

Abstract
We identify two novel mutations in acetylcholine receptor (AChR) causing a slow-channel congenital myasthenia syndrome (CMS) in three unrelated patients (Pts). Pt 1 harbors a heterozygous βV266A mutation (p.Val289Ala) in the second transmembrane domain (M2) of the AChR β subunit (CHRNB1). Pts 2 and 3 carry the same mutation at an equivalent site in the ε subunit (CHRNE), εV265A (p.Val285Ala). The mutant residues are conserved across all AChR subunits of all species and are components of a valine ring in the channel pore which is positioned four residues above the leucine ring. Both βV266A and εV265A reduce the amino acid size and lengthen the channel opening bursts by 4.0-fold by enhancing gating efficiency by approximately 30-fold. Substitution of alanine for valine at the corresponding position in the δ and α subunit prolongs the burst duration 4- and 8-fold, respectively. Replacing valine at ε codon 265 either by a still smaller glycine or by a larger leucine also lengthens the burst duration. Our analysis reveals that each valine in the valine ring contributes to channel kinetics equally, and the valine ring has been optimized in the course of evolution to govern channel gating.
Keywords: ion channel, acetylcholine receptors, patch-clamp, congenital myasthenic syndrome, slow-channel syndrome
Introduction
Congenital myasthenic syndromes (CMS) are heterogeneous disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Although 24 CMS disease genes have been identified (CHRNA1, MIM# 100690; CHRNB1, MIM# 100710; CHRND, MIM# 100720; CHRNE, MIM# 100725; RAPSN, MIM# 601592; AGRN, MIM# 103320; MUSK, MIM# 601296; LRP4, MIM# 604270; DOK7, MIM# 610285; SCN4A, MIM# 603967; PLEC, MIM# 601282; GFPT1, MIM# 138292; DPAGT1, MIM# 191350; ALG2, MIM# 607905; ALG14, MIM# 612866; GMPPB, MIM# 615320; PREPL, MIM# 609557; SLC25A1, MIM# 190315; LAMB2, MIM# 150325; COLQ, MIM# 603033; CHAT, MIM# 118490; SYT2, MIM# 600104; SNAP25B, MIM# 600322; COL13A1, MIM# 120350), approximately one-half of CMS stem from mutations in subunits of the acetylcholine receptor (AChR), which are encoded by CHRNA1, CHRNB1, CHNRD, and CHRNE (Engel et al., 2015). AChR is a pentameric ligand-gated cationic ion channel and composed of homologous subunits with stoichiometry (α1)2β1δγ in embryonic type and (α1)2β1δε in adult type. Adult-type ε-AChR has a higher conductance and a shorter open time compared to embryonic γ-AChR and the open channel is non-selectively cation permeable. The slow-channel congenital myasthenic syndromes (SCCMS) are caused by prolonged opening episodes of AChR due to dominant gain-of-function mutations in subunit genes of the receptor (Ohno et al., 1995; Ohno et al., 2014). In this disease, signal transmission at the motor endplate is compromised by multiple mechanisms: First, staircase summation of prolonged endplate potentials (EPP), each arising in wake of a preceding EPP, inactivates the postsynaptic voltage-gated sodium channels. Second, slow-channel AChRs are prone to become desensitized (Milone et al., 1997), which reduces the number of AChRs that can be opened by ACh. Third, the prolonged opening episodes of AChR allow excessive influx of extracellular calcium that causes an endplate myopathy with focal degeneration of the junctional folds and loss of AChR as well as apoptosis of some of the junctional nuclei (Groshong et al., 2007). If the prolonged EPP exceeds the refractory period of the muscle fiber, it can evoke one or more muscle fiber action potentials.
Twenty-four slow-channel mutations in different domains of the AChR α, β, δ, and ε subunits have been reported to date (Suppl. Table S1). Mutations near the ACh-binding site at the extracellular domain (e.g. αG153S (Sine et al., 1995)) and in the N-terminal part of the first transmembrane domain (M1) (e.g. αN271K (Wang et al., 1997) and εL221F (Hatton et al., 2003)) increase affinity for ACh and cause repeated reopenings of the channel during prolonged occupancy by ACh. Mutations in the second transmembrane domain (M2) that lines the ion channel pore (Fig. 1E) promote the open state by slowing the channel-closing rate (α) and sometimes by facilitating the channel-opening rate (β) (e.g. αV249F (Milone et al., 1997), βV266M (Engel et al., 1996), εT264P (Ohno et al., 1995), and εL269F (Engel et al., 1996)). In addition, αV249F (Milone et al., 1997), εT264P (Ohno et al., 1995), and εL269F (Engel et al., 1996), but not βV266M (Engel et al., 1996), increase affinity for ACh. In the adult-type ε-AChR, 7% of the endplate current is carried by Ca2+ (Fucile et al., 2006). Two SCCMS mutations (εT264P (Ohno et al., 1995) and εV259F (Fidzianska et al., 2005)) increase the Ca2+ permeability of 1.5- to 2.0-fold (Di Castro et al., 2007), which further worsens the endplate myopathy. A total of 12 mutations were found in the M2 domain, and most mutations introduce a larger amino acid except for βT265S (Chaouch et al., 2012) which reduces amino acid size by 22% (Creighton, 1993) and εT264P which kinks the α-helix that contributes the lining of the ion channel (Azuma et al., 2015; Ohno et al., 1995). Introduction of a larger amino acid into the channel-lining M2 domain may hinder closure of the AChR channel whereas introduction of a smaller amino acid may or may not have this effect. As βT265S was not functionally analyzed (Chaouch et al., 2012), we examined the kinetic effect of introducing a smaller amino acid into the M2 domain.
Fig. 1.

A. Pedigrees of patients 1, 2, and 3. The mother of patients 3 is mosaic for εV265A. Arrows point to the proband. B. Multiple alignment of the M2 of AChR subunits. βV266 and εV265 are conserved across all human AChR subunits and across AChR subunits of different species. C. Haplotype analysis of patients 2 and 3. SNPs on an allele carrying εV265A (star mark) are determined by analyzing the parents' DNA. Intron 6 to Intron 8 of CHRNE encoded on the reverse strand are drawn to scale. Note that haplotypes are not shared between patients 2 and 3. D. Sequencing chromatograms of the mother, and patient 3 of CHRNE exon 8. Note that the mother is mosaic for εV265A and the mutant peak in the mother is lower than that in patient 3. E. Scheme of SCCMS mutations at the M2. The M2 domains of five subunits (α1 × 2, β1, δ, and ε subunits) form α helices and constitute the ion channel pore that is opened by binding of two ACh molecules to the extracellular domains of these subunits. The smallest blue circle in the middle represents the narrowest leucine ring in the channel pore. The valine ring is 4 residues above the leucine ring and is indicated in red. The two large gray rings represent the amino acids margining the M2. The previously reported amino acid-substituting mutations in the M2 domains are indicated by closed symbols. βV266A and εV265A are shown in red. Mutations are indicated according to the legacy nomenclature, in which the first codon is the N-terminal end of a mature peptide. Add 20, 23, 21, and 20 codons for AChR α, β, δ, and ε subunits, respectively, to convert the legacy nomenclature to the HGVS nomenclature.
We here report three patients harboring slow-channel mutations; one harbors βV266A and two carry εV265A in a corresponding position of the ε subunit. The substituting alanine reduces amino acid size by 36% (Creighton, 1993).
Materials and Methods
Patients
All human studies were in accord with and approved by the Institutional Review Boards of Mayo Clinic, Nagoya University Graduate School of Medicine, and National Center Hospital of Neurology and Psychiatry in Japan. All human studies were performed in accordance with relevant guidelines. After obtaining the appropriate informed consent, venous blood samples were obtained for genetic studies from patients and relatives. Genomic DNA was isolated with the QIAamp Blood DNA kit (Qiagen) according to the manufacturer's recommendations.
Mutation analysis by Sanger sequencing
For patients 1 and 2, the AChR subunit genes were sequenced directly using genomic DNA (Ohno et al., 1995). PCR-amplified fragments were cleared of unincorporated dNTPs and primers by incubating at 37°C for 60 min with ExoSap-It (United States Biochemical). The enzymes were then inactivated at 80°C for 15 min. PCR products were sequenced with an ABI377 DNA sequencer or 3730xl DNA analyzer (Applied Biosystems).
Exome capture resequencing analysis
For patient 3, we enriched exonic fragments using the SureSelect Human All Exon v5 (Agilent Technologies) and sequenced 150 bp in paired-end directions using HiSeq2500 (Illumina). The sequencing fragments were mapped to the human genome GRCh37/hg19 using BWA 0.7.5a (Li and Durbin, 2010) and BLAT v35 at http://genome.ucsc.edu/. SNVs/indels were called by Isaac (Illumina), Avadis NGS 1.5.0 (Strand), and VarScan 2.3 (Koboldt et al., 2012). The observed mutation was confirmed by Sanger sequencing using ABI Prism 3100 in patient 3 and her parents.
Legacy and HGVS nomenclatures
Mutations are indicated according to the legacy nomenclature, and the HGVS nomenclature is indicated in parallel as much as possible. With the legacy nomenclature, codon number +1 is the first codon of the N-terminal end of a mature peptide. With the HGVS nomenclature, codon number +1 is the initiation codon of NM_000079.3, NM_000747.2, NM_000751.2, and NM_000080.3 for CHRNA1, CHRNB1, CHRND, and CHNRE, respectively. To convert the legacy nomenclature to the HGVS nomenclature, 20, 23, 21, and 20 codons are added for AChR α, β, δ, and ε subunits, respectively.
Analyses of mosaicism and haplotypes
For analysis of mosaicism in the mother of patient 3, a 384-bp DNA segment spanning εV265A was amplified with primers 5′-ATCGTGCCCTGTGTGCTCATC-3′ on exon 7 and 5′-CCACCCAGCGTCCGAATAAA-3′ on intron 8 by PCR, and was digested with AciI (R0551, New England Biolabs). AciI digestion gave rise to two fragments of 53 and 45 bp from the mutant allele, and a single fragment of 98 bp from the wild-type allele. Intensities of these fragments on a 6% NuSieve 3:1 agarose gel (50091, Lonza) were quantified by ImageJ to estimate the ratio of the mutant to the wild-type allele.
For haplotype analysis of patients 2 and 3, a 384-bp DNA segment spanning εV265A was amplified by PCR, as stated above. An amplicon of patient 2 and was cloned into pGEM-T (Promega) and was sequenced by 3730xl DNA analyzer to identify an allele carrying εV265A. Amplicons of patient 3 and her parents were directly sequenced by 3730xl DNA analyzer, and heterozygous SNPs shared by patient 3 and her mother were searched for to identify SNPs on the mutant allele.
Construction and expression of wild-type and mutant AChRs
Constructions of mammalian expression vectors carrying human CHRNA1, CHRNB1, CHRND, and CHRNE were previously described (Ohno et al., 1996). Patient's and artificial mutations were engineered into CRHNB1 and CHRNE encoding the AChR β and ε subunit, respectively, using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The presence of each mutation and absence of artifacts were confirmed by sequencing the entire inserts. HEK293 cells were transfected with vectors comprising pRBG4-α, β, δ, ε, and pEGFP-N1 at a ratio of 2:1:1:1:1, using FuGene 6 transfection reagent (Roche) (Shen et al., 2005).
α-Bungarotoxin and ACh binding measurements
The total numbers of 125I-α-bungarotoxin (125I-α-bgt) sites of intact or saponin-permeabilized HEK293 cells were determined as previously described (Ohno et al., 1996). The ACh binding properties of wild-type and mutant AChRs expressed on the surface of HEK293 cells were examined by measuring ACh binding in competition against the initial rate of 125I-α-bgt binding (Ohno et al., 1996). The surface competition measurements were analyzed according to the following Hill equation.
where Y is fractional occupancy by ACh, n is the Hill coefficient, KOV, is an overall dissociation constant for a monophasic binding profile.
Single-channel patch clamp recordings
Single-channel recordings were obtained in the cell-attached configuration at a membrane potential of −80 mV at 22°C and with bath and pipette solutions containing (in mM): KCl, 142; NaCl, 5.4; CaCl2, 1.8; MgCl2, 1.7; HEPES, 10, pH 7.4. Single-channel currents were recorded using an Axopatch 200A amplifier (Axon Instruments) at a bandwidth of 50 kHz, digitized at 5-μs intervals using a Digidata 1322A (Axon Instruments) and recorded to hard disk using the program Clampex 8.2 (Axon Instruments). Recordings obtained with ACh were analyzed at a uniform bandwidth of 11.7 kHz with dead time of 15.3 μs imposed by using TACx4.0.9 software (Bruxton). Dwell-time histograms were plotted on a logarithmic abscissa and fitted by the sum of exponentials by maximum likelihood (Sigworth and Sine, 1987).
To estimate rate constants underlying AChR activation, we employed desensitizing concentrations of partial agonist choline, which has been used widely to study the kinetics of slow-channel mutations (Zhou et al., 1999; Grosman and Auerbach, 2000; Shen et al., 2006). High concentrations of agonist cause events from a single receptor channel to cluster into sequences of identifiable activation episodes (Sakmann et al., 1980). Channel events elicited by low concentrations of choline (20 to 50 μM) were analyzed at 11.7 kHz. Clusters of channel events elicited by 0.5 to 20 mM choline were analyzed at 4 kHz due to frequent, brief channel blockages that reduced the single-channel current amplitude. Clusters were identified as a series of closely spaced openings preceded and followed by closed intervals greater than a defined critical time. The critical time was determined by a method that misclassifies an equal number of events between 2 adjacent closed-time components (Colquhoun and Sakmann, 1985). For each receptor, the critical time that provided the best fit for the closed-time histogram was chosen for the final analysis. Clusters with fewer than 5 openings were excluded from analysis. Individual clusters were examined for homogeneity based on the mean Popen and open duration for each cluster, and clusters within 2 SDs of the mean were accepted for further analysis (Qin et al., 1997; Shen et al., 2002). The resulting global set of open and closed dwell times from wild-type and mutant AChRs were analyzed using the program MIL (QuB suite; http://www.qub.buffalo.edu/wiki/index.php/main_page), which uses an interval-based maximum likelihood method that also corrects for missed events to yield fitted rate constants in a kinetic scheme for receptor activation (Qin et al., 1997).
Results
Characteristics of CMS Patients
Patient 1 was a 3-week-old Caucasian boy who had bilateral eyelid ptosis, facial weakness, severe hypotonia, and respiratory insufficiency requiring assisted ventilation since birth. EMG studies revealed that the low amplitude of compound muscle action potentials (CMAP) was smaller than the lower limit in control subjects of the same age, and the low CMAP amplitude was not increased by edrophonium. There was a decremental response of the CMAP on 3 Hz repetitive stimulation indicated by a reduced amplitude of the fourth compared to the first evoked CMAP. No repetitive CMAPs were detected, likely because of the low initial CMAP amplitude. Like other slow-channel patients, he failed to respond to pyridostigmine. He was started on therapeutic doses of quinidine sulfate, a long-lived open-channel blocker of AChR (Harper and Engel, 1998) but the response could not be evaluated because he was lost for follow-up.
Patient 2 was a 21-year-old woman with severe myasthenic symptoms since birth. She could not climb stairs and could walk only for a short distance. The parents were not consanguineous and there no similarly affected family members. She had selectively severe weakness of the cervical and limb muscles with sparing of the quadriceps and gastrocnemius muscles. Repetitive nerve stimulation at 3 Hz showed a 20-40% decrement of the evoked CMAP and she had repetitive CMAPs. This patient responded incompletely to cholinesterase inhibitors.
Patient 3 was an 8-year-old female with severe myasthenic symptoms since birth. Her father is Japanese and her mother Chinese. She can only climb stairs by holding a handrail and walk without assistance only in the morning. Repetitive nerve stimulation at 3 Hz showed a 30-65% decrement of CMAPs and repetitive CMAPs.
None of the patients have similarly affected family members and all tested negatively for anti-AChR antibodies.
Mutation Analysis
We directly sequenced four AChR subunit genes in patients 1 and 2, and searched for candidate mutations by exome capture resequencing analysis in patient 3.
Patient 1 had a heterozygous T-to-C mutation in CHRNB1 exon 8 at position 7,357,661 (GRCh37/hg19) on chromosome 17, which predicted a valine-to-alanine substitution at amino acid 266 of the mature peptide (βV266A). The mutated residue is positioned in the second transmembrane domain (M2) of the AChR β subunit and faces the lumen of ion channel (Fig 1E). The human genome variation society (HGVS) annotation of the mutation was c.866T>C (NM_000747.2) in CHRNB1 predicting p.Val289Ala. Parents of patient 1 carry no mutation (Fig. 1A). We previously reported the βV266A mutation in Patient 1 in an abstract (Shen et al., 2003). The βV266A mutation was submitted to ClinVar at http://www.ncbi.nlm.nih.gov/clinvar/.
Patients 2 and 3 had a heterozygous T-to-C mutation in CHRNE exon 8 at position 7,404,124 (GRCh37/hg19) on chromosome 17, which predicted a valine-to-alanine substitution at the 265th amino acid of the mature peptide (εV265A) in M2 of the AChR ε subunit. The HGVS annotation of the mutation was c.854T>C in CHRNE predicting p.Val285Ala. We previously reported the εV265A mutation in patient 2 in an abstract (Ohno et al., 1998). The εV265A mutation was submitted to ClinVar at http://www.ncbi.nlm.nih.gov/clinvar. Parents of patient 2 and the father of patient 3 carried no mutation, but the mother of patient 3 carried εV265A (Fig. 1A). As the mutant peak predicting εV265A in the sequencing chromatogram of the mother's blood DNA was not as high as that in patient 3 (Fig. 1D), we analyzed mosaicism by AciI restriction enzyme analysis of the PCR amplicon and found that only 13% of the genome in mother's blood carried εV265A. The mother had no episodes of muscle weakness and no neurological deficits even on detailed neurological evaluation. Both βV266 and εV265 are conserved across all human AChR subunits and across AChR β and ε subunits in all species (Fig. 1B). For both βV266A and εV265A, the mutated valine faces the channel pore and is four residues above the leucine ring, which forms the narrowest part of the channel pore (Fig. 1E). Haplotype analysis of patients 2 and 3 revealed that each carries a unique SNP and that there was no founder effect for the εV266A mutation (Fig.1C).
Cell surface expression of mutant AChR on HEK293 cells
We engineered the βV266A mutation into CHRNB1 cDNA and the εV265A mutation into CHRNE cDNA, and coexpressed each mutant cDNA with complementary wild-type AChR subunit cDNAs in HEK293 cells. Measurement of [125I]α-bungarotoxin binding showed that surface expressions of βV266A-AChR and εV265A-AChR were 114.0 ± 5.4% (mean and SD, n = 3) and 86.7 ± 8.4% (mean and SD, n = 6) of wild-type AChR, respectively (Fig. 2A). To compare the apparent affinity for ACh of the mutant relative to the wild-type AChR, we measured ACh binding at steady state by competition against the initial rate of [125I]α-bungarotoxin binding to intact cells. The apparent dissociation constants of βV266A-AChR and εV265A-AChR were 3.9- and 16.8-fold smaller than that of wild-type AChR (Fig. 2B), suggesting that the mutations promote functional states with high affinity for the agonist, such as the open-channel or desensitized states. Fig. 2A and B show the surface expressions and the apparent affinities for ACh of the εV265G and εV265L mutants. These mutations are further discussed below, under the section of expression studies of mutant AChRs at ε265 with altered residue size.
Fig. 2.

A. Specific [125I]α-bungarotoxin binding to surface AChRs on intact HEK293 cells transfected with the indicated AChR subunits. Values are normalized for α-bungarotoxin binding to wild-type AChR and also for total amount of proteins in each experiment. Mean and SD of three to six experiments are indicated. B. ACh binding to intact HEK cells transfected with indicated AChR subunits determined by competition against the initial rate of [125I]α-bgt binding. Curves are fitted to the Hill equation 1-Y = 1 / (1+ ([ACh] / Kov)n), whereas Y is fractional occupancy by ACh, Kov is overall apparent dissociation constant (in units of M), n is the Hill coefficient. Values of Kov (x E-07) / n are 7.51 / 1.20, 1.92 / 1.08, 0.45 / 1.08, 0.50 / 0.70, and 3.15 / 1.00 in wild-type, βV266A, εV265A, εV265G, and εV265L, respectively. Mutations are indicated according to the legacy nomenclature.
Single-channel recordings of mutant AChRs expressed on HEK293 cells
To evaluate the kinetic consequences of βV266A and εV265A, we recorded single-channel currents activated by a limiting low concentration of ACh applied to HEK293 cells expressing wild-type, βV266A-, or εV265A-AChRs (Fig. 3). For both wild-type and mutant AChRs, single-channel currents appear as isolated single openings or as bursts of several openings in quick succession. Histograms of open and burst durations exhibit three exponential components (Fig. 3 and Table 1), which were attributed to two brief monoliganded open states and one long diliganded open state. Relative to the wild-type AChR, βV266A and εV265A increased the mean duration of the major burst component 4.0- and 4.3-fold (Table 1), respectively, whereas the longest component of open intervals in both increase about twofold, indicating that both mutants are slow-channel mutations.
Fig. 3.

Left: AChR channel events elicited by 50 nM ACh on HEK293 cells transfected with indicated AChR subunits. Right: Burst duration histograms fitted to the sum of exponentials. Mutations are indicated according to the legacy nomenclature.
Table 1. Open intervals and burst durations of wild-type and mutant AChRs on HEK cells.
| Open Intervals | Bursts | |||||
|---|---|---|---|---|---|---|
| τ1, ms | τ2, ms | τ3, ms | τ1, ms | τ2, ms | τ3, ms | |
| (a1) | (a2) | (a3) | (a1) | (a2) | (a3) | |
| Wild type | 0.037 ± 0.033a | 0.31 ± 0.050 | 1.35 ± 0.051 | 0.036 ± 0.0017b | 0.47 ± 0.059 | 3.31 ± 0.12 |
| (0.17 ± 0.022) | (0.27 ± 0.038) | (0.67 ± 0.042) | (0.24 ± 0.021) | (0.21 ± 0.027) | (0.58 ± 0.038) | |
| βV266A | 0.068 ± 0.0072 | 0.85 ± 0.087 | 2.27 ± 0.10 | 0.073 ± 0.0068 | 0.88 ± 0.21 | 13.35 ± 0.87 |
| (p.Val289Ala) | (0.20 ± 0.0050) | (0.55 ± 0.031) | (0.25 ± 0.030) | (0.41 ± 0.041) | (0.20 ± 0.045) | (0.39 ± 0.046) |
| εV265A | 0.035 ± 0.0025 | 0.31 ± 0.030 | 2.61 ± 0.010 | 0.048 ± 0.0070 | 0.44 ± 0.020 | 14.33 ± 0.31 |
| (p.Val285Ala) | (0.18 ± 0.0080) | (0.18 ± 0.027) | (0.65 ± 0.019) | (0.43 ± 0.020) | (0.29 ± 0.033) | (0.28 ± 0.014) |
| εV265G | 0.080 ± 0.011 | 1.34 ± 0.33 | 5.30 ± 0.38 | 0.082 ± 0.0026 | 1.60 ± 0.39 | 24.46 ± 3.07 |
| (p.Val285Gly) | (0.090 ± 0.013) | (0.30 ± 0.026) | (0.61 ± 0.022) | (0.13 ± 0.013) | (0.24 ± 0.063) | (0.64 ± 0.050) |
| εV265L | 0.032 ± 0.0079 | 0.52 ± 0.039 | 3.80 ± 0.66 | 0.035 ± 0.0090 | 0.39 ± 0.025 | 10.82 ± 1.10 |
| (p.Val285Leu) | (0.36 ± 0.061) | (0.21 ± 0.028) | (0.44 ± 0.071) | (0.52 ± 0.056) | (0.21 ± 0.048) | (0.28 ± 0.095) |
| αV255A | 0.046 ± 0.0052 | 0.34 ± 0.022 | 6.31 ± 0.48 | 0.079 ± 0.012 | 0.55 ± 0.083 | 26.59 ± 2.70 |
| (p.Val275Ala) | (0.23± 0.020) | (0.09 ± 0.0040) | (0.67 ± 0.028) | (0.32 ± 0.055) | (0.12 ± 0.017) | (0.56 ± 0.053) |
| δV269A | 0.042 ± 0.0034 | 0.35 ± 0.073 | 4.72 ± 0.77 | 0.047± 0.0076 | 0.59 ± 0.090 | 13.90 ± 1.89 |
| (p.Val290Ala) | (0.22 ± 0.057) | (0.11 ± 0.028) | (0.67 ± 0.079) | (0.30 ± 0.096) | (0.10 ± 0.012) | (0.60 ± 0.099) |
Values indicate mean ± SE of 21 patches for wild-type and at least 3 patches for all mutants. All patches with ACh 50 nM at final bandwidth 11.7 -12 kHz. MP = -80 mV.
τ1 components of opening intervals and bursts were not detected at 12 and 3 patches, respectively, in wild-type AChR. HGVS nomenclature is shown in parentheses.
To understand the kinetic steps altered by the mutations, we recorded single-channel currents activated by a range of choline concentrations in which individual AChR channels activate in clusters of many channel openings in quick succession before entering desensitized state. Single channel currents activated by a saturating concentration of choline (20 mM) appeared in clusters of closely spaced openings flanked by prolonged closed periods (Fig. 4). The reciprocal of the mean of the intra-cluster closed intervals, which directly represents the channel opening rate constant β (Zhou et al., 1999; Grosman and Auerbach, 2000; Shen et al., 2006), showed that βV266A and εV265A speeded the rate constant of channel opening by 4- and 6-fold, respectively. However, at the saturating concentration of 20 mM choline rapidly blocked the open channel, as indicated by reduced single-channel current amplitude (Fig. 4), preventing direct determination of the channel closing rate constant. We fitted the following minimal kinetic scheme to sequences of open and closed dwell times obtained over a range of increased subsaturating choline concentrations:
Fig. 4.

Kinetics of activation of wild-type AChR (A), βV266A-AChR (B), and εV265A-AChR (C). Left column, individual clusters of single-channel currents elicited by the indicated choline concentrations on HEK293 cells. Center and right columns show histograms of closed and open durations, respectively, with superimposed probability density functions for kinetic scheme of receptor activation for the entire range of choline concentrations. The rate constants are shown in Table 2. Mutations are indicated according to the legacy nomenclature.
In this scheme, two agonists (A) bind to the receptor (R) with association rate constants k+1 and k+2, and dissociate with rate constants k–1 and k–2. The doubly-occupied receptor opens with a rate constant β and closes with a rate constant α, and ACh blocks the open channel with a rate constant k+b, and unblocks from the open channel with a rate constant k–b. Owing to bandwidth limitations, the previously identified closed state between A2R and A2R*, known as flip or primed (Mukhtasimova et al., 2009; Lape et al., 2008), is not included in this scheme, thus the fitted rates β, α, and k–2 are apparent rate constants. Also, a desensitized state is not included because each cluster begins following recovery from desensitization and ends upon return to the desensitized state, so that closings within clusters represent transitions between activatable states. We determined the rate constants in the kinetic scheme under the simplifying assumptions that the association and dissociation rate constants are equivalent at each of the two binding sites, and the rate constant for channel opening is the reciprocal of the mean of intracluster closed intervals obtained at a saturating concentration of choline. The analysis showed that βV266A and εV265A increased the gating equilibrium constant (ϴ), or β/α, by 33- and 25-fold, respectively, owing to increased opening rate (β) and decreased closing rate (α) (Fig. 4, Table 2). The dissociation rate constant (K) in either mutation had no significant change.
Table 2. Kinetic parameters of wild-type and mutant AChRs expressed in HEK cells with choline as agonist.
| Rate constants | Wild-type | βV266A (p.Val289Ala) | εV265A (p.Val285Ala) |
|---|---|---|---|
| k+1 | 684 ± 179 | 910 ± 74 | 2,148 ± 380 |
| k-1 | 459 ± 122 | 316 ± 26 | 1,298 ± 235 |
| K1 (mM) | 0.67 | 0.35 | 0.60 |
| k+2 | 342 ± 89 | 455 ± 37 | 1,074 ± 190 |
| k-2 | 918 ± 245 | 632 ± 53 | 2,596 ± 471 |
| K2 (mM) | 2.68 | 1.38 | 2.42 |
| β | 50 ± 1 | 210 ± 13 | 300 ± 13 |
| α | 1,094 ± 18 | 138 ± 2 | 266 ± 8 |
| θ | 0.046 | 1.52 | 1.13 |
| Predicted Burst length (ms) | 0.96 | 9.65 | 4.19 |
| Recorded Burst length (ms) | 0.82 | 8.58 | 3.69 |
Association rates are in mM-1s-1; all other rate constants are in s-1. The intrinsic dissociation constant of each site (Kn) is k-n/k+n. The gating equilibrium constant θ = β/α. β was derived from currents elicited by 20 mM choline, which were fitted by a subset of the sequential kinetic scheme 1 containing only fully occupied states; the resulting estimate of β was constrained for the global fit of the full kinetic scheme to data obtained at 1 and 2 mM choline in wild-type, 0.5 to 2 mM choline in mutants. HGVS nomenclature is shown in parentheses.
To validate the fitted rate constants, we measured channel burst durations for wild-type and mutant AChRs in the presence of limiting low concentrations of choline (20 μM for mutant, 50 μM for wild-type) and compared them with burst durations predicted by the fitted rate constants. At low concentrations of choline channel openings appear as bursts of several openings in quick succession, with each burst arising from a different channel. The predicted burst durations by (β/K-2+1)/α for wild-type (0.96 ms), βV266A (9.65 ms), and εV265A (4.19 ms) AChR agreed with the measured burst durations (0.82, 8.58, and 3.69 ms, respectively) (Table 2). Thus the dominant effects of both mutations are due to a markedly enhanced efficiency of channel gating.
Expression studies of mutant AChRs at ε265 with altered residue size
Among the 24 mutations causing SCCMS, 14 mutations are in the M2 domain. Among the 14 mutations, three mutations introduce a smaller amino acid (βT265S, βV266A, and εV265A) and the others introduce a larger amino acid or proline causing a kink in the α helix (Suppl. Table S1). In contrast to βT265S, the currently reporting βV266A and εV265A face the ion channel pore (Fig. 1E). To investigate the effect of the volume of the side-chain of a channel-lining residue, we introduced a still smaller glycine residue (εV265G) and a larger leucine residue (εV265L) at εV265. The van der Waals volumes of the side-chains of glycine and leucine are 0.46- and 1.18- fold of that of valine, respectively.
Cell surface expressions of εV265G- and εV265L-AChRs were 100.8 ± 5.9% and 97.3 ± 11.0% (mean and SD, n = 3) of wild-type AChR, respectively (Fig. 2A). The apparent dissociation constants for ACh of εV265G- and εV265L-AChRs were 15.2- and 2.4-fold smaller than that of wild-type AChR, respectively (Fig. 2B), suggesting that both εV265G and εV265L cause prolonged openings of ion channels. Indeed single-channel recordings of εV265G- and εV265L-AChRs expressed on HEK293 cells revealed that the mean durations of the major burst component were prolonged 7.4- and 3.3-fold (Fig. 3, Table 1), respectively. Thus, either increased (εV265L) or reduced (εV265A, εV265G) volume of the side-chain of the residue at ε265 causes slow-channel kinetics of AChR.
Expression studies of mutant AChRs with mutation at equivalent position of α and δ subunits
To determine the kinetic consequences of mutations of the equivalent valines in M2 of α and δ subunits, αV255A and δV269A, we again recorded single channel currents elicited by 50 nM ACh and found that the lengths of the major burst components was prolonged 8.0- and 4.1-fold (Fig. 3, Table 1), respectively. Thus, the substitution of Ala for Val in the AChR M2 domain prolongs the length of channel openings, and when the mutation occurs in alpha subunit, the effect on channel opening length is twice of that observed with the mutation occurring in other subunits. To determine whether the enhanced effect of the αV255A mutation on burst length was due to presence of two mutant α subunits, we transfected the HEK cells with cDNAs encoding αV255A, wt α, wt β, wt δ, and wt ε at a ratio of 1:1:1:1:1. With this approach, three types of AChRs, expressing 2 mutant, 2 wild type, and one wild-type and one mutant α subunit, can be expected. We recorded single channel currents from the transfected cells under the same condition as from wild-type and αV255A-AChRs. The histograms of the opening burst lengths of the combined receptors were best fitted with 5 components in 3 patches (Fig. 3, Table 1). The time constants of the first and the second components are similar to the first and second components of wild-type and other AChR mutants. The time constant of the third component (3.07 ms) is similar to that of longest component of wild-type (3.31 ms). The time constant of the fourth component (13.38 ms) is similar to the longest component of βV266A-, εV265A-, δV269A-AChR (13.35 ms, 14.33 ms, 13.90 ms, respectively). The time constant of the fifth component (27.78 ms) is similar to that of longest component of αV255A-AChR (26.59 ms).
Discussion
We traced the cause of congenital myasthenic syndromes (CMS) in three unrelated patients harboring dominant missense mutations in the M2 transmembrane domain of AChR. In each patient a valine residue is replaced by a smaller alanine. In two patients the mutation resides in the ε subunit (εV265A) and in the third patient it occurs at an equivalent position in the β subunit (βV266A).
Among the 12 previously reported SCCMS mutations in M2 (Suppl. Table S1), only βT265S introduces a smaller amino acid (Chaouch et al., 2012). The other mutations introduce a larger amino acid (Engel et al., 2015) or a kink in the α helix (εT264P) (Ohno et al., 1995). βT265S was reported in familial SCCMS in a Serbian family, but single-channel recordings were not obtained (Chaouch et al., 2012). Our results show that both βV266A and εV265A markedly increase the gating efficiency by increasing the opening rate and decreasing the closing rate of the receptor, but have no effect on agonist affinity or surface expression. The overall results indicate that the kinetic abnormality caused by βV266A and εV265A determine the patient phenotypes.
Both βV266 and εV265 are conserved across all AChR subunits of all species and are components of a valine ring, positioned four residues above the leucine ring, in the channel pore (see Fig. 1E). In the mouse AChR ε subunit, mutation of valine ring residue has a greater effect on gating efficiency and the free energy change than any other mutations introduced into the M2 domain (Jha et al., 2009). Thus, εVal265 is a key contributor to channel opening. In the closed form of the Torpedo AChR channel, the leucine and valine rings are at the narrowest regions of the channel pore (Miyazawa et al., 2003). However, during channel opening the pore widens substantially at the level of the valine ring (Unwin, 2013). To better understand the role of the valine ring, we introduced a smaller (glycine) or larger (leucine) residue into ε-265 and found that either mutation prolonged the channel opening events (Fig 3 and Table 1). That introduction of other amino acids at ε265 destabilizes the AChR channel indicates that the valine ring is optimized and critical for regulating the channel gating.
We also examined the kinetic consequences of mutating valine to alanine at equivalent positions in the α and δ subunits. This revealed that the channel opening bursts of αV255A mutant were twice as long as those of the βV266A, δV269A and εV265A mutants. Finally, co-expression of wild-type and mutant α subunits indicated that the enhanced effect of the α mutant is due to the presence of two mutant α subunits. Thus, each valine from different subunits in the valine-ring contributes equally to the gating of muscle AChR.
Supplementary Material
Scheme 1.
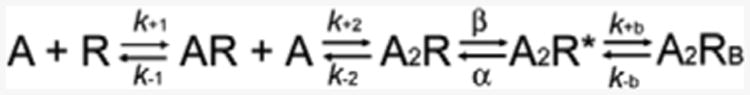
Acknowledgments
This work was supported by NIH Grant NS6277 (to AGE) and Grants-in-Aid from the MEXT, MHLW, and AMED of Japan (to KO and HK).
Abbreviation
- CMS
congenital myasthenic syndrome
- AChR
acetylcholine receptor
- M1, M2, M3, and M4
1st, 2nd, 3rd, and 4th transmembrane domains of AChR
Footnotes
Contributors: Study conception and design: XMS, KiO, AGE; acquisition, analysis, and interpretation of biochemical and physiological data: XMS, MM, AGE; acquisition, analysis, and interpretation of genetic data: TO, KeO, KiO, XMS; acquisition, analysis, and interpretation of patients' data: MM, KT, HK, EG, AGE; preparation of the manuscript: XMS, TO, KiO, AGE.
Disclosure statement: The authors have no conflicts of interest to declare.
Patient consent: Written informed consent was obtained for genetic testing and the case reports comply with the regulations of the Institutional Review Board of the Mayo Clinic and Ethical Review Committee of Nagoya University.
References
- Azuma Y, Nakata T, Tanaka M, Shen XM, Ito M, Iwata S, Okuno T, Nomura Y, Ando N, Ishigaki K, Ohkawara B, Masuda A, et al. Congenital myasthenic syndrome in Japan: ethnically unique mutations in muscle nicotinic acetylcholine receptor subunits. Neuromuscul Disord. 2015;25:60–69. doi: 10.1016/j.nmd.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Chaouch A, Muller JS, Guergueltcheva V, Dusl M, Schara U, Rakocevic-Stojanovic V, Lindberg C, Scola RH, Werneck LC, Colomer J, Nascimento A, Vilchez JJ, et al. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J Neurol. 2012;259:474–481. doi: 10.1007/s00415-011-6204-9. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton TE. Physical Interactions that determine the properties of proteins. In: Creighton TE, editor. Proteins: Structures and Molecular properties. New York: W. H. Freeman and Company; 1993. pp. 139–169. [Google Scholar]
- Di Castro A, Martinello K, Grassi F, Eusebi F, Engel AG. Pathogenic point mutations in a transmembrane domain of the epsilon subunit increase the Ca2+ permeability of the human endplate ACh receptor. J Physiol. 2007;579:671–677. doi: 10.1113/jphysiol.2007.127977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Milone M, Wang HL, Nakano S, Bouzat C, Pruitt JN, Hutchinson DO, Brengman JM, Bren N, Sieb JP, Sine SM. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1996;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015;14:420–434. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidzianska A, Ryniewicz B, Shen XM, Engel AG. IBM-type inclusions in a patient with slow-channel syndrome caused by a mutation in the AChR epsilon subunit. Neuromuscul Disord. 2005;15:753–759. doi: 10.1016/j.nmd.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Fucile S, Sucapane A, Grassi F, Eusebi F, Engel AG. The human adult subtype ACh receptor channel has high Ca2+ permeability and predisposes to endplate Ca2+ overloading. J Physiol. 2006;573:35–43. doi: 10.1113/jphysiol.2006.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groshong JS, Spencer MJ, Bhattacharyya BJ, Kudryashova E, Vohra BP, Zayas R, Wollmann RL, Miller RJ, Gomez CM. Calpain activation impairs neuromuscular transmission in a mouse model of the slow-channel myasthenic syndrome. J Clin Invest. 2007;17:2903–2912. doi: 10.1172/JCI30383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Auerbach A. Asymmetric and independent contribution of the second transmembrane segment 12′ residues to diliganded gating of acetylcholine receptor channels: a single-channel study with choline as the agonist. J Gen Physiol. 2000;115:637–651. doi: 10.1085/jgp.115.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CM, Engel AG. Safety and efficacy of quinidine sulfate in slow-channel congenital myasthenic syndrome. Ann N Y Acad Sci. 1998;841:203–206. doi: 10.1111/j.1749-6632.1998.tb10929.x. [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Shelley C, Brydson M, Beeson D, Colquhoun D. Properties of the human muscle nicotinic receptor, and of the slow-channel myasthenic syndrome mutant epsilonL221F, inferred from maximum likelihood fits. J Physiol. 2003;547:729–760. doi: 10.1113/jphysiol.2002.034173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Purohit P, Auerbach A. Energy and structure of the M2 helix in acetylcholine receptor-channel gating. Biophys J. 2009;96:4075–4084. doi: 10.1016/j.bpj.2009.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor alpha subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459:451–454. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Hutchinson DO, Milone M, Brengman JM, Bouzat C, Sine SM, Engel AG. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit. Proc Natl Acad Sci U S A. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Milone M, Brengman JM, LoMonaco M, Evoli A, Tonali PA, Engel AG. Slow-channel congenital myasthenic syndrome caused by a novel mutation in the acetylcholine receptor ε subunit. Neurology. 1998;50:A432. [Google Scholar]
- Ohno K, Ohkawara B, Ito M, Engel AG. Molecular Genetics of Congenital Myasthenic Syndromes. eLS: John Wiley & Sons, Inc; 2014. [DOI] [Google Scholar]
- Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, Engel AG. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Maximum likelihood estimation of aggregated Markov processes. Proc Biol Sci. 1997;264:375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- Shen XM, Deymeer F, Sine SM, Engel AG. Slow-channel mutation in acetylcholine receptor alphaM4 domain and its efficient knockdown. Ann Neurol. 2006;60:128–136. doi: 10.1002/ana.20861. [DOI] [PubMed] [Google Scholar]
- Shen XM, Ohno K, Fukudome T, Tsujino A, Brengman JM, De Vivo DC, Packer RJ, Engel AG. Congenital myasthenic syndrome caused by low-expressor fast-channel AChR delta subunit mutation. Neurology. 2002;59:1881–1888. doi: 10.1212/01.wnl.0000042422.87384.2f. [DOI] [PubMed] [Google Scholar]
- Shen XM, Ohno K, Milone M, Brengman J, Tsujino A, Engel AG. Effect of residue side-chain mass on channel kinetics in second trasmembrane domain of muscle AChR. Mol Biol Cell. 2003;14:223a. [Google Scholar]
- Shen XM, Ohno K, Sine SM, Engel AG. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3-M4 linker. Brain. 2005;128:345–355. doi: 10.1093/brain/awh364. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt JN, Engel AG. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Unwin N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: insights from Torpedo postsynaptic membranes. Q Rev Biophys. 2013;46:283–322. doi: 10.1017/S0033583513000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Engel AG, Auerbach A. Serum choline activates mutant acetylcholine receptors that cause slow channel congenital myasthenic syndromes. Proc Natl Acad Sci U S A. 1999;96:10466–10471. doi: 10.1073/pnas.96.18.10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.