it should be noticed that both systems do not fully represent a complete virus infection cycle. The development of an efficient in vitro replication system for HuNoVs will be required to validate results from this study and provide further insight into the innate immune response to HuNoV replication in the intestinal epithelium to better understand host control of these notorious viruses.
ACKNOWLEDGMENTS
We acknowledge the assistance of the Integrated Microscopy Core of Baylor College of Medicine, which is supported by P30 DK-56338 (to H. El-Serag), P30 CA-125123 (to C. K. Osborne), and the Dan L. Duncan Cancer Center at Baylor College of Medicine. This work is supported by NIH Pedi GI Training Grant T32 DK 07664 (to L.Q.), PO1 AI 057788 (to M.K.E.), P30 DK 56338, which supports the Texas Medical Center Digestive Disease Center, and Agriculture and Food Research Initiative competitive grant 2011-68003-30395 (to M.K.E.) from the USDA National Institute of Food and Agriculture.
We thank Frederick Neill and Baijun Kou for technical assistance and Sue Crawford and Sasirekha Ramani for critical readings of the manuscript.
REFERENCES
- 1.Ramani S, Atmar RL, Estes MK. 2014. Epidemiology of human noroviruses and updates on vaccine development. Curr Opin Gastroenterol 30:25–33. doi: 10.1097/MOG.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Payne DC, Vinje J, Szilagyi PG, Edwards KM, Staat MA, Weinberg GA, Hall CB, Chappell J, Bernstein DI, Curns AT, Wikswo M, Shirley SH, Hall AJ, Lopman B, Parashar UD. 2013. Norovirus and medically attended gastroenteritis in U.S. children. N Engl J Med 368:1121–1130. doi: 10.1056/NEJMsa1206589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koo HL, Neill FH, Estes MK, Munoz FM, Cameron A, Dupont HL, Atmar RL. 2013. Noroviruses: the most common pediatric viral enteric pathogen at a large university hospital after introduction of rotavirus vaccination. J Pediatric Infect Dis Soc 2:57–60. doi: 10.1093/jpids/pis070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N Engl J Med 361:1776–1785. doi: 10.1056/NEJMra0804575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noel JS, Fankhauser RL, Ando T, Monroe SS, Glass RI. 1999. Identification of a distinct common strain of “Norwalk-like viruses” having a global distribution. J Infect Dis 179:1334–1344. doi: 10.1086/314783. [DOI] [PubMed] [Google Scholar]
- 6.Leshem E, Wikswo M, Barclay L, Brandt E, Storm W, Salehi E, DeSalvo T, Davis T, Saupe A, Dobbins G, Booth HA, Biggs C, Garman K, Woron AM, Parashar UD, Vinje J, Hall AJ. 2013. Effects and clinical significance of GII.4 Sydney norovirus, United States, 2012–2013. Emerg Infect Dis 19:1231–1238. doi: 10.3201/eid1908.130458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan MC, Lee N, Hung TN, Kwok K, Cheung K, Tin EK, Lai RW, Nelson EA, Leung TF, Chan PK. 2015. Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat Commun 6:10061. doi: 10.1038/ncomms10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Graaf M, van Beek J, Vennema H, Podkolzin AT, Hewitt J, Bucardo F, Templeton K, Mans J, Nordgren J, Reuter G, Lynch M, Rasmussen LD, Iritani N, Chan MC, Martella V, Ambert-Balay K, Vinje J, White PA, Koopmans MP. 2015. Emergence of a novel GII.17 norovirus—end of the GII.4 era? Euro Surveill 20(26):pii=21178 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kavanagh O, Estes MK, Reeck A, Raju RM, Opekun AR, Gilger MA, Graham DY, Atmar RL. 2011. Serological responses to experimental Norwalk virus infection measured using a quantitative duplex time-resolved fluorescence immunoassay. Clin Vaccine Immunol 18:1187–1190. doi: 10.1128/CVI.00039-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czako R, Atmar RL, Opekun AR, Gilger MA, Graham DY, Estes MK. 2012. Serum hemagglutination inhibition activity correlates with protection from gastroenteritis in persons infected with Norwalk virus. Clin Vaccine Immunol 19:284–287. doi: 10.1128/CVI.05592-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ajami NJ, Barry MA, Carrillo B, Muhaxhiri Z, Neill FH, Prasad BV, Opekun AR, Gilger MA, Graham DY, Atmar RL, Estes MK. 2012. Antibody responses to norovirus genogroup GI.1 and GII.4 proteases in volunteers administered Norwalk virus. Clin Vaccine Immunol 19:1980–1983. doi: 10.1128/CVI.00411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Souza M, Cheetham SM, Azevedo MS, Costantini V, Saif LJ. 2007. Cytokine and antibody responses in gnotobiotic pigs after infection with human norovirus genogroup II.4 (HS66 strain). J Virol 81:9183–9192. doi: 10.1128/JVI.00558-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Souza M, Azevedo MS, Jung K, Cheetham S, Saif LJ. 2008. Pathogenesis and immune responses in gnotobiotic calves after infection with the genogroup II.4-HS66 strain of human norovirus. J Virol 82:1777–1786. doi: 10.1128/JVI.01347-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW 4th. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- 15.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. 2004. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol 2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lay MK, Atmar RL, Guix S, Bharadwaj U, He H, Neill FH, Sastry KJ, Yao Q, Estes MK. 2010. Norwalk virus does not replicate in human macrophages or dendritic cells derived from the peripheral blood of susceptible humans. Virology 406:1–11. doi: 10.1016/j.virol.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taube S, Kolawole AO, Hohne M, Wilkinson JE, Handley SA, Perry JW, Thackray LB, Akkina R, Wobus CE. 2013. A mouse model for human norovirus. mBio 4:e00450-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karandikar UC, Crawford SE, Ajami NJ, Murakami K, Kou B, Ettayebi K, Papanicolaou GA, Jongwutiwes U, Perales MA, Shia J, Mercer D, Finegold MJ, Vinje J, Atmar RL, Estes MK. 12 July 2016. Detection of human norovirus in intestinal biopsies from immunocompromised transplant patients. J Gen Virol doi: 10.1099/jgv.0.000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alhatlani B, Vashist S, Goodfellow I. 2015. Functions of the 5′ and 3′ ends of calicivirus genomes. Virus Res 206:134–143. doi: 10.1016/j.virusres.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glass PJ, White LJ, Ball JM, Leparc-Goffart I, Hardy ME, Estes MK. 2000. Norwalk virus open reading frame 3 encodes a minor structural protein. J Virol 74:6581–6591. doi: 10.1128/JVI.74.14.6581-6591.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang X, Wang M, Wang K, Estes MK. 1993. Sequence and genomic organization of Norwalk virus. Virology 195:51–61. doi: 10.1006/viro.1993.1345. [DOI] [PubMed] [Google Scholar]
- 23.Vongpunsawad S, Venkataram Prasad BV, Estes MK. 2013. Norwalk virus minor capsid protein VP2 associates within the VP1 shell domain. J Virol 87:4818–4825. doi: 10.1128/JVI.03508-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorne LG, Goodfellow IG. 2014. Norovirus gene expression and replication. J Gen Virol 95:278–291. doi: 10.1099/vir.0.059634-0. [DOI] [PubMed] [Google Scholar]
- 25.Kawai T, Akira S. 2007. Antiviral signaling through pattern recognition receptors. J Biochem 141:137–145. [DOI] [PubMed] [Google Scholar]
- 26.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Rauch I, Muller M, Decker T. 2013. The regulation of inflammation by interferons and their STATs. JAKSTAT 2:e23820. doi: 10.4161/jkst.23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Weerd NA, Nguyen T. 2012. The interferons and their receptors—distribution and regulation. Immunol Cell Biol 90:483–491. doi: 10.1038/icb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thackray LB, Duan E, Lazear HM, Kambal A, Schreiber RD, Diamond MS, Virgin HW. 2012. Critical role for interferon regulatory factor 3 (IRF-3) and IRF-7 in type I interferon-mediated control of murine norovirus replication. J Virol 86:13515–13523. doi: 10.1128/JVI.01824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW, Colonna M. 2008. MDA-5 recognition of a murine norovirus. PLoS Pathog 4:e1000108. doi: 10.1371/journal.ppat.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. 2015. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. 2015. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347:269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guix S, Asanaka M, Katayama K, Crawford SE, Neill FH, Atmar RL, Estes MK. 2007. Norwalk virus RNA is infectious in mammalian cells. J Virol 81:12238–12248. doi: 10.1128/JVI.01489-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katayama K, Murakami K, Sharp TM, Guix S, Oka T, Takai-Todaka R, Nakanishi A, Crawford SE, Atmar RL, Estes MK. 2014. Plasmid-based human norovirus reverse genetics system produces reporter-tagged progeny virus containing infectious genomic RNA. Proc Natl Acad Sci U S A 111:E4043–4052. doi: 10.1073/pnas.1415096111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Ramani S, Hill H, Ferreira J, Graham DY. 2014. Determination of the 50% human infectious dose for Norwalk virus. J Infect Dis 209:1016–1022. doi: 10.1093/infdis/jit620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Graham DY. 2008. Norwalk virus shedding after experimental human infection. Emerg Infect Dis 14:1553–1557. doi: 10.3201/eid1410.080117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asanaka M, Atmar RL, Ruvolo V, Crawford SE, Neill FH, Estes MK. 2005. Replication and packaging of Norwalk virus RNA in cultured mammalian cells. Proc Natl Acad Sci U S A 102:10327–10332. doi: 10.1073/pnas.0408529102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang X, Wang M, Graham DY, Estes MK. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J Virol 66:6527–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharp TM. 2010. Norwalk virus nonstructural protein p22 is a novel inhibitor of cellular protein secretion. PhD dissertation. Baylor College of Medicine, Houston, TX. [Google Scholar]
- 40.Kou B, Crawford SE, Ajami NJ, Czako R, Neill FH, Tanaka TN, Kitamoto N, Palzkill TG, Estes MK, Atmar RL. 2015. Characterization of cross-reactive norovirus-specific monoclonal antibodies. Clin Vaccine Immunol 22:160–167. doi: 10.1128/CVI.00519-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qu L, Vongpunsawad S, Atmar RL, Prasad BV, Estes MK. 2014. Development of a Gaussia luciferase-based human norovirus protease reporter system: cell type-specific profile of Norwalk virus protease precursors and evaluation of inhibitors. J Virol 88:10312–10326. doi: 10.1128/JVI.01111-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li K, Chen Z, Kato N, Gale M Jr, Lemon SM. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J Biol Chem 280:16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 43.Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K, Lemon SM. 2011. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog 7:e1002169. doi: 10.1371/journal.ppat.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rohayem J, Robel I, Jager K, Scheffler U, Rudolph W. 2006. Protein-primed and de novo initiation of RNA synthesis by norovirus 3Dpol. J Virol 80:7060–7069. doi: 10.1128/JVI.02195-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCormick CJ, Salim O, Lambden PR, Clarke IN. 2008. Translation termination reinitiation between open reading frame 1 (ORF1) and ORF2 enables capsid expression in a bovine norovirus without the need for production of viral subgenomic RNA. J Virol 82:8917–8921. doi: 10.1128/JVI.02362-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noyce RS, Taylor K, Ciechonska M, Collins SE, Duncan R, Mossman KL. 2011. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J Virol 85:10926–10931. doi: 10.1128/JVI.00862-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim MJ, Latham AG, Krug RM. 2002. Human influenza viruses activate an interferon-independent transcription of cellular antiviral genes: outcome with influenza A virus is unique. Proc Natl Acad Sci U S A 99:10096–10101. doi: 10.1073/pnas.152327499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brownell J, Bruckner J, Wagoner J, Thomas E, Loo YM, Gale M Jr, Liang TJ, Polyak SJ. 2014. Direct, interferon-independent activation of the CXCL10 promoter by NF-κB and interferon regulatory factor 3 during hepatitis C virus infection. J Virol 88:1582–1590. doi: 10.1128/JVI.02007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colamonici OR, Domanski P, Sweitzer SM, Larner A, Buller RM. 1995. Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon alpha transmembrane signaling. J Biol Chem 270:15974–15978. doi: 10.1074/jbc.270.27.15974. [DOI] [PubMed] [Google Scholar]
- 50.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez-Vega V, Sosnovtsev SV, Belliot G, King AD, Mitra T, Gorbalenya A, Green KY. 2004. Norwalk virus N-terminal nonstructural protein is associated with disassembly of the Golgi complex in transfected cells. J Virol 78:4827–4837. doi: 10.1128/JVI.78.9.4827-4837.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ettayebi K, Hardy ME. 2003. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP-A and prevents cell surface expression of vesicular stomatitis virus G protein. J Virol 77:11790–11797. doi: 10.1128/JVI.77.21.11790-11797.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A 104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hidmark AS, McInerney GM, Nordstrom EK, Douagi I, Werner KM, Liljestrom P, Karlsson Hedestam GB. 2005. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J Virol 79:10376–10385. doi: 10.1128/JVI.79.16.10376-10385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hare DN, Collins SE, Mukherjee S, Loo YM, Gale M Jr, Janssen LJ, Mossman KL. 2016. Membrane perturbation-associated Ca2+ signaling and incoming genome sensing are required for the host response to low-level enveloped virus particle entry. J Virol 90:3018–3027. doi: 10.1128/JVI.02642-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spiegel M, Weber F. 2006. Inhibition of cytokine gene expression and induction of chemokine genes in non-lymphatic cells infected with SARS coronavirus. Virol J 3:17. doi: 10.1186/1743-422X-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang KO, George DW. 2007. Interferons and ribavirin effectively inhibit Norwalk virus replication in replicon-bearing cells. J Virol 81:12111–12118. doi: 10.1128/JVI.00560-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang KO, Sosnovtsev SV, Belliot G, King AD, Green KY. 2006. Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 353:463–473. doi: 10.1016/j.virol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 59.Changotra H, Jia Y, Moore TN, Liu G, Kahan SM, Sosnovtsev SV, Karst SM. 2009. Type I and type II interferons inhibit the translation of murine norovirus proteins. J Virol 83:5683–5692. doi: 10.1128/JVI.00231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindesmith L, Moe C, Lependu J, Frelinger JA, Treanor J, Baric RS. 2005. Cellular and humoral immunity following Snow Mountain virus challenge. J Virol 79:2900–2909. doi: 10.1128/JVI.79.5.2900-2909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gonzalez-Hernandez MB, Liu T, Blanco LP, Auble H, Payne HC, Wobus CE. 2013. Murine norovirus transcytosis across an in vitro polarized murine intestinal epithelial monolayer is mediated by M-like cells. J Virol 87:12685–12693. doi: 10.1128/JVI.02378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalez-Hernandez MB, Liu T, Payne HC, Stencel-Baerenwald JE, Ikizler M, Yagita H, Dermody TS, Williams IR, Wobus CE. 2014. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J Virol 88:6934–6943. doi: 10.1128/JVI.00204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kolawole AO, Gonzalez-Hernandez MB, Turula H, Yu C, Elftman MD, Wobus CE. 2016. Oral norovirus infection is blocked in mice lacking Peyer's patches and mature M cells. J Virol 90:1499–1506. doi: 10.1128/JVI.02872-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sillanpaa M, Kaukinen P, Melen K, Julkunen I. 2008. Hepatitis C virus proteins interfere with the activation of chemokine gene promoters and downregulate chemokine gene expression. J Gen Virol 89:432–443. doi: 10.1099/vir.0.83316-0. [DOI] [PubMed] [Google Scholar]
- 66.Versteeg GA, Bredenbeek PJ, van den Worm SH, Spaan WJ. 2007. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology 361:18–26. doi: 10.1016/j.virol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou H, Perlman S. 2007. Mouse hepatitis virus does not induce beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J Virol 81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roth-Cross JK, Martinez-Sobrido L, Scott EP, Garcia-Sastre A, Weiss SR. 2007. Inhibition of the alpha/beta interferon response by mouse hepatitis virus at multiple levels. J Virol 81:7189–7199. doi: 10.1128/JVI.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neufeldt CJ, Joyce MA, Van Buuren N, Levin A, Kirkegaard K, Gale M Jr, Tyrrell DL, Wozniak RW. 2016. The hepatitis C virus-induced membranous web and associated nuclear transport machinery limit access of pattern recognition receptors to viral replication sites. PLoS Pathog 12:e1005428. doi: 10.1371/journal.ppat.1005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uchida L, Espada-Murao LA, Takamatsu Y, Okamoto K, Hayasaka D, Yu F, Nabeshima T, Buerano CC, Morita K. 2014. The dengue virus conceals double-stranded RNA in the intracellular membrane to escape from an interferon response. Sci Rep 4:7395. doi: 10.1038/srep07395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koetzner CA, Kuo L, Goebel SJ, Dean AB, Parker MM, Masters PS. 2010. Accessory protein 5a is a major antagonist of the antiviral action of interferon against murine coronavirus. J Virol 84:8262–8274. doi: 10.1128/JVI.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]