

Abstract
Ras proteins are small GTPases, cycling between inactive GDP-bound and active GTP-bound states. Through these switches they regulate signaling that controls cell growth and proliferation. Activating Ras mutations are associated with approximately 30% of human cancers, which are frequently resistant to standard therapies. Over the past few years, structural biology and in silico drug design, coupled with improved screening technology, led to a handful of promising inhibitors, raising the possibility of drugging Ras proteins. At the same time, the invariable emergence of drug resistance argues for the critical importance of additionally honing in on signaling pathways which are likely to be involved. Here we overview current advances in Ras structural knowledge, including the conformational dynamic of full-length Ras in solution and at the membrane, therapeutic inhibition of Ras activity by targeting its active site, allosteric sites, and Ras–effector protein-protein interfaces, Ras dimers, the K-Ras4B/calmodulin/PI3Kα trimer, and targeting Ras with siRNA. To mitigate drug resistance, we propose signaling pathways that can be co-targeted along with Ras and explain why. These include pathways leading to the expression (or activation) of YAP1 and c-Myc. We postulate that these and Ras signaling pathways, MAPK/ERK and PI3K/Akt/mTOR, act independently and in corresponding ways in cell cycle control. The structural data are instrumental in the discovery and development of Ras inhibitors for treating RAS-driven cancers. Together with the signaling blueprints through which drug resistance can evolve, this review provides a comprehensive and innovative master plan for tackling mutant Ras proteins.
Graphical abstract

In this review we overview current advances in Ras structural knowledge and the signaling pathways that can be co-targeted along with Ras to mitigate drug resistance.
1 Introduction
Ras, a small GTPase, is an essential component of signaling networks controlling signal transduction pathways and promoting cell proliferation and survival.1–5 The Ras family includes H-Ras, N-Ras, and splice variants of K-Ras, K-Ras4A and K-Ras4B.6,7 They operate as binary switches in signal transduction pathways, cycling between inactive GDP-bound and active GTP-bound Ras states. The intrinsic hydrolysis of GTP by Ras is slow, but it is significantly accelerated by GTPase-activating proteins (GAPs),8–10 which serve as negative regulators of Ras activity by expediting GTP hydrolysis, leading to turning off Ras signaling. Ras activation is regulated by guanine nucleotide exchange factors (GEFs),11–13 which function as positive regulators of Ras activity through catalyzing the release of GDP and binding the more abundant GTP. In the active GTP-bound state, Ras proteins can associate with numerous downstream effector proteins,14–19 including phosphatidylinositol 3-kinase (PI3K), Raf kinase, and Ral guanine nucleotide dissociation stimulator (RalGDS), to regulate a diverse range of cellular processes.
Malfunctions in Ras proteins are common in tumorigenesis and, thus far, tumors initiated by Ras mutations have been among the most difficult to treat.20–22 In fact, the Catalogue of Somatic Mutations in Cancer (COSMIC)23 dataset has unraveled that oncogenic mutations in Ras account for approximately 30% of human cancers, with the mutations frequently found in lung, colon, and pancreatic cancers, and the vast majority of oncogenic mutations are concentrated on three hot spot residues (Gly12, Gly13 and Gln61).24 Oncogenic mutations that impair GTPase activity are resistant to GAP-mediated GTP hydrolysis, locking mutant Ras proteins in their active, GTP-bound state.25–28 Under these circumstances, mutant Ras proteins can excessively initiate downstream effector proteins signaling through pathways such as mitogen-activated Raf/MEK/ERK (MAPK) and PI3K/Akt,29–36 which promote cancer cell growth and survival. Analyses of Ras isoform mutational status in cancer emphasize that KRAS is the most frequently mutated isoform in RAS-driven cancers followed by NRAS and HRAS.24 Moreover, mutated isoforms tend to associate with particular tumor types:24,37,38 KRAS mutations are frequently linked to pancreatic, colon, and lung cancers; NRAS mutations are commonly detected in skin and hematological cancers; HRAS mutations are the most frequent in head and neck cancer, and cancers of the urinary tract.
Owing to the central role of Ras in oncogenesis, therapeutic inhibition of Ras provides a promising strategy for the development of targeted therapies for RAS-driven cancers.39–54 However, despite more than three decades of Ras-focused research, to date no drugs are available for clinical use, to directly bind and block these carcinogenic proteins, rendering the Ras proteins still “undruggable”.55 This is ascribed to the picomolar affinity of Ras for their GTP and GDP substrates and the lack of deep hydrophobic pockets on Ras catalytic domain for high affinity small molecule binders.56 Farnesyltransferase inhibitors, the first class of drugs to target Ras indirectly, hinder the transfer of the farnesyl group to Ras, which is of prime importance for Ras’ association with the membrane.57–60 However, the inhibitory effects of these inhibitors were not effective in advanced solid tumors because of the existence of an alternative – prenylation and geranylgeranylation – of K-Ras4B and N-Ras catalyzed by geranylgeranyl transferase I.61 Although past failures in targeting Ras have defeated the impetus for the development of Ras inhibitors, the renewed interest in Ras as a therapeutic target in recent years has promoted efforts to understand in depth the structural biology of Ras and identify novel Ras inhibitors.62,63 For example, recent data have indicated that the disordered hypervariable region (HVR) of Ras prefers to interact with the catalytic domain of the GDP-bound rather than GTP-bound states and that the orientations of the catalytic domain of Ras with respect to the membrane surface are influenced by the bound nucleotide, GDP or GTP.64–67 Understanding the specific HVR–catalytic domain interactions could thus be helpful for the development of Ras inhibitors to stabilize the inactive state of Ras. Nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography pinpointed that GTP-bound Ras exists in dynamic equilibrium between the inactive and active conformations.68–72 Recent studies have identified novel allosteric sites on Ras that interact with small molecule ligands to stabilize the inactive GTP-bound state.73–75 This has spurred intense interest and effort to discover Ras-specific allosteric inhibitors using structure-based drug design. More significantly, the discovery of small molecule inhibitors covalently bound to the allosteric switch II pocket of G12C K-Ras has inspired the Ras community.76–78 These inhibitors allosterically stabilize the inactive GDP-bound G12C K-Ras and subsequently inhibit this oncogenic protein, raising the possibility of drugging G12C K-Ras. The identified high affinity small molecule inhibitor, rigosertib, dramatically, impedes the interaction of Ras proteins with their downstream effector proteins, offering an attractive approach for inactivation of Ras signaling by targeting at Ras–effector protein-protein interfaces.79 Table 1 enumerates the current status of promising Ras inhibitors in various development states. A key aspect considered in the development of Ras inhibitors is toxicity. Inhibitors that target all Ras isoforms have the potential to be toxic. This is due to the high structural similarity among the catalytic domains of the Ras isoforms. Currently, it is extremely challenging to develop a Ras-isoform-specific drug.80 However, recent studies revealed that the HVR of K-Ras4B and PI3Kα interacting with calmodulin (CaM) can achieve isoform selectivity with small molecule inhibitors, making the K-Ras4B/CaM/PI3Kα trimer an attractive potential drug target for adenocarcinoma.81–84
Table 1.
Current promising Ras inhibitors in various stages of development
| Inhibitor (company/academia) |
Structure | Mechanism | Phase |
|---|---|---|---|
| Acrylamide University of California, San Fransico |
 |
Allosteirc G12C K-Ras inhibitor (Disulfide-based tethering) |
Preclinical |
| ARS-109 University of Texas Southwestern Medical Center at Dallas |
 |
Allosteirc G12C K-Ras inhibitor (Disulfide-based tethering) |
Preclinical |
| ARS-853 Memorial Sloan Kettering Cancer Center (New York); Wellspring Biosciences (La Jolla, California) |
 |
Allosteirc G12C K-Ras inhibitor (Disulfide-based tethering) |
Preclinical |
| SML-8-73-1 University of Texas Southwestern Medical Center at Dallas |
 |
Othosteric G12C K-Ras inhibitor (Disulfide-based tethering) |
Preclinical |
| Rigosertib Icahn School of Medicine at Mount Sinai (New York) |
 |
Inhibition of Ras–effector association |
Preclinical |
| Andrographolide University of Texas Health Science Center (Houston, Texas) |
 |
Allosteric K-Ras inhibitor; Inhibition of nucleotide exchange |
Preclinical |
|
Aminopiperidine indole Vanderbilt University |
 |
Inhibition of nucleotide exchange |
Preclinical |
| Maleimide AstraZeneca |
 |
Inhibition of nucleotide exchange |
Preclinical |
| Kobe0065 Kobe University Graduate School of Medicine (Kobe, Japan) |
 |
Stabilization of inactive Ras–GTP |
Preclinical |
| ML210 Columbia University; Kyras Therapeutics |
 |
Ras-selective lethal compound |
Preclinical; Phase I targeted in 2018 |
| Deltarasin Max Planck Institute of Molecular Physiology, Lead Discovery Center (Dortmund) |
 |
Inhibition of K-Ras localization; Small-molecule inhibitor of phosphodiesterase δ |
Preclinical |
|
Polyketides–FK506-binding proteins complex Warp Drive Bio; Sanofi |
Inhibition of Ras–effector association |
Preclinical; Phase I targeted in 2018 |
|
| Tipifarnib Janssen Pharmaceutica BV; Eiger BioPharmaceuticals Inc; Kura Oncology Inc |
 |
Inhibition of Ras farnesylation |
Phase II |
| GGTI-2418 University of Pittsburgh; Prescient Therapeutics Ltd |
 |
Inhibition of Ras farnesylation |
Phase I |
| PRLX-93936 Whitehead Institute for Biomedical Research; Rines Therapeutics Ltd |
 |
Inhibition of Ras GTPase | Phase I |
| Monoterpene perillyl alcohol Hospital Universitario Antonio Pedro; NeOnc Technologies Inc |
 |
Inhibition of Ras GTPase | Phase II |
In this review, we focus on recent advances in the regulation of Ras function from a structural perspective. We first introduce the associations of the HVR with the catalytic domain of Ras in solution and the orientations of the catalytic domain with respect to the membrane surface, followed by the description of inhibition of Ras activity by targeting the active site, allosteric sites, Ras–effector protein-protein interfaces, Ras dimers, and small interfering RNA (siRNA). We also discuss CaM acting specifically in full activation of PI3Kα in KRAS4B-driven cancers, raising the possibility that a trimer can be an attractive drug target for KRAS4B-driven cancers. Finally, parallel pathways will be highlighted to increase the awareness of using drug combinations in RAS-directed therapies. We particularly focus on the Hippo/Wingless-type mouse mammary tumor virus integration site family member (Wnt) pathways and the proteins that they regulate, corresponding Yes-associated protein 1 (YAP1) and β-catenin, and their independence and correspondence with major Ras pathways, MAPK and PI3K/Akt. Insights into Ras structural biology are critical to the discovery and development of Ras inhibitors using protein-structure-based design approaches.
2 Nucleotides and oncogenic mutations affect the conformational dynamics of the HVR and the orientations of the catalytic domain
Ras proteins are composed of the catalytic domain (residues 1–166) and the C-terminal membrane-associated HVR (residues 167–189 for H-Ras, K-Ras4A, and N-Ras; 167–188 for K-Ras4B) (Fig. 1); the latter is responsible for trafficking, subcellular localization, and lateral nanoclusters segregation.85–88 All share approximately 90% sequence identity in their catalytic domain but exhibit appreciable differences in their HVR. The sequence homology between the HVR of Ras isoforms is less than 20%. In addition to distinct HVR sequences, another distinguishing feature is the differences in their HVR post-translational modification (PTM) states.4,20 The HVR of K-Ras4B is highly positively charged (multiple lysine residues). This, coupled with the farnesylation at Cys185, recruits K-Ras4B to the inner leaflet of acidic plasma membrane (PM). In contrast, because of the scarcity of polybasic residues, H-Ras, K-Ras4A, and N-Ras require additional palmitoylation in the HVR (Cys181 and Cys184 for H-Ras; Cys180 for K-Ras4A; Cys181 for N-Ras) to enhance the lipophilicity of the HVR and subsequently allow the HVR to effectively engage with the PM. K-Ras4A may exist in two states: palmitoylated and farnesylated and only farnesylated, even though the non-palmitoylated state may not interact as stably as K-Ras4B due to the lesser basic charge of its HVR.89 The diversity of HVR sequences and PTM states is indicative of isoform-specific differences in network signaling and oncogenic potential.90
Fig. 1.

Multiple sequence alignment of the amino acids in the H-Ras, N-Ras, K-Ras4A, and K-Ras4B. In the sequence, hydrophobic, polar/glycine, positively charged, and negatively charged residues are colored black, green, blue, and red, respectively. The non-identity of residues in the alignment is indicated by red circles. In the hypervariable region (HVR) sequences, the purple boxes denote the palmiltoylated cysteines and orange boxes indicate the farnesylated cysteines. A distinguishing feature of the HVR of K-Ras4B is bearing a polybasic stretch that is highlighted by a red box. Modified with permission from ref 193. Copyright 2016 Informa Healthcare.
The HVR plays a fundamental role in Ras signaling, but structures of full-length Ras are currently unavailable. In fact, Ras structures are solved exclusively with a truncated catalytic domain. When full-length Ras was attempted by X-ray crystallography, no HVR electron density was obtained, which portends the high flexibility of the HVR disordered state.91–93 However, NMR chemical shift perturbations (CSPs) in concert with molecular modeling simulations can provide useful insights into the associations of the HVR with the catalytic domain of Ras in solution and at the membrane.
2.1 In solution
To validate the engagement of the HVR with the catalytic domain, NMR experiments of full-length K-Ras4B1–188 and of the truncated domain K-Ras4B1–166 in their GDP- and GTP-γ-S-bound states were carried out in solution and the CSPs induced by the HVR were compared.65 In the GDP-bound state, remarkable CSPs for backbone amides occur for residues in the effector-binding region, switch I region and strand β2. This indicates that the HVR associates extensively with the catalytic domain. In contrast, in the GTP-γ-S-bound state, only a small fraction of changes occurs in the effector-binding region. This suggests the disengagement of the HVR from the catalytic domain. Calcium modulator protein CaM binds specifically to K-Ras4B predominantly by virtue of the HVR.94 To test the hypothesis that the conformational state of the HVR depends on Ras’ nucleotide-bound state (GDP or GTP), NMR and isothermal titration calorimetry (ITC) experiments were performed with full-length K-Ras4B–GDP/–GTP-γ-S and CaM. The results showed that no significant binding was observed between K-Ras4B–GDP and CaM, whereas K-Ras4B–GTP-γ-S can interact with CaM with micromolar affinity.95 Together, these data suggest that the HVR is sequestered by the catalytic domain of K-Ras4B in the GDP-bound state and released from the catalytic domain in the GTP-bound state.
In order to obtain structural details of the HVR interaction with the catalytic domain, based on the NMR CSPs data we constructed four initial structures of each full-length K-Ras4B–GDP/–GTP with a covalently connected HVR to H166 through interactive molecular dynamics (MD) simulations.65 Analysis of the interaction energy between the HVR and the catalytic domain of each structure led us to select two conformations of each K-Ras4B–GDP/–GTP. In both K-Ras4B–GTP models, the HVR interacts with the switch I region of the catalytic domain (Fig. 2A and Fig. 2B). However, in addition to the interaction with the switch I region, the HVR can interact with the switch II region of the catalytic domain in both K-Ras4B–GDP models (Fig. 2C and Fig. 2D). Compared to the K-Ras4B–GTP models, the additional HVR contacts with the switch II region in K-Ras4B–GDP are feasible owing to conformational rearrangements occurring in the GDP-bound state. Analysis of the interaction energy between the HVR and the catalytic domain based on each MD trajectory of K-Ras4B–GDP/–GTP showed that the tight interaction of the HVR with the catalytic domain renders the K-Ras4B–GDP in its autoinhibited state, whereas the loose association of the HVR with the catalytic domain in the K-Ras4B–GTP may facilitate the release of the HVR from the catalytic domain.64 Consistently, an assessment of binding affinity of a K-Ras4B HVR synthetic analog (Ac-KEKLNSKDGKKKKKKSKTK-NH2) with the K-Ras4B catalytic domain demonstrated that the interaction of the HVR with the catalytic domain in the GDP-bound state (Kd, 250.0 ± 33.4 nM) is much stronger (by ~75-fold) than that of the HVR with the catalytic domain in the GTP-bound state (Kd, 18.6 ± 0.9 µM).65
Fig. 2.

Cartoon and surface representations of the models 1 (A) and 2 (B) of full-length GTP-bound K-Ras4B and the models 1 (C) and 2 (D) of full-length GDP-bound K-Ras4B. The catalytic domain, HVR, switch I and switch II regions are colored in gray, cyan, pink, and blue, respectively. GTP/GDP and Mg2+ are depicted by stick and sphere models, respectively. Modified with permission from ref 129. Copyright 2016 American Chemical Society.
To further figure out the effect of frequent oncogenic mutations (G12C, G12D, G12V, G13D, and Q61H) and infrequent mutations (E37K and R164Q) on the conformational dynamics of HVR, MD simulations and NMR experiments were carried out.64 The results revealed that specific oncogenic mutations (G12D, G12V, and E37K) weaken the interaction of the HVR with the catalytic domain in both GDP- and GTP-bound states, which may contribute to decouple the HVR from the catalytic domain in a nucleotide-independent manner.64 Indeed, P-loop mutations (G12D and G12V) and switch I mutation E37K are away from and directly at the switch I/effector-binding region, respectively. In this context, G12D/G12V and E37K mutations resulted in the dissociation of the HVR from the catalytic domain by allosteric and direct interactions, respectively, which were supported by the NMR CSPs of the most oncogenic G12D mutation, and the E37K mutation.64
Over the past few years, there has been a flurry of drug discovery activity to develop small molecule inhibitors or peptide mimetics that are capable of inhibiting Ras activity.39,96–98 However, apart from thiol-reactive agents that covalently modify the mutant cysteine of G12C K-Ras,76,99–101 the remaining candidates cannot be differentiated between the target oncogenic mutant Ras and the wild-type (WT) Ras, leading to the inhibition of both oncogenic and WT Ras. This effect has the potential to cause significant toxic side effects. However, on the plus side, compared to the WT K-Ras4B, oncogenic mutations disengage the contacts between the catalytic domain and the HVR, shifting the equilibrium toward exposed effector-binding region, thereby dysregulating oncogenic Ras signaling. Thus, the different preferred conformational states of the HVR between the WT and oncogenic mutant Ras can be exploited to design inhibitors or antibodies that preferentially bind to the effector-binding region of the mutant, leaving the WT Ras in normal cells largely unaffected. In fact, Tanaka et al.102 previously designed a single domain antibody fragment, iDab#6, which can interact with oncogenic H-Ras–GTP with mutations at either Gly12 or Gln61. More importantly, the binding affinity of iDab#6 to the oncogenic mutant H-Ras–GTP is two orders of magnitude stronger than to the WT H-Ras–GTP. One possible explanation for this appreciable difference in the binding affinities of iDab#6 to the oncogenic mutant and WT Ras is attributed to the exposure of the effector-binding region in the oncogenic mutants, which may facilitate iDab#6 binding to the catalytic domain of the mutants. The determination of the co-crystal structure between the catalytic domain of G12V H-Ras–GTP and the anti-Ras intrabody in an Fv format consisting of the variable heavy (VH) chain and the variable light (VL) chain domains (PDB ID: 2UZI) revealed that the VH chain and VL chain domains of anti-Ras intrabody bind mainly to the switch I and switch II regions of H-Ras, respectively, overlapping with the Ras effector-binding region (Fig. 3).102 Thus, the anti-Ras intrabody provides a promising new paradigm to directly interfere with the oncogenic Ras function in human cancers.
Fig. 3.

Cartoon (A) and surface (B) representations of the crystal structure of the H-RasG12V–GTP–anti-Ras single domain complex (PDB ID 2UZI). H-RasG12V–GTP is shown in gray, the switch I region in pink, and the switch II region in blue. The variable heavy (VH) chain and the variable light (VL) chain domains of anti-Ras intrabody are shown in limon and lime, respectively. GTP is depicted by stick models. The effector-binding region is shown by a dashed circle on the H-Ras protein.
Due to the conspicuous differences of the HVR sequences, the character of the catalytic domain–HVR interactions in Ras isoforms is distinct. Based on NMR spectroscopy, Thapar et al.93 elucidated the behavior of the HVR of H-Ras observing that it transiently interacts with the catalytic domain, which is clearly suggestive of the weaker catalytic domain–HVR interactions in H-Ras compared to K-Ras4B. Most remarkably, recent MD simulations of full-length K-Ras4A showed that unlike the extended HVR of K-Ras4B, the HVR of K-Ras4A is collapsed in both GDP- and GTP-bound states and unable to cover the effector-binding region, suggesting the instability of K-Ras4A autoinhibited states.103 However, the potential catalytic domain–HVR interactions in N-Ras are unclear. Taken together, these results on the catalytic domain–HVR interactions of K-Ras4B, K-Ras4A, and H-Ras unveil dynamical differences, which may affect Ras activation and interactions with downstream effectors.104
2.2 At the membrane
Association with the PM is a requirement for Ras proteins to exert their biological functions.105 Insights into how the orientations of Ras with respect to the membrane surface and the interactions between Ras and the PM are thus critical to understand signaling specificity among Ras isoforms and for the design of selective anticancer drugs.106–111
Recently, Mazhab-Jarari et al.66 deployed solution NMR experiments to uncover the dynamic interactions between full-length K-Ras4B and nanodisc lipid bilayer composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), and the thiol-reactive lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidomethyl)cyclohexane-carboxamide] (PE-MCC) (molar ratio 15:4:1), as well as the associations of K-Ras4B with the Ras-binding domains (RBDs) of effector proteins at the surface of the lipid bilayer. Nanodisc provides a stable lipid environment to constitute membrane proteins for structural and functional studies, which is widely employed in solution NMR and SPR experiments.112,113 The results of the paramagnetic relaxation enhancement (PRE) measurements of K-Ras4B tethered to Gd3+-containing nanodiscs suggested that K-Ras4B exists in two major orientations at the membrane surface. Based on PRE distance restraints from the lipid bilayer to the GDP- and GppNHp-bound K-Ras4B, two distinct clusters of membrane-associated complexes of each GDP-/GppNHp-bound K-Ras4B were modeled using the high ambiguity driven biomolecular docking (HADDOCK) simulations. In the GDP-bound state, the major population (64%) of K-Ras4B orients their helices parallel to the lipid bilayer and the K-Ras4B–membrane interface is derived mainly from the N-terminus of β1 (N-β1), α4, β6, α5, and the loop connecting from β2 to β3. This orientation of K-Ras4B in relation to the bilayer plane is called the α-interface, which exposes the effector-binding region (switch I and β2) that can interact with downstream effector proteins. In sharp contrast, in the GppNHp-bound state, the preferred population (45%) of K-Ras4B orients their helices semi-perpendicular to the bilayer plane and the K-Ras4B–membrane interface is derived mainly from the C-terminus of switch I (C-switch I), β1-β3, α2, and the C-terminal of α3. This cluster is called the β-interface. It buries the effector-binding region at the membrane surface that occludes the binding of downstream effector proteins to K-Ras4B. However, in addition to the β-interface, another cluster of α-interface representing the 32% of population was also observed in the GppNHp-bound K-Ras4B. These data suggest orientational equilibrium of K-Ras4B at the membrane and that although K-Ras4B is in the GTP-bound form, it is unable to initiate signaling when it adopts the β-interface at the membrane surface because the membrane surface sequesters the K-Ras4B binding site for effector proteins.66 Importantly, recently we have shown that the preferred interfaces are isoform-specific.114
To further shed light on the effect of RBD of the Raf isoform A-Raf on the orientation of GppNHp-bound K-Ras4B at the membrane surface, NMR measurements were carried out to obtain PRE distance restraints from the lipid bilayer to both K-Ras4B and A-Raf-RBD and the resulting HADDOCK models of the ternary complex among the lipid bilayer, K-Ras4B, and A-Raf-RBD were obtained based on PRE-derived restraints. In this complex, the predominant orientation (87%) of the catalytic domain of K-Ras4B was in a semiexposed orientation intermediate between the exposed (α-interface) and occluded (β-interface) orientations, with a small fraction of exposed orientations (12%). Interestingly, replacement of A-Raf-RBD with RBD of RalGDS (RalGDS-RBD) shifts the population from the occluded toward a completely exposed orientation of the catalytic domain at the membrane surface. Cumulatively, these results suggest that the orientations of the catalytic domain of K-Ras4B in the K-Ras4B–effector complexes at the membrane surface depend on the features of the effector proteins. Furthermore, the impact of the oncogenic G12D mutation and two Noonan syndrome mutations (K5N and D153V) on the orientations of K-Ras4B were explored. Comparison of PRE profiles between WT and mutants showed that these mutations shift the population of the catalytic domain of mutant K-Ras4B from the occluded β-interface to the exposed α-interface, therefore derepressing the membrane-dependent autoinhibition of K-Ras4B thereby potentiating signaling. Together, these data provide an instrumental tactic in taming Ras isoform-specific inhibition for therapeutic intervention by sandwiching the autoinhibitory associations of the HVR between oncogenic catalytic K-Ras domain and the membrane.
To further investigate the influence of the membrane on the conformational dynamics of the HVR, we simulated the farnesylated K-Ras4B on the surface of an anionic lipid bilayer consisting of DOPC:DOPS (mole ratio 4:1).67 Based on the NMR CSP data of K-Ras4B binding to nanodiscs,65 four different initial configurations of each GDP- and GTP-bound WT K-Ras4B with the farnesylation on Cys185 were placed on the membrane surface to represent the diversity of K-Ras4B–membrane interactions. The simulated results revealed significant differences among the HVR–catalytic domain interactions on the membrane. In the GDP-bound state (Fig. 4A), the HVR interacts with the catalytic domain in three configurations, retaining the autoinhibited state (configurations 1, 3, and 4), whereas it is released from the catalytic domain in configuration 2 and subsequently inserts its farnesyl group into the membrane. In contrast, in the GTP-bound state (Fig. 4B), the HVR decouples with the catalytic domain in three configurations (configurations 1, 3, and 4), but is still autoinhibited in configuration 2 with the HVR covering the effector-binding region. Despite the release of the HVR from the catalytic domain in configuration 2 of GDP-bound K-Ras4B (Fig. 4A), the effector-binding region orients to and interacts with the lipids, thereby preventing it from recruiting downstream effector proteins. In all configurations, GDP-bound K-Ras4B represents an inactive form with the allosteric lobes oriented parallel (~180°) to the membrane surface. However, in configurations 1, 3, and 4 of the GTP-bound K-Ras4B (Fig. 4B), the disassociation of the HVR from the catalytic domain exposes the effector-binding region, leading to perpendicular (~90°) alignments of the allosteric lobes relative to the membrane surface. In these orientations, K-Ras4B represents an active form and can interact with downstream effector proteins. However, in configuration 2 of the GTP-bound K-Ras4B, the allosteric lobe orients in parallel (~180°) to the membrane surface, yielding a GDP-like behavior. This orientation represents an inactive form on the membrane. Collectively, these data suggest that GTP binding may not propel K-Ras4B into an active state; instead, we propose that a complex process, including GDP/GTP exchange, HVR sequestration, farnesyl insertion, and orientation of the catalytic domain at the membrane, determines the active or inactive state of K-Ras4B.67 It is noted that in vitro systems involving nanodisc and computational lipid bilayers are synthetic model membranes that are reconstituted with only few lipid compositions. In contrast, in vivo plasma membranes consist of a number of different entities, where organizations of different types of lipids at the microdomains result in different physical properties.105 Although the experimental and computational model membranes were able to substitute the role of the plasma membrane in supporting the conformations and orientations of the catalytic domain of Ras, they were limited in presenting lateral lipid segregation through the dynamic formations of microdomains with different phases. Membrane-associating Ras proteins utilize these microdomains as signaling platforms for their functional regulations.
Fig. 4.
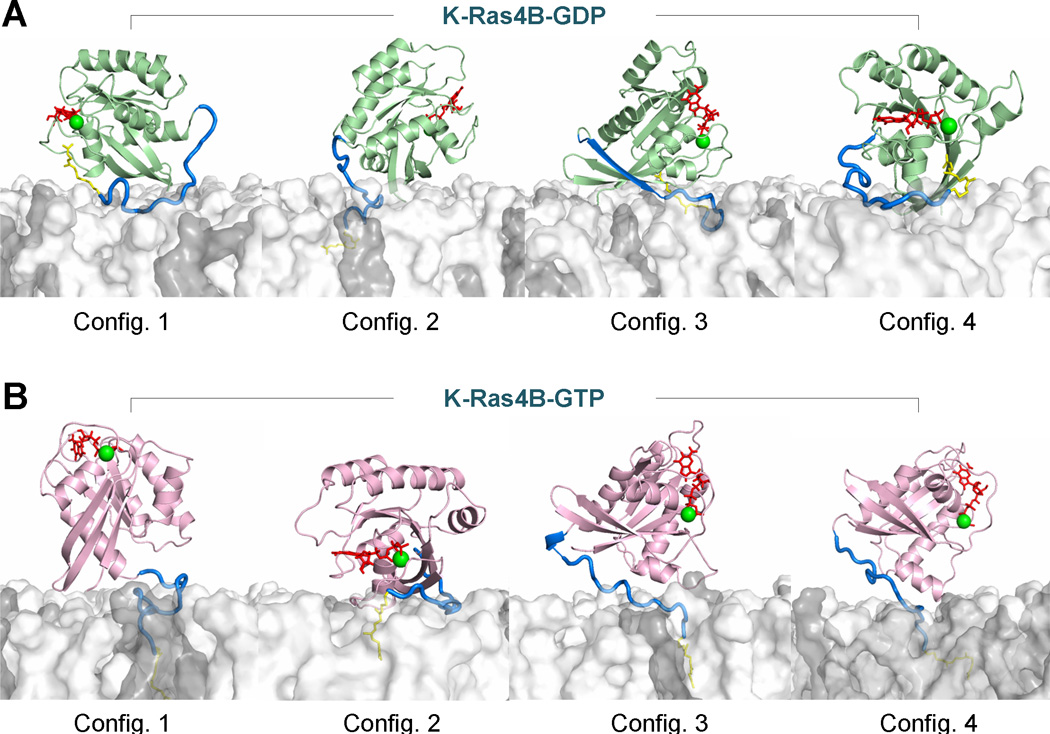
Snapshots representing the K-Ras4B–membrane interactions with the anionic lipid bilayer composed of DOPC:DOPS (mole ratio 4:1) for the (A) GDP-bound and (B) GTP-bound states. Cartoons for the catalytic domains are shown in green and pink for the GDP-bound and GTP-bound states, respectively. The HVR in the tube representation is colored in blue, and the farnesyl as a stick is colored yellow. In the catalytic domain, the red sticks and green spheres represent the nucleotide and Mg2+ ions, respectively. For the lipid bilayer, white surface denotes DOPC and gray surface represent DOPS. In the GDP-bound state, the HVR autoinhibition can be observed in configurations 1, 3, and 4, while the HVR is released from the effector lobe in configuration 2. In the GTP-bound state, the HVR still binds the effector lobe in configuration 2, retaining the autoinhibition state, while other configurations release the HVR from the catalytic domain. Configurations 3 and 4 of K-Ras4B-GDP are the most representative of the inactive K-Ras4B. Configurations 1 and 2 of K-Ras4B-GDP are also in the inactive state due to the inaccessibility of the Raf-binding effector region. Configurations 1, 3 and 4 of K-Ras4B-GTP are the most representative of the active K-Ras4B, while configuration 2 of K-Ras4B-GTP behaves like the inactive GDP-bound state, showing inaccessible Raf-binding effector region. Modified with permission from ref 67. Copyright 2015 The American Society for Biochemistry and Molecular Biology.
Recently, Prakash et al.115 performed large-scale MD simulations of full-length GTP-bound G12D K-Ras4B embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/palmitoyl-oleoyl phosphatidylserine (POPC/POPS, mole ratio 4:1) bilayer to probe how oncogenic mutations affect the orientations of the catalytic domain relative to the membrane surface at the atomic level. Two major orientations of the catalytic domain K-Ras4BG12D–GTP relative to the membrane surface were observed.115 In one orientation (OS1), helices α3 and α4 of the catalytic domain are involved in the interactions with the membrane lipids, exposing the effector-binding region. The OS1 orientation of K-Ras4BG12D–GTP at the membrane can interact with downstream effector proteins such as Raf kinase. In an alternative orientation (OS2), strands β1–β3 and helix α2 of the catalytic domain contribute to contact with the membrane lipids, burying the effector-binding region. The OS2 orientation of K-Ras4BG12D–GTP at the membrane is thus incapable of interacting with downstream effector proteins, but it has the potential to form Ras–Ras dimerization via the proposed helical interfaces.116–119 However, how the oncogenic mutations affect the orientations of the catalytic domain of GDP-bound K-Ras4B relative to the membrane surface at the atomic level remains unclear.
MD simulations of full-length GDP- and GTP-bound H-RasG12V embedded in a 1,2-dimyristoylglycero-3-phosphocholine (DMPC) bilayer also demonstrated that the orientations of the catalytic domain relative to the membrane surface depend on the nucleotide types.120 However, the orientations of H-RasG12V are significantly different from K-Ras4B. In the GDP-bound state, the catalytic domain of H-RasG12V is in an orientation perpendicular to the membrane surface, with strands β2 and β3 interacting with lipids.120 In contrast, in the GTP-bound state, the catalytic domain of H-RasG12V is oriented in parallel to the membrane surface, with helices α4 and α5 directly interacting with lipids.120 Thus, the different paradigms of the interactions between the Ras isoforms89 and the membrane may provide a potential venue for a structure-based design of selective anticancer drugs.86,110
3 Direct inhibition of Ras by targeting its active site
The high intracellular concentrations of GTP and GDP and their picomolar affinity for Ras render the design of competitive inhibitors targeting the active site on Ras daunting. However, a naturally occurring oncogenic mutation on Ras, G12C, introduces a solvent-accessible cysteine on the P-loop adjacent to the active site (Fig. 5A), which can serve as a reactive protein side chain to form covalent inhibitors with small molecules that bear reactive functional groups. This strategy can be used to distinguish the G12C mutant from WT Ras and other mutants.101
Fig. 5.

(A) Cartoon representation of the crystal structure of GDP-bound G12C K-Ras (PDB ID 4LDJ). The P-loop, switch I, and switch II regions are colored lime, pink, and blue, respectively. GDP and Cys12 are depicted by stick models and Mg2+ by a green sphere. (B) Chemical structure of a GDP analog, SML-8-73-1 (SML, 1). (C) Cartoon representation of the crystal structure of SML-bound G12C K-Ras (PDB ID 4NMM). (D) Backbone superimposition of the crystal structures of GDP-bound WT (PDB ID 4OBE, cyan) and G12C K-Ras (pink) onto that of SML-bound G12C Ras (orange).
Based on the homology model of G12C K-Ras, Lim et al.100 designed a GDP analogue, SML-8-73-1 (SML, 1) (Fig. 5B), which has a high likelihood of forming a covalent inhibitor with Cys12. Subsequent mass spectroscopy (MS) experiments confirmed that SML was able to covalently modify G12C K-Ras, but did not label WT K-Ras. To further illuminate the binding mode between SML and G12C K-Ras, Hunter et al.99 solved the co-crystal structure of G12C K-Ras bound to SML (PDB ID 4NMM, Fig. 5C) as well as the structures of WT (PDB ID 4OBE) and G12C (PDB ID 4LDJ) GDP-bound K-Ras. Backbone superimposition of the crystal structures of K-RasG12C–GDP and K-RasWT–GDP onto that of K-RasG12C–SML exhibits that the overall conformation of K-Ras is extremely similar among three structures and that the orientation of SML in the active site of K-RasG12C is also nearly identical to those of GDP in the K-RasG12C and K-RasWT (Fig. 5D), ruling out the possibility that covalent modification of Cys12 by SML results in extensive conformational remodeling of the switch I and switch II regions. Thus, the resulting conformation of K-RasG12C is in the open, inactive conformation upon SML binding, which is incapable of interacting with downstream effector proteins. To test this hypothesis, the structure of K-RasG12C–SML was superimposed onto those of H-Ras–GppNHp in complex with the RBD of Raf kinase (Raf-RBD) (PDB ID 4G0N)121 and PI3Kγ (PDB ID 1HE8),122 respectively. Remarkably, compared to the formation of exquisite salt bridge networks between the switch I region of H-Ras–GppNHp and Raf-RBD and PI3Kγ, no salt bridges were formed between the switch I region of K-RasG12C–SML and Raf-RBD (Fig. 6A) and PI3Kγ (Fig. 6B). Moreover, a steric conflict between the switch I region of K-RasG12C–SML and the β-strands of Raf-RBD and PI3Kγ was observed. Functional biochemical assay through the AlphaScreen technology was performed to assess the affinity between K-RasG12C–GppNHp/–GDP and Raf-RBD in the presence or absence of SML.100 The results showed that K-RasG12C–GppNHp strongly interacts with Raf-RBD with an EC50 of 6.6 nM, which has a higher affinity than the interaction between K-RasG12C–GDP and RafRBD with an EC50 of 22.6 nM. The AlphaScreen assay further revealed that the affinity of K-RasG12C–SML for RafRBD is nearly identical to that of K-RasG12C–GDP for Raf-RBD. These data suggest that SML binding to the active site of Ras is likely to prevent the activation of Ras downstream signaling.
Fig. 6.

(A) Backbone superimposition of the crystal structure of SML-bound G12C K-Ras (PDB ID 4NMM, lime) onto that of GppNHp-bound H-Ras (pink) in complex with RafRBD (light blue) (PDB ID 4G0N). The two enlarged figures show the different interactions between the switch I residues of K-RasG12C–SML, H-RasWT–GppNHp and RafRBD. In the H-Ras–GppNHp–RafRBD, residues Glu31 and Asp33 of H-Ras form electrostatic interactions with residue Lys84 of RafRBD, and residues Glu37 and Asp38 of H-Ras form electrostatic interactions with residues Arg59, Arg67, and Arg89 of RafRBD. (B) Backbone superimposition of the crystal structure of SML-bound G12C K-Ras (lime) onto that of GppNHp-bound H-Ras (pink) in complex with PI3Kγ (light blue) (PDB ID 1HEB). In the H-Ras–GppNHp–PI3Kγ, residues Asp33, Glu37, and Asp38 of H-Ras form electrostatic interactions with residues Lys251, Lys255, Gln231, and Lys223 of PI3Kγ.
4 Allosteric inhibition of Ras by targeting its allosteric sites
Because of the extremely high affinity of Ras proteins for their GDP and GTP substrate, it is challenging to develop competitive substrate inhibitors to abrogate Ras function. An alternative approach is the design of small molecules that bind to allosteric sites of Ras in order to disrupt Ras nucleotide exchange or stabilize the inactive GTP-bound state of Ras, thereby resulting in growth inhibition and cancer cell apoptosis. Allostery, an intrinsic property of a protein, regulates protein function through the binding of an effector to an allosteric site topographically distinct from the active site.123–127 Thus, allosteric modulators do not compete with orthosteric ligands that occupy the active site. Rather, they typically exert theirs effects in concert with the orthosteric ligands in the active site.128 Ras is an allosteric protein and several allosteric sites on Ras have been validated by X-ray or NMR spectroscopy experiments.129–139 Targeting allosteric sites, as a novel tactic, is gaining increasing recognition in taming Ras inhibition for therapeutic intervention. Here, we recapitulate the identification of allosteric sites on Ras proteins with the exception of Ras–effector or Ras–GEF protein-protein interfaces.
4.1 Helix α3, helix α4, and loop 7
Buhrman et al.140 determined the crystal structure of GppNHp-bound H-RasWT grown in 200 mM calcium chloride (CaCl2) (PDB ID 2RGE). Inspection of this structure revealed that the switch II region is disordered with no electron density for the catalytic residue Q61. Replacing CaCl2 by calcium acetate [Ca(OAc)2], the authors solved another crystal structure of GppNHp-bound H-RasWT (PDB ID 3K8Y).141 Significantly, in the presence of 200 mM Ca(OAc)2, the electron density of the switch II region is clearly visible. In three-dimensional space, Ca(OAc)2 binds between helix α3, helix α4, and loop 7 at the allosteric lobe of Ras (Fig. 7A), which is distant from the switch II region with no direct interaction. This suggests that the behavior of the conformational ordering in the switch II region is through an allosteric switch in response to Ca(OAc)2 binding. To elucidate the effect of Ca(OAc)2 binding on the ordering of the switch II region, the backbone of the crystal structure of H-RasWT–GppNHp–Ca(OAc)2 was superposed onto that of H-RasWT–GppNHp–CaCl2. Despite the overall structural similarity between the two crystals, some regions in the structure of H-RasWT–GppNHp–Ca(OAc)2 exhibit appreciable differences from those in the structure of H-RasWT–GppNHp–CaCl2. These regions encompass loop 7, helix α3, and the C-terminal switch II region (Fig. 7B). Owing to the ordered switch II region in the presence of Ca(OAc)2, the catalytic residue Gln61 in the switch II region can interact with the bridging water molecule (W189), which in turn interacts with the side chain OH group of Tyr32 and the γ-phosphate. Moreover, the catalytic water (W175) in the active site is proximal to the γ-phosphorus and hydrogen-bonded to the backbone NH group of Gln61 and the side chain OH group of Thr35 (Fig. 7C). The different architectures between the active sites of H-RasWT–GppNHp–Ca(OAc)2 and H-RasWT–GppNHp–CaCl2 underpin Ca(OAc)2’s binding at the allosteric site triggering a disorder to order transition in the switch II region with alignment of Gln61 for catalysis, while in the presence of CaCl2 the switch II region is still in a disordered conformation.142,143 On the basis of these observations, the structure of H-RasWT–GppNHp–Ca(OAc)2 represents a catalytically competent state (“on” conformation). In contrast, the structure of H-RasWT–GppNHp–CaCl2 represents a catalytically incompetent state (“off” conformation). Recently, Holzapfel et al.144 further demonstrated that the dynamic conformational ensembles between the on and off states of H-Ras–GTP in solution can be modulated by small molecules, dithioerythritol and dithiothreitol, and bulk solvent composition. Although the Ca(OAc)2 binding site at the allosteric lobe of H-Ras represents a potential allosteric site, no small molecules have currently been identified to bind to this site. One possible explanation is that the small volume of Ca(OAc)2 binding site makes it difficult to develop small molecule inhibitors to this allosteric site (Fig. 7D).
Fig. 7.

(A) Cartoon representation of H-RasWT–GppNHp showing the binding of calcium acetate [Ca(OAc)2] in the allosteric site lined by helix α3, helix α4, and loop 7 (PDB ID 3K8Y). (B) Backbone superimposition of the crystal structure of H-RasWT–GppNHp–Ca(OAc)2 (pink) onto that of H-RasWT–GppNHp–CaCl2 (PDB ID 2RGE, light blue). (C) The active site of H-RasWT–GppNHp–Ca(OAc)2 showing Tyr32, Gln61, and the catalytic (W175) and bridging (W189) water molecules near the γ-phosphate. (D) Surface representation of H-RasWT–GppNHp–Ca(OAc)2 showing the Ca(OAc)2 binding site on the allosteric lobe of H-Ras. Ca(OAc)2 is depicted by spheres.
4.2 Helix α5, loop 7, and loop 9
The conformational transitions between the inactive GDP-bound and the active GTP-bound Ras are of fundamental importance to the understanding of the mechanisms underlying Ras signaling transduction.145–147 These nucleotide-dependent conformational transitions have the potential to expose hidden pockets or allosteric sites,148–150 which provide a valuable new venue for the development of small molecule allosteric Ras inhibitors. Based on this notion, Grant et al.151 collected a comprehensive ensemble of Ras to account for conformational heterogeneity. This ensemble includes Ras crystallographic structures and additional conformers derived from MD simulations. Three different approaches for the identification of potential binding sites, including fragment, grid, and ligand based binding-site mapping methods, were subsequently performed on the ensemble of Ras structures. These identified four non-nucleotide binding pockets from the available Ras structures and MD conformers. Remarkably, one of the four pockets locates in the allosteric lobe of Ras encased by the C-terminal helix α5, loop 7, and loop 9 (Fig. 8A and Fig. 8B), which is the most distal to the active site with a distance of approximately 25 Å. This pocket is conserved in all ensemble conformers. Virtual screening against this novel binding site using compounds from the NCIDS II and Zinc drugs-now subset was carried out and 19 ligands were chosen as promising leads after Lipinski-filtering of the initial hits, which were further evaluated by experimental testing. Cell-based extracellular signal regulated kinase 1/2 (ERK1/2) phosphorylation assays showed that candidate inhibitors are able to inhibit the downstream signaling activity of Ras. However, this computationally identified allosteric site on the Ras C-terminal is needed to be validated by X-ray crystallography to make this site a viable target serving as a starting point for the development of small molecule allosteric Ras inhibitors.
Fig. 8.

(A) Cartoon and (B) surface representations of GppNHp-bound H-Ras (PDB ID 5P21) showing the allosteric site on the allosteric lobe of H-Ras formed by the C-terminal helix α5 (red), loop 7 (pink), and loop 9 (blue).
31P NMR and X-ray crystallographic data from Shima et al.,69–71 together with MD simulations from our group,152 showed that GppNHp-bound H-Ras in solution contains two interconverting conformations, “inactive” state 1 and “active” state 2; the former has a weak binding affinity for effector proteins, while the latter has a strong binding affinity for effector proteins.68,153 31P NMR spectroscopy revealed that the conformational ensemble of GppNHp-bound H-Ras in solution shifts towards the weak binding state 1 upon binding of Zn2+–1,4,7,10-tetraazacyclodecane (Zn2+–cyclen, 2) (Fig. 9A) or Cu2+–cyclen (3) (Fig. 9B) to H-Ras.74,154 Comparisons of NMR CSPs of H-Ras in the presence and absence of Zn2+/Cu2+–cyclen suggested the existence of two distinct binding sites for Zn2+/Cu2+–cyclen on the surface of H-Ras.74 The first binding site (binding site 1) is adjacent to the γ-phosphate of the GppNHp and the major differences of the CSPs for backbone amines occur mostly for residues in the P-loop (Gly13), switch I (Tyr32) and switch II (Ala59, Gly60, and Gln61) regions. The second binding site (binding site 2) is formed by residues from the negatively charged loop 7 (Asp105, Ser106, Asp107, Asp108, Val109, and Met111) and the C-terminal helix α5 (Glu162, Gln165, and His166). The determination of the crystal structure of H-Ras–GppNHp in complex with Zn2+–cyclen (PDB ID 3L8Y) showed that Zn2+–cyclen can be detected only at binding site 2 (Fig. 9C), which is located in proximity to loop 7 and the C-terminal helix α5. To confirm the stabilization of the weak binding state 1 of the active GTP-bound H-Ras by the transition metal-cyclen complexes, the effect of Zn2+– or Cu2+–cyclen on the binding affinity between GppNHp-bound H-Ras and Raf-RBD was assessed by using ITC. The measurements showed that the binding affinity between GppNHp-bound H-Ras and Raf-RBD decreased significantly in the presence of Zn2+– or Cu2+–cyclen.74,155 This result suggested that Zn2+– or Cu2+–cyclen binds especially to the conformational state 1 of active GTP-bound Ras and thereby results in population shift of the conformational ensemble of GTP-bound Ras towards the weak binding state 1. In fact, aside from transition metal-cyclen complexes, 31P NMR showed that Zn2+–bis(2-picolyl)amine (Zn2+–BPA, 4) (Fig. 9D) can also bind to the weak binding state 1 of GppNHp-bound H-Ras and subsequently shifts the conformational ensemble of GppNHp-bound H-Ras towards this state.75
Fig. 9.

Chemical structures of Zn2+–cyclen (2) (A) and Cu2+–cyclen (3) (B). (C) Surface representation of the crystal structure of GppNHp-bound H-Ras in complex with Zn2+–cyclen (PDB ID 3L8Y). The second binding site of Zn2+–cyclen is adjacent to loop 7 and the C-terminal helix α5. GppNHp and cyclen are depicted by sticks and Zn2+ by a green sphere. (D) Chemical structure of Zn2+–BPA (4).
4.3 Switch II pocket (S-IIP)
Based on the oncogenic G12C K-Ras4B mutant that introduces a cysteine residue in the P-loop, Ostrem et al.76 used protein MS to screen a library of 480 disulfide-containing small molecule compounds against G12C K-Ras4B–GDP. Two fragments, 6H05 (5) and 2E07 (6) (Fig. 10), were identified to significantly modify G12C K-Ras4B–GDP, leaving WT K-Ras4B–GDP unaffected. In order to improve the binding affinity, the structure of fragment 6H05 was modified, resulting in identification of compound 7 (Fig. 10), which showed a 4.2-fold higher potency compared to unmodified fragment 6H05. To uncover the structural details of the binding between compound 7 and G12C K-Ras4B, the X-ray structure of G12C K-Ras4B–GDP in complex with compound 7 was determined (PDB ID 4LUC). Unlike the SML compound that binds to the active site of K-Ras and covalently modifies G12C K-Ras, compound 7 does not bind to the active site but extends from Cys12 into an adjacent pocket that is opposite to the active site (Fig. 11A and Fig. 11B). This pocket, named the switch II pocket (S-IIP), is formed by residues from the central strand β1, the switch II region, and helix α3. However, S-IIP is composed largely from the switch II region, particularly the helix α2. To examine the impact of the binding of compound 7 on the conformation of K-Ras4BG12C–GDP, the crystal structures of K-Ras4BG12C–GDP–7 and K-Ras4BG12C–GDP were compared. As shown in Fig. 11C, in the structure of K-Ras4BG12C–GDP (PDB ID 4L9S), the N-terminus (residues 61–67) of the switch II region is disordered. However, after compound 7 binding to S-IIP, the N-terminus of the switch II region becomes ordered and the electron density is clearly visible. This observation suggests that binding of compound 7 to K-Ras4BG12C–GDP contributes to ordering the switch II region. To further reveal the origin of S-IIP, backbone superimposition of the crystal structures of K-Ras4BG12C–GppNHp (PDB ID 4L9W) and K-Ras4BG12C–GDP onto that of K-Ras4BG12C–GDP–7 was performed. As shown in Fig. 11D, arguably the most significant feature is that the position occupied by helix α2 from the switch II region in the structure of K-Ras4BG12C–GDP–7 is between those in the structures of K-Ras4BG12C–GppNHp and K-Ras4BG12C–GDP. Moreover, the N-terminus of the switch II region in the structure K-Ras4BG12C–GppNHp has a steric conflict with compound 7, which implies that S-IIP does not exist in the structure of K-Ras4BG12C–GppNHp. In the structure of K-Ras4BG12C–GDP helix α2 is displaced outward with respect to that in the structure of K-Ras4BG12C–GDP–7. In fact, its position in the structure of K-Ras4BG12C–GDP is similar to that of K-Ras4BWT–GDP (PDB ID 4LPK) (Fig. 11E). On the basis of these observations, S-IIP may be in the conformational reaction path of Ras during the GTP-to-GDP hydrolysis. In addition to disulfide-based compounds, the authors attempted to use carbon-based electrophiles, vinyl sulphonamides (8, Fig. 10) and acrylamides (9 and 10, Fig. 10) to covalently modify the C12 of K-Ras4BG12C–GDP. Similar to the compound 7 bound to S-IIP, the determination of co-crystal structures between K-Ras4BG12C–GDP and compounds 8 (PDB ID 4LYF) and 9 (PDB ID 4M21) showed that both compounds 8 and 9 bind to the same pocket (Figs. 11E–11I). Plate-based assay was performed to determine the relative affinity of K-Ras4BG12C for GDP or GTP in the presence of S-IIP binding compounds. The results showed that binding of these inhibitors to K-Ras4BG12C shifts the relative nucleotide affinities of Ras to prefer GDP over GTP, which leads to the accumulation of K-Ras4BG12C in its inactive state.
Fig. 10.

Chemical structures of 6H05 (5), 2E07 (6), compound 7, vinyl sulphonamides (8), acrylamides (9, 10), ARS-109 (11), and ARS-853 (12). Modified with permission from ref 129. Copyright 2016 American Chemical Society.
Fig. 11.

(A) Cartoon and (B) surface representations of K-Ras4BG12C–GDP–7 (PDB ID 4LUC) showing the compound 7 bound to the switch II pocket, S-IIP, formed by the central sheet β1, (cyan), switch II region (pink), and helix α3 (red). (C) Backbone superimposition of the crystal structure of K-Ras4BG12C–GDP (PDB ID 4L9S, cyan) onto that of K-Ras4BG12C–GDP–7 (pink). The N-terminal switch II region is disordered in the structure of K-Ras4BG12C–GDP. (D) Backbone superimposition of the crystal structures of K-Ras4BG12C–GppNHp (PDB ID 4L9W, orange) and K-Ras4BG12C–GDP (cyan) onto that of K-Ras4BG12C–GDP–7 (pink). (E) Backbone superimposition of the crystal structure of K-Ras4BWT–GDP (PDB ID 4LPK, purple) onto that of K-Ras4BG12C–GDP (cyan). (F) Cartoon and (G) surface representations of K-Ras4BG12C–GDP–8 (PDB ID 4LYF) showing the compound 8 bound to S-IIP. (H) Cartoon and (I) surface representations of K-Ras4BG12C–GDP–9 (PDB ID 4LYF) showing the compound 9 bound to S-IIP.
Recently, based on the compound 10, Patricelli et al.,77 performed iterative structure-based design of covalent K-Ras4BG12C inhibitors. Linker modification of compound 10 identified ARS-109 (11, Fig. 10). Further structural modification of ARS-109 identified the most potent compound, ARS-853 (12, Fig. 10), which shows an IC50 of 1.6 µM for K-Ras4BG12C and a more than 600-fold improvement against compound 10. The determination of the co-crystal structure of K-Ras4BG12C–GDP in complex with ARS-853 (PDB ID 5F2E) confirmed that ARS-853 covalently labels Cys12 and occupies the previously characterized S-IIP (Fig. 12A and Fig. 12B). Compared to the structures of K-Ras4BG12C–GDP in complex with corresponding compounds 7–9, binding of ARS-853 to the S-IIP leads to a marked rotation of helix α2 (Figure 12C). Biochemical assays revealed that ARS-853 interacts selectively with K-Ras4BG12C–GDP, but not with K-Ras4BG12C–GTP, consistent with compounds 7–9. To more precisely unveil the mechanism of K-Ras4BG12C inhibition by ARS-853, Lito et al.78 performed thermal stability assays using differential scanning fluorimetry. The results suggest that ARS-853 preferentially binds to inactive, GDP-bound K-Ras4BG12C, in agreement with the structural and biochemical data of Patricelli and co-workers.77 The cellular effects of ARS-853 on Ras-mediated signaling in cells were further explored. Both groups showed that ARS-853 is capable of inhibiting downstream signaling of K-Ras4BG12C,77,78 including C-Raf, ERK and Akt. Surprisingly, cell-based assays by Lito et al.78 found that GTPase activity is required for ARS-853 to interact with and inhibit K-Ras4BG12C, which contradicts the previously long-held view that mutated Ras proteins persist in the active, GTP-bound form, leading to a sustained oncogenic signal.25,156,157 Collectively, these data suggest that targeting the allosteric site, S-IIP, to stabilize the inactive, GDP-bound RasG12C provides a framework for generating new anti-Ras therapeutics.158
Fig. 12.

(A) Cartoon and (B) surface representations of K-Ras4BG12C–GDP–ARS-853 (PDB ID 5F2E) showing ARS-853 bound to S-IIP. (C) Backbone superimposition of the crystal structures of K-Ras4BG12C–GDP–7 (PDB ID 4LUS, pink), K-Ras4BG12C–GDP–8 (PDB ID 4LYF, orange), and K-Ras4BG12C–GDP–9 (PDB ID 4M21, cyan) onto that of K-Ras4BG12C–GDP–ARS-853 (purple).
4.4 Helix α1, switch I, and strand β2
Large-scale MD simulations of K-Ras4BQ61H–GTP found that the conformational ensemble of K-Ras4BQ61H–GTP contained active GTP-, intermediate GTP-, inactive GDP-bound, and nucleotide-free states.159 Rational design of small molecule inhibitors that especially interact with the inactive states of GTP-bound Ras and shift the conformational ensemble towards the inactive states can inhibit Ras signaling. Based on this notion, Hocker et al.160 docked andrographolide (AGP, 13)–a bicyclic diterpenoid lactone–and its benzylidene derivatives (SRJ09 (14), SRJ10 (15), and SRJ23 (16)) (Fig. 13) to an ensemble of 75 structures. Three major pockets were observed for binding of these ligands: p1, p2, and p3. Pocket 1 (p1) is lined by the switch I region, strand β2, and several residues in helix α1; pocket 2 (p2) contains residues from the core β1 and β2, part of the effector loop, and the switch II region; pocket 3 (p3) includes helix α5 and the N-terminal and preceding loop residues of stands β5 and β6. Neither pocket overlaps the nucleotide-binding site. To test the stability of the binding of ligands to these distinct pockets, MD simulations of K-Ras4B–GTP in complex with SRJ23 were performed. The results showed that p1 is the preferred pocket for the SRJ23 (Fig. 14). Subsequently, in vitro assays demonstrated that SRJ compounds bind to Ras and prevent GTP loading. This effect blocked GDP–GTP exchange in live cells and inhibited cancer cell growth. Although the p1 represents a potential allosteric site for SRJ compounds binding, this computationally determined pocket is still required to be validated by X-ray crystallography or NMR experiments.
Fig. 13.

Chemical structures of andrographolide (AGP) (13) and its benzylidene derivatives SRJ09 (14), SRJ10 (15), and SRJ23 (16).
Fig. 14.

The location of pocket 1 (p1) of SRJ derivatives on the surface of the inactive state 1 crystal structure of H-RasT35S–GppNHp (PDB ID 3KKN).
5 Isoform-specific inhibition of Ras dimers
Ras can form dimers.116,118,161–165 Dimerization via the helical interface is critical for Raf’s dimerization and activation.116 Recent computational and experimental studies on the B-Raf kinase indicate that phosphorylation of the N-terminal acidic (NtA) motif promotes B-Raf dimerization as a result of the formation of several interprotomer salt bridges between the NtA motif of one of the protomers and the positively charged C-terminal end of the αC-helix of the other protomer. These revealed the asymmetric transactivation mechanism of Raf kinases.166,167 The Ras dimer interface appears highly dynamic.114 Recently, we have shown that as expected, Ras self-association is dynamic and can involve multiple interfaces;114 however, importantly, the interfaces are isoform selective: H-Ras dimer interface differs from the K-Ras4B dimer interface. This reflects the composition and environment of the isoforms. Altered membrane microdomain composition and organization can further drive formation of isoform-specific Ras dimers. Oncogenic mutations may also redistribute the dimers’ ensemble resulting in different interfaces preferentially occupied at the membrane. Currently, data increasingly support Ras dimerization and higher oligomerization which would and promote Raf’s activation thus contribute to increased signaling output.117,168–170 Thus, even though proximal monomeric Ras can activate Raf, its dimerization enhances downstream signaling, likely reflecting the higher degrees of freedom of the membrane-associate monomer, thus lower efficiently.
Oncogenic Ras isoforms are differentially expressed across different cancer types.89 The additional different landscapes of the dimeric states among the isoforms raise the possibility of isoform-specific drug development.114 Nonetheless, noteworthy, all dimeric states exist for the different isoforms; only their distributions vary.
6 Inhibition of Ras by targeting Ras–effector protein-protein interfaces
The diverse cellular processes initiated by Ras proteins are mediated by their downstream effector proteins.171 Currently, more than 10 distinct Ras effector proteins have been identified,14 including the most prominent and best characterized Raf kinase, PI3K, and RalGDS, as well as Ras and Rab interactor 1 (RIN1), T-cell lymphoma invasion and metastasis-inducing protein (TIAM), Af6, Novel Ras effector 1A (NORE1A), Bry2 kinase, phospholipase C (PLC), and growth factor receptor 14 (Grb14). These effector proteins exclusively interact with the active, GTP-bound Ras to trigger different downstream signaling cascades.172,173 To date, several crystal structures of Ras in complex with Ras effector proteins in the GppNHp-bound states have been determined. However, in these Ras–effector complexes, the H-Ras isoform, as a prototype in studies of Ras biology, was the only one reported. The solved crystal structures of H-Ras complexed with Ras effector proteins include Raf (PDB ID 4G0N),121 PI3Kγ (PDB ID 1HE8),122 RalGDS (PDB ID 1LFD),174 NORE1A (PDB ID 3DDC),175 PLCε (PDB ID 2C5L),176 Grb14 (PDB ID 4K81),177 and Bry2 (PDB ID 1K8R).178 Representative structures of H-Ras–effector complexes are shown in Fig. 15. Structural analysis of these complexes reveals that the mode of interaction is highly similar for all Ras effector proteins. These effector proteins associate with the catalytic domain of H-Ras through a conserved structural domain (RBD). Analysis of binding interfaces between H-Ras and Ras effector proteins shows that the major contributions to the interaction from effector proteins are derived from the strand β2 of RBD, while the effector-binding region from H-Ras depends on the types of Ras effector proteins. In the H-Ras–Raf-RBD complexes, strand β2 and the switch I region of H-Ras interact mainly with the strand β2 of Raf-RBD. For the remaining H-Ras–effector complexes, aside from strand β2 and the switch I region, the switch II region of H-Ras is involved in the interaction.
Fig. 15.

Active, GTP-bound Ras interacts with downstream effector proteins. Most effector proteins interact with the effector-binding region or switch I region (dashed circle) on the protein through their Ras binding domains (RBDs) or Ras-associating (RA, also known as RBD) domains. RafRBD (pink, PDB ID: 4G0N), PI3Kγ (brown, PDB ID: 1HE8), RalGDS-RBD (olive, PDB ID 1LFD), PLCε-RBD (wheat, PDB ID: 2C5L), NORE1A-RA (orange, PDB ID: 3DDC), Byr2RBD (palegreen, PDB ID: 1K8R), and Grb14 (lightblue, PDB ID: 4K81) bind to the effector-binding region on H-Ras–GppNHp (cyan, PDB ID: 5P21). In the H-Ras–RalGDS complex, the crystal contains two molecules of each H-Ras and RalGDS-RBD in the asymmetric unit.
Taking into account the fact that Ras proteins interact with downstream effector proteins through protein-protein interactions (PPIs), targeting Ras–effector PPIs gains new momentum with the design of inhibitors bound to the effector-binding region of Ras to block their interaction with downstream effector proteins.179–185 This effect results in growth inhibition and apoptosis of cancer cells. Because of the large interface area of Ras–effector PPIs and the relatively flat PPI interfaces, it is highly challenging to design small molecule inhibitors bound to PPI interfaces. Alternatively, designed peptides with relatively large sizes have the potential to occupy the flat protein surfaces. To date, several peptide mimetics and cyclic peptides were developed to occupy the effector-binding region of Ras,186–190 thereby inhibiting Ras signaling. These cyclic peptides specifically blocked Ras–Raf PPIs.
Recently, Athuluri-Divakar and co-workers, for the first time, identified a small-molecule inhibitor, rigosertib (RGS, 17) (Fig. 16), which interacts with the RBDs of a number of Ras effector proteins.79 RGS, a styrylbenzyl sulfone, is in phase III clinical trials for myelodysplastic syndrome.191 RGS was attached to a biotin; this RGS–biotin conjugate acts as an affinity matrix to identify a series of RGS-binding proteins, including A-Raf, B-Raf, c-Raf, Hsp27, Hsp73, and FUBP3.79 Differential scanning fluorimetry and western analysis of c-Raf–RGS interaction validated RGS binding especially to c-Raf-RBD; not to c-Raf kinase domain. Microscale thermophoretic analysis of RGS association with c-Raf-RBD and B-Raf-RDB showed that RGS interacts with both RBDs of c-Raf and B-Raf strongly, with Kd values of 0.71 nM and 0.18 nM, respectively. In addition to Raf family proteins, the authors also found that RGS can bind to the RBDs of RalGDS and four class I PI3Ks (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ), suggesting that RGS is a general inhibitor bound to the RBDs of Ras effector proteins. The determination of the solution structure of B-Raf-RBD–RGS complex ascertained that RGS is bound to strands β1 and β2 and helix α1 of Raf-RBD, at the same location to which the switch I region of Ras binds, indicating that RGS binding to Raf can interfere with Ras–Raf interaction. Biochemical analysis further revealed that addition of RGS to HeLa cells inhibits the heterodimerization of c-Raf/B-Raf, thereby inhibiting the activation of MEK/ERK pathway. In vivo efficacy studies based on xenograft models of colorectal, lung, and pancreatic cancers, displayed that treatment of RGS significantly inhibits Ras-mediated transformation and tumor growth. Moreover, the phosphorylation levels of ERK and Akt decreased markedly in response to RGS treatment, indicating that RGS treatment impairs Ras-mediated signaling. Cumulatively, these data suggest the feasibility of RGS in the treatment of RAS-driven cancers.
Fig. 16.

Chemical structure of rigosertib (RGS) (17).
7 K-Ras4B/calmodulin/PI3Kα complexes as a potential new adenocarcinoma-specific drug target
Calcium-bound calmodulin (CaM/Ca2+) is established to bind only to K-Ras4B; however, recent data suggest that it may also bind to farnesylated but depalmitoylated K-Ras4A.89 It does not bind to the H-Ras or N-Ras isoforms. Binding takes place through the involvement of the highly positively charged, farnesylated HVR, uniquely present in the K-Ras isoform.192 Binding of CaM can promote KRAS-driven cancers. Due to its importance as a target in drug discovery determining the structure of the K-Ras4B/CaM complex has been a long sought-out aim; however, crystallography or NMR of the complex has proven challenging, likely due to its flexibility. The many lysine residues on the K-Ras4B HVR and the negatively charged CaM linker suggest that the HVR–CaM interaction can take place in several ways. In addition, even though the interaction between the farnesylated HVR and CaM is likely to be tight, the inherent fluctuations of the HVR with respect to the catalytic domain coupled with the apparently weak interactions between CaM and the K-Ras4B catalytic domain and the likelihood that several CaM–catalytic domain interaction states may be involved compound the difficulties. Based on our mechanistic view of how CaM/Ca2+ acts to promote adenocarcinomas – through full activation of PI3Kα in KRAS-driven cancers – we proposed that it forms a K-Ras4B/CaM/PI3Kα complex.94,193 We reasoned that under normal physiological signaling, receptor tyrosine kinase (RTK) collaborates with K-Ras in recruiting and activating PI3K at the membrane. In contrast, oncogenic constitutive K-Ras4B signaling proceeds in the absence of a stimulated RTK. Under these circumstances, CaM substitutes for the RTK’s role. CaM/Ca2+ recruit PI3Kα to the membrane and promote oncogenic GTP-bound K-Ras4B activation of PI3Kα. Orthosteric or allosteric blocking the CaM/PI3Kα binding in an oncogenic K-Ras4B/CaM/PI3Kα complex could be adenocarcinoma-specific – thus with tolerable toxicity – therapeutic strategy.
This concept provides a plausible new target. Its main advantage is adenocarcinoma specificity. Even though the modeled trimer does not permit detailed suggestions for drug discovery, it does suggest focusing on the tighter interface between CaM and the cSH2 domain of the p85 subunit of PI3Kα in the trimer K-Ras4B/CaM/PI3Kα which acts to activate PI3Kα. CaM might also interact with the nSH2 domain, which would relieve nSH2’s autoinhibitory action on the p110 catalytic domain of PI3K; however targeting the nSH2 surface may activate PI3Kα rather than blocking it.
8 Targeting Ras with siRNA
The exploration of small interfering RNA (siRNA)-based RNA interference (RNAi) for cancer gene therapy has gained increasing attention because it can silence cancer targets that otherwise may not be effectively inhibited by means of conventional approaches, like the “undruggable oncogenic Ras proteins.194–196 RNAi is involved in sequence-specific post-transcriptional gene silencing conferred by a double-stranded siRNA with a sequence complementary to the target gene. A major concern in the development of RNAi therapy is the feasibility of using efficient carriers for delivering siRNAs through the cell membranes. Recently, based on a synthetic nanoparticles that deliver single siRNA KRas or combined siRNAs KRas and PI3K to KRas tumor xenografts, Yuan et al.194 revealed that nanoparticle-mediated delivery of siKRAS to KRAS-mutant tumors impaired tumor growth; however, combination of siRNAs K-Ras and PI3K increased efficacy. Consistently, based on a polymer-based 7C1 nanoparticle, Xue et al.197 showed that combination of siRNAs targeting miR-34a, a p53-regulated tumor suppressor miRNA, and KRas impaired KRAS-driven lung adenocarcinomas in vivo. Using different delivery systems, such as cationic poly(lactic acid)-based degradable nanocapsules and miniature biodegradable polymeric matrix, Lin et al.198 and Khvalevsky et al.199 unveiled that siRNA KRasG12D inhibited tumor growth in pancreatic cancer cells. Bäumer et al.200 observed that anti-EGFR antibody-mediated delivery of siRNA KRas significantly inhibited tumor growth and overcame therapy resistance in colon cancer in xenograft mouse models. Together, these studies suggest that siRNA strategies provide an alternative approach to direct targeting of Ras.
9 Drug resistance
Drug resistance mechanisms vary. They reflect the hallmarks of cancer.201,202 The eight hallmarks underlie the organizing principle that helps in understanding the complexity cancer. The hallmarks include sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis. The genome instability foments multiple hallmark functions that dominate drug resistance mechanisms. Broadly, these mechanisms include mechanisms through which the pharmacological actions can fail, such as increased rates of drug efflux, e.g. through cell membrane transporter proteins,203–205 DNA damage repair or cell death, which can induce cell cycle arrest,206,207 alterations in the tumor microenviroment208,209 and in drug metabolism e.g. via oxidative stress210,211, emergence of cancer stem cells which can display higher levels of drug efflux proteins, anti-apoptotic proteins, and DNA damage repair212–215 and mutations of drug targets, as in the case of B-Raf and EGFR,211–214 and cell death inhibition205,211,216,217 Therapeutic strategies aim to halt proliferative signaling and enhance growth suppressors; promote cell death and prevent the hallmark of replicative immortality, suppress cancer cell angiogenesis, which may relate to nutrition, and inactivate invasion and metastasis.
10 Inhibiting parallel pathways in tumor initiation
Drug resistance via signaling, can take place by mutations downstream of an inhibited pathway; it also often takes place through mutations that result in parallel pathways taking over.218 This mode of drug resistance is common and challenging and raises the question whether we can we predict a priori which parallel pathways – and proteins within these – would be involved in RAS-driven cancers. Sustaining proliferative growth is a fundamental hallmark of cancer201,202 and thus of drug resistance.211,219 An ability to forecast these – which could lead to multi-component prophylactic treatments – is of vast importance. This challenge requires insight into cellular signaling mechanisms to figure out which independent pathways eventually fulfil the same – i.e. corresponding – roles in cell cycle control in tumor initiation. Based on the available literature, recently we proposed that there are two independent pathways in tumor initiation:220 the first involves two K-Ras pathways, MAPK and PI3K. The second involves the pathways leading to the expression (or activation) of YAP1 and c-Myc. YAP1 is regulated by the Hippo pathway; c-Myc by a number of pathways, including Wnt (β-catenin), Notch, Hedgehog, the eukaryotic translation initiation factor 4E (eIF4E)221–224 and more. YAP1 and ERK (in the MAPK pathway), and β-catenin and PI3K/Akt, accomplish similar roles in cell cycle control despite the fact that they are stimulated by different cues: cell-cell contact/mechanical strain and growth factor/hormone signaling.221,225,226 Thus, even though different types of events differentially turn on signaling through the Hippo, Wnt, MAPK, and PI3K pathways, their functions in cell cycle control and tumor initiation are analogous. This can explain how YAP1 rescues K-Ras or B-Raf ablation,227,228 and why drug resistance to PI3Kα inhibitors can involve overexpression/activation of β-catenin.229 It can also explain the clinical data of the increase in aggressiveness of lung tumors when both oncogenic K-Ras and β-catenin signaling take place. Overexpression/activation of YAP1 and β-catenin (or broadly c-Myc) is able to rescue tumor cells in Ras drug resistance, because they act consecutively in the G1 (Gap 1) phase through the S (Synthesis) cell cycle restriction point analogous to MAPK/ERK and PI3K/Akt. Together, this mechanistic insight may pave the road to pathway-based drug discovery. The implications of this proposition are powerful. They suggest that to avert drug resistance in KRAS-driven cancer initiation,230,231 pathways that control the same cell cycle action and their four combinations (MAPK + PI3K; YAP1 + β-catenin; MAPK + β-catenin; PI3K + YAP1) should be prophylactically co-targeted. A single combination can result in cell cycle dysregulation, thus sustaining proliferative signaling. When trying various combinations these could account for their corresponding functions in the cell. This might open new horizons in drug regimes. The strategy that we have outlined here the four equivalent combinations of the two pathways through which drug resistance can emerge may provide blueprints toward this challenging aim.
11 Conclusions and perspectives
The critical role of Ras in a broad spectrum of cancers and the current lack of an effective drug has challenged the scientific and pharma community to tackle RAS-driven cancers. To come up with therapeutic strategies, we, as a community, need to understand Ras structural biology and its complex signaling behavior.
All Ras proteins are typically small GTPases, which are devoid of evident pockets on their catalytic domains in which small molecules can bind. They also have high affinities for their GDP and GTP substrates. Together, these obstacles underscore the difficulties in direct Ras attack. To date, only several GDP analogs covalently modify the G12C Ras at the active site. The catalytic domains of all isoforms are highly homologous. However, the C-terminal HVRs of Ras are disordered and their sequences and charge properties are significantly different. NMR spectroscopy and MD simulations revealed that the HVR is autoinhibited by shielding the effector binding site in the GDP-bound K-Ras4B and is released upon GTP binding and in response to certain oncogenic mutations. However, the structural and dynamic characteristics of the HVR in K-Ras4B are not shared by K-Ras4A, H-Ras, and N-Ras, suggesting that autoinhibition is a unique feature of K-Ras4B. Moreover, the orientations of the catalytic domain of Ras with respect to the membrane surface are also distinct between Ras isoforms. Because of the dynamic differences of the HVR between oncogenic and WT Ras as well as between Ras isoforms, it may be possible to design small molecule inhibitors or antibodies that preferentially target an oncogenic Ras mutant and a specific Ras isoform. For example, an antibody fragment, iDab#6, can identify the full-length H-Ras–GTP with oncogenic mutations at Gly12 or Gln61.102
Ras is an allosteric enzyme. Allosteric inhibition is the preferred option to achieve enhanced selectivity or reduced toxicity.232 Accumulating evidence indicates an allosteric communication between the Ras active site and its membrane-facing surface and existence of multiple conformational substates [Ras-GTP state 1 (inactive state) and Ras-GTP state 2 (active state)].130,135 These observations may open new possibilities of inhibiting Ras activity and provide clues into Ras potential druggability. Thus far, four allosteric ligand binding sites on Ras have been identified by the discovery of a number of allosteric Ras binders, which were further confirmed by NMR or X-ray crystal structures of Ras in complex with small-molecule ligands. Two of the four pockets are located near the functional switch regions and the remaining two pockets are located between loop 7, loop 9 and helix 5 as well as loop 7 and helix 3, helix 4. Most importantly, the discovery of an allosteric S-II pocket in proximity to the switch II region has greatly encouraged the Ras community. Small molecule inhibitors target G12C K-Ras selectively without any effect on the WT form and have the potential to advance as clinical candidates.
PPIs are of prime importance for all biological processes. Signaling is conveyed through PPIs, pathways and pathway cross talk, traveling across the entire cellular network.181,233–235 The unique PPI network in a pathway defines signaling specificity, thus precise functional control.236–238 Despite the challenge in targeting PPIs, however, new strategies and successes as in the cases of p53, HIV-1, and Bcl-2 family have increasingly strengthened the notion that PPI interfaces can provide potential pockets for compound binding.239–242 Ras proteins interact with their downstream protein effectors and can form Ras–Ras dimers, providing an excellent example. The recent discovery of the promising small molecule RGS inhibitor as a Ras-mimetic to inhibit Ras signaling opens a new horizon for the future of PPI drug discovery based on Ras–effector protein-protein interfaces.79 Additionally, our recent identification of the two interfaces of K-Ras4B dimers, a β-sheet and α-helical, indicates that the helical interface may be an additional new drug target.116 Disruption of Ras–Ras dimers by targeting this helical interface would inhibit Ras dimerization and thus downregulate Raf activation and MAPK signaling.
A key aim in drug discovery is to obtain isoform-specific drugs. Achieving this aim would lessen toxicity. None of the drugs designed to date targets a specific isoform. This is because the sequences and structures of the catalytic domains of the isoforms are highly similar. Even though their HVR sequences are highly variable and flexible, their apparent lack of specificity argues against serving as direct targets. The distinct features of the HVR involve their overall charge, which plays a role in their lipid post-translational modifications and membrane attachment preferences. The farnesyl is present in all isoforms, which is not the case for the palmitoyl, which is absent in the most oncogenic isoform, K-Ras4B. Thus, to date, efforts focused on inhibiting farnesylation. This however resulted in setbacks: toxicity (farnesyl transferase farnesylates additional proteins) and the taking over by geranylgeranylation. Recently, we proposed that CaM acts specifically in full activation of PI3Kα in KRAS4B-driven cancers, but not in H-Ras or N-Ras cancers, suggesting that a trimer could be an attractive drug target for KRAS4B-driven cancers. However, a crystal structure or high resolution electron microscopy or at the very least more rigorous modeling of the K-Ras4B/CaM/PI3Kα trimer should be determined in order to illuminate the details of the possible interface where a drug can target.
Notwithstanding, whichever drugs are designed and deployed unfortunately it can be expected that drug resistance will develop. The idea of drug combinations is not new. The key – and to date still unsolved – question is which type of combinations to select. The combinations can target the same protein, other proteins in the same pathway or proteins in different pathways. The latter may well be the most challenging because it requires biological insight into which pathways compensate each other in cell proliferation and growth. To avert drug resistance in RAS-driven cancers, we propose that efforts should particularly focus on the Hippo/Wnt pathways and the proteins that they regulate, correspondingly YAP1 and β-catenin, and their independent and corresponding major Ras pathways, MAPK and PI3K/Akt. Constructing libraries of combinations of such drugs appears a meritorious and significant aim. We believe that charting the complete signaling map of independent core pathways, initiating from cell surface receptors, down to the respective cell cycle actions and the activated transcription factors – should be a major aim in cancer biology and pharmaceutics. The current pathway diagrams do not allow such in-depth understanding.
In summary, we expect that increasingly, comprehensive mechanistic, signaling structural, biochemical and clinical data in Ras-focused research will provide valuable venues toward drugging Ras – and no less important for longer-term success – overcome drug resistance.
Acknowledgments
We thank Dr. Chung-Jung Tsai for discussions. This work was supported by National Basic Research Program of China (973 Program) (2015CB910403); National Natural Science Foundation of China (81322046, 81302698, 81473137); Shanghai Rising-Star Program (13QA1402300); Program for New Century Excellent Talents in University (NCET-12-0355); Shanghai Health and Family Planning Commission (20154Y0058); Collaborative Innovation Center of Systems Biomedicine. This project has also been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Biographies

Shaoyong Lu received his undergraduate Applied Chemistry degree in Hangzhou Normal University, China, in 2007. After which he obtained the Ph.D. degree of Chemistry in Zhejiang University, China, in 2012. Then, he continued the postdoctoral research at Shanghai Jiao Tong University, School of Medicine, China, in the Prof. Jian Zhang’s group, and became as a researcher assistant in the same group in 2014. During the 2014–2015, he studied as a visiting scholar in the Prof. Ruth Nussinov’s group at the National Cancer Institute-Frederick. His main interests include the study of allosteric regulation of Ras and the development of allosteric methods and applications.

Hyunbum Jang earned his Ph.D. in physics from the Department of Physics at the University of Ackland, New Zealand in 1999, where he studied the thin magnetic films with competing surface fields by performing a series of Monte Carlo (MC) simulations of Heisenberg spin systems with anisotropy. He joined the Hall lab at the North Carolina State University as a postdoctoral research associate in 2000, where his research focused on the thermodynamic and kinetic properties of isolated β-sheet proteins and β-sheet complex using discontinuous molecular dynamics (DMD) technique. Moving towards more complex biomolecular systems, he joined the Woolf lab at the Johns Hopkins University as a postdoctoral fellow in 2002, where his research focused on the membrane proteins using all-atom molecular dynamics (MD) simulations with the CHARMM program. In 2005, he accepted a position as a computational scientist at the National Cancer Institute of the NIH, Leidos Biomedical Research, Inc. He is a member of American Association for Cancer Research.

Shuo Gu is a Associate Professor and Vice Chief Doctor of Pediatric Surgery at Shanghai Children’s Medical Center Affiliated to Shanghai Jiaotong University School of Medicine, China. He received his Ph.D. degree from Shanghai Second Medical University. He has a broad interest in children disease, including congenital malformation and cancers, his current research focus on pathological mechanisms of nervous system like medulloblastoma and MB pathogenesis as well as siRNA.

Jian Zhang received a B.M. degree in Pharmacology in 2002 from Peking University and a Ph.D. in 2007 from Shanghai Institute of Materia Medica, Chinese Academy of Sciences (under the tutelage of Prof. Hualiang Jiang). After receiving his Ph.D., he moved to University of Michigan to carry out postdoctoral research in Prof. Shaomeng Wang’s group. In 2009, he joined the Shanghai Jiao Tong University, School of Medicine, where he is a full researcher and doctoral supervisor. Now, he is also the director of Medicinal Bioinformatics Center. His fields of research include drug design and chemical biology that mainly pertain to the repertoire of allostery, in particular the construction of allosteric database (ASD), the development of computational approaches for predicting allosteric sites (Allosite, Alloscore and ASBench), and the identification of allosteric modulators for proteins. His website gives further details. http://mdl.shsmu.edu.cn/.

Ruth Nussinov received her Ph.D. in 1977 from Rutgers University and did post-doctoral work in the Structural Chemistry Department of the Weizmann Institute. Subsequently she was at the Chemistry Department at Berkeley, the Biochemistry Department at Harvard, and a visiting scientist at the NIH. In 1984 she joined the Department of Human Genetics, at the Medical School at Tel Aviv University. In 1985, she accepted a concurrent position at the National Cancer Institute of the NIH, Leidos Biomedical Research, where she is a Senior Principal Scientist and Principle Investigator heading the Computational Structural Biology Section at the NCI. She has authored over 500 scientific papers. She is the Editor-in-Chief in PLoS Computational Biology and Associate Editor and on the Editorial Boards of a number of journals. She is a frequent speaker in Domestic and International meetings, symposia and academic institutions, won several award and elected fellow of several societies. Her National Cancer Institute website gives further details. https://ccr.cancer.gov/ruth-nussinov.
References
- 1.Barbacid M. Annu. Rev. Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. Nat. Rev. Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 3.Karnoub AE, Weinberg RA. Nat. Rev. Mol. Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox AD, Der CJ. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. Trends Biochem. Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rojas AM, Fuentes G, Rausell A, Valencia A. J. Cell Biol. 2012;196:189–201. doi: 10.1083/jcb.201103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colicelli J. Sci. STKE. 2004;2004:RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schweins T, Geyer M, Scheffzek K, Warshel A, Kalbitzer HR, Wittinghofer A. Nat. Struct. Biol. 1995;2:36–44. doi: 10.1038/nsb0195-36. [DOI] [PubMed] [Google Scholar]
- 9.Scheffzek K, Lautwein A, Kabsch W, Ahmadian MR, Wittinghofer A. Nature. 1996;384:591–596. doi: 10.1038/384591a0. [DOI] [PubMed] [Google Scholar]
- 10.Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A. Nat. Struct. Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 11.Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D. Mol. Cell. Biol. 1998;18:880–886. doi: 10.1128/mcb.18.2.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-sagi D, Kuriyan J. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 13.Boriack-sjodin PA, Margarit SM, Bar-sagi D, Kuriyan J. Nature. 1998;394:337–343. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- 14.Mott HR, Owen D. Crit. Rev. Biochem. Mol. Biol. 2015;50:85–133. doi: 10.3109/10409238.2014.999191. [DOI] [PubMed] [Google Scholar]
- 15.Rajalingam K, Schreck R, Rapp UR, Albert S. Biochim. Biophys. Acta. 2007;1773:1177–1195. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Herrmann C. Curr. Opin. Struct. Biol. 2003;13:122–129. doi: 10.1016/s0959-440x(02)00007-6. [DOI] [PubMed] [Google Scholar]
- 17.Smith MJ, Ikura M. Nat. Chem. Biol. 2014;10:223–230. doi: 10.1038/nchembio.1435. [DOI] [PubMed] [Google Scholar]
- 18.McCormick F, Wittinghofer A. Curr. Opin. Biotechnol. 1996;7:449–456. doi: 10.1016/s0958-1669(96)80123-6. [DOI] [PubMed] [Google Scholar]
- 19.Akasaka K, Tamada M, Wang F, Kariya K, Shima F, Kikuchi A, Yamamoto M, Shirouzu M, Yokoyama S, Kataoka T. J. Biol. Chem. 1996;271:5353–5360. doi: 10.1074/jbc.271.10.5353. [DOI] [PubMed] [Google Scholar]
- 20.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. Nat. Rev. Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gysin S, Salt M, Young A, McCormick F. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stephen AG, Esposito D, Bagni RK, McCormick F. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prior IA, Lewis PD, Mattos C. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bos JL, Rehmann H, Wittinghofer A. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 27.Smith MJ, Neel BG, Ikura M. Proc. Natl. Acad. Sci. U. S. A. 2013;110:4574–4579. doi: 10.1073/pnas.1218173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clausen R, Ma B, Nussinov R, Shehu A. PLoS Comput. Biol. 2015;11:e1004470. doi: 10.1371/journal.pcbi.1004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samatar AA, Poulikakos PI. Nat. Rev. Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 30.Downward J. Nat. Rev. Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 31.Downward J. Nat. Med. 2008;14:1315–1316. doi: 10.1038/nm1208-1315. [DOI] [PubMed] [Google Scholar]
- 32.Castellano E, Sheridan C, Thin MZ, Nye E, Spencer-Dene B, Diefenbacher ME, Moore C, Kumar MS, Murillo MM, Grönroos E, Lassailly F, Stamp G, Downward J. Cancer Cell. 2013;24:617–630. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holderfield M, Deuker MM, McCormick F, McMahon M. Nat. Rev. Cancer. 2014;14:455–467. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCormick F. Expert Opin. Ther. Targets. 2015;19:451–454. doi: 10.1517/14728222.2014.1000304. [DOI] [PubMed] [Google Scholar]
- 35.Nussinov R, Tsai C, Muratcioglu S, Jang H, Gursoy A, Keskin O. Expert Rev. Proteomics. 2015;12:669–682. doi: 10.1586/14789450.2015.1100079. [DOI] [PubMed] [Google Scholar]
- 36.De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. Expert Opin. Ther. Targets. 2012;16:S17–S27. doi: 10.1517/14728222.2011.639361. [DOI] [PubMed] [Google Scholar]
- 37.Castellano E, Santos E. Genes Cancer. 2011;2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernández-Medarde A, Santos E. Genes Cancer. 2011;2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Kaiser CE, Frett B, Li H. J. Med. Chem. 2013;56:5219–5230. doi: 10.1021/jm3017706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PIH, Waldmann H. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 41.Leshchiner ES, Parkhitko A, Bird GH, Luccarelli J, Bellairs JA, Escudero S, Opoku-Nsiah K, Godes M, Perrimon N, Walensky LD. Proc. Natl. Acad. Sci. U. S. A. 2015;112:1761–1766. doi: 10.1073/pnas.1413185112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winter JJG, Anderson M, Blades K, Brassington C, Breeze AL, Chresta C, Embrey K, Fairley G, Faulder P, Finlay MRV, Kettle JG, Nowak T, Overman R, Patel SJ, Perkins P, Spadola L, Tart J, Tucker JA, Wrigley G. J. Med. Chem. 2015;58:2265–2274. doi: 10.1021/jm501660t. [DOI] [PubMed] [Google Scholar]
- 43.Evelyn CR, Biesiada J, Duan X, Tang H, Shang X, Papoian R, Seibel WL, Nelson S, Meller J, Zheng Y. J. Biol. Chem. 2015;290:12879–12898. doi: 10.1074/jbc.M114.634493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adjei AA. J. Natl. Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 45.Shima F, Matsumoto S, Yoshikawa Y, Kawamura T, Isa M, Kataoka T. J. Biochem. 2015;158:91–99. doi: 10.1093/jb/mvv060. [DOI] [PubMed] [Google Scholar]
- 46.Brock EJ, Ji K, Reiners JJ, Mattingly RR. Mini Rev. Med. Chem. 2016;16:358–369. doi: 10.2174/1389557515666151001154002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Upadhyaya P, Bedewy W, Pei D. Mini Rev. Med. Chem. 2016;16:376–382. doi: 10.2174/1389557515666151001141713. [DOI] [PubMed] [Google Scholar]
- 48.Quah SY, Tan MS, Yuan HT, Stanslas J. Pharmacol. Ther. 2016;162:35–57. doi: 10.1016/j.pharmthera.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 49.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Proc. Natl. Acad. Sci. U. S. A. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evelyn CR, Biesiada J, Duan X, Tang H, Shang X, Papoian R, Seibel WL, Nelson S, Meller J, Zheng Y. Chem. Biol. 2015;290:12879–12898. doi: 10.1074/jbc.M114.634493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burns MC, Sun Q, Daniels RN, Camper D, Kennedy JP, Phan J, Olejniczak ET, Lee T, Waterson AG, Rossanese OW, Fesik SW. Proc. Natl. Acad. Sci. U. S. A. 2014;111:3401–3406. doi: 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schöpel M, Jockers KFG, Düppe PM, Autzen J, Potheraveedu VN, Ince S, Yip KT, Heumann R, Herrmann C, Scherkenbeck J, Stoll R. J. Med. Chem. 2013;56:9664–9672. doi: 10.1021/jm401291q. [DOI] [PubMed] [Google Scholar]
- 53.Fanelli F, Raimondi F. Curr. Pharm. Des. 2013;19:4214–4225. doi: 10.2174/1381612811319230006. [DOI] [PubMed] [Google Scholar]
- 54.Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Angew. Chem. Int. Ed. Engl. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Nat. Rev. Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ledford H. Nature. 2015;520:278–280. doi: 10.1038/520278a. [DOI] [PubMed] [Google Scholar]
- 57.Arozarena I, Calvo F, Crespo P. Genes Cancer. 2011;2:182–194. doi: 10.1177/1947601911409213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmick M, Kraemer A, Bastiaens PIH. Trends Cell Biol. 2015;25:190–197. doi: 10.1016/j.tcb.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Weise K, Kapoor S, Denter C, Nikolaus J, Opitz N, Koch S, Triola G, Herrmann A, Waldmann H, Winter R. J. Am. Chem. Soc. 2011;133:880–887. doi: 10.1021/ja107532q. [DOI] [PubMed] [Google Scholar]
- 60.Li Z, Janosi L, Gorfe AA. J. Am. Chem. Soc. 2012;134:17278–17285. doi: 10.1021/ja307716z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. J. Biol. Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 62.Sheridan C. Nat. Biotechnol. 2016;34:217–218. doi: 10.1038/nbt0316-217. [DOI] [PubMed] [Google Scholar]
- 63.Tan Z, Zhang S. Mini Rev. Med. Chem. 2016;16:345–357. doi: 10.2174/1389557515666151001154111. [DOI] [PubMed] [Google Scholar]
- 64.Lu S, Banerjee A, Jang H, Zhang J, Gaponenko V, Nussinov R. J. Biol. Chem. 2015;290:28887–28900. doi: 10.1074/jbc.M115.664755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chavan TS, Jang H, Khavrutskii L, Abraham SJ, Banerjee A, Freed BC, Johannessen L, Tarasov SG, Gaponenko V, Nussinov R, Tarasova NI. Biophys. J. 2015;109:2602–2613. doi: 10.1016/j.bpj.2015.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mazhab-Jafari MT, Marshall CB, Smith MJ, Gasmi-Seabrook GMC, Stathopulos PB, Inagaki F, Kay LE, Neel BG, Ikura M. Proc. Natl. Acad. Sci. U. S. A. 2015;112:6625–6630. doi: 10.1073/pnas.1419895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jang H, Banerjee A, Chavan TS, Lu S, Zhang J, Gaponenko V, Nussinov R. FASEB J. 2016;30:1643–1655. doi: 10.1096/fj.15-279091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spoerner M, Nuehs A, Herrmann C, Steiner G, Kalbitzer HR. FEBS J. 2007;274:1419–1433. doi: 10.1111/j.1742-4658.2007.05681.x. [DOI] [PubMed] [Google Scholar]
- 69.Araki M, Shima F, Yoshikawa Y, Muraoka S, Ijiri Y, Nagahara Y, Shirono T, Kataoka T, Tamura A. J. Biol. Chem. 2011;286:39644–39653. doi: 10.1074/jbc.M111.227074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, Matsumoto K, Yamamoto N, Sugimoto T, Yoshikawa Y, Kumasaka T, Yamamoto M, Tamura A, Kataoka T. J. Biol. Chem. 2010;285:22696–22705. doi: 10.1074/jbc.M110.125161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye M, Shima F, Muraoka S, Liao J, Okamoto H, Yamamoto M, Tamura A, Yagi N, Ueki T, Kataoka T. J. Biol. Chem. 2005;280:31267–31275. doi: 10.1074/jbc.M505503200. [DOI] [PubMed] [Google Scholar]
- 72.O’Connor C, Kovrigin EL. Biochemistry. 2008;47:10244–10246. doi: 10.1021/bi801076c. [DOI] [PubMed] [Google Scholar]
- 73.Prakash P, Gorfe AA. Biochim. Biophys. Acta. 2013;1830:5211–5218. doi: 10.1016/j.bbagen.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosnizeck IC, Graf T, Spoerner M, Tränkle J, Filchtinski D, Herrmann C, Gremer L, Vetter IR, Wittinghofer A, König B, Kalbitzer HR. Angew. Chem. Int. Ed. Engl. 2010;49:3830–3833. doi: 10.1002/anie.200907002. [DOI] [PubMed] [Google Scholar]
- 75.Rosnizeck IC, Spoerner M, Harsch T, Kreitner S, Filchtinski D, Herrmann C, Engel D, König B, Kalbitzer HR. Angew. Chem. Int. Ed. Engl. 2012;51:10647–10651. doi: 10.1002/anie.201204148. [DOI] [PubMed] [Google Scholar]
- 76.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patricelli MP, Janes MR, Li L-S, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 78.Lito P, Solomon M, Li L, Hansen R, Rosen N. Science. 2016;351:604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Athuluri-divakar SK, Carpio RV, Dutta K, Guha C, Baker SJ. Cell. 2016;165:643–655. doi: 10.1016/j.cell.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hobbs GA, Der CJ, Rossman KL. J. Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCormick F. Clin. Cancer Res. 2015;21:1797–1801. doi: 10.1158/1078-0432.CCR-14-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 83.Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, Downward J. J. Clin. Invest. 2014;124:3601–3611. doi: 10.1172/JCI74134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang M-T, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, McCormick F. Cell. 2015;163:1237–1251. doi: 10.1016/j.cell.2015.10.041. [DOI] [PubMed] [Google Scholar]
- 85.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Nat. Rev. Mol. Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cox AD, Der CJ, Philips MR. Clin. Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chiu VK, Silletti J, Dinsell V, Wiener H, Loukeris K, Ou G, Philips MR, Pillinger MH. J. Biol. Chem. 2004;279:7346–7352. doi: 10.1074/jbc.M311602200. [DOI] [PubMed] [Google Scholar]
- 88.Parker JA, Mattos C. Mol. Cancer Res. 2015;13:595–603. doi: 10.1158/1541-7786.MCR-14-0535. [DOI] [PubMed] [Google Scholar]
- 89.Nussinov R, Tsai C, Chakrabarti M, Jang H. Cancer Res. 2016;76:18–24. doi: 10.1158/0008-5472.CAN-15-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nussinov R, Jang H, Tsai C-J. Biol. Rev. 2015;90:587–598. doi: 10.1111/brv.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banerjee A, Jang H, Nussinov R, Gaponenko V. Curr. Opin. Struct. Biol. 2016;36:10–17. doi: 10.1016/j.sbi.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, Nussinov R, Gaponenko V. J. Biol. Chem. 2015;290:9465–9477. doi: 10.1074/jbc.M114.620724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thapar R, Williams JG, Campbell SL. J. Mol. Biol. 2004;343:1391–1408. doi: 10.1016/j.jmb.2004.08.106. [DOI] [PubMed] [Google Scholar]
- 94.Nussinov R, Muratcioglu S, Tsai C-J, Jang H, Gursoy A, Keskin O. Mol. Cancer Res. 2015;13:1265–1273. doi: 10.1158/1541-7786.MCR-15-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abraham SJ, Nolet RP, Calvert RJ, Anderson LM, Gaponenko V. Biochemistry. 2009;48:7575–7583. doi: 10.1021/bi900769j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cromm PM, Spiegel J, Grossmann TN, Waldmann H. Angew. Chem. Int. Ed. Engl. 2015;54:13516–13537. doi: 10.1002/anie.201504357. [DOI] [PubMed] [Google Scholar]
- 97.Milroy L, Ottmann C. ACS Chem. Biol. 2014;9:2447–2458. doi: 10.1021/cb500555h. [DOI] [PubMed] [Google Scholar]
- 98.Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H. Nat. Chem. Biol. 2014;10:613–622. doi: 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- 99.Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD. Proc. Natl. Acad. Sci. U. S. A. 2014;111:8895–8900. doi: 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Jänne PA, Meyerson M, Marto JA, Engen JR, Gray NS. Angew. Chem. Int. Ed. Engl. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rudolph J, Stokoe D. Angew. Chem. Int. Ed. Engl. 2014;53:3777–3779. doi: 10.1002/anie.201400233. [DOI] [PubMed] [Google Scholar]
- 102.Tanaka T, Williams RL, Rabbitts TH. EMBO J. 2007;26:3250–3259. doi: 10.1038/sj.emboj.7601744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chakrabarti M, Jang H, Nussinov R. J. Phys. Chem. B. 2016;120:667–679. doi: 10.1021/acs.jpcb.5b11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR. Proc. Natl. Acad. Sci. U. S. A. 2015;112:779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chavan TS, Muratcioglu S, Marszalek R, Jang H, Keskin O, Gursoy A, Nussinov R, Gaponenko V. Cell. Logist. 2016;5:e1136374. doi: 10.1080/21592799.2015.1136374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gorfe AA, Babakhani A, Mccammon JA. 2007:12280–12286. doi: 10.1021/ja073949v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abankwa D, Gorfe AA, Hancock JF. Semin. Cell Dev. Biol. 2007;18:599–607. doi: 10.1016/j.semcdb.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abankwa D, Gorfe AA, Hancock JF. Cell Cycle. 2014;7:2667–2673. doi: 10.4161/cc.7.17.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Abankwa D, Hanzal-Bayer M, Ariotti N, Plowman SJ, Gorfe AA, Parton RG, McCammon JA, Hancock JF. EMBO J. 2008;27:727–735. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gorfe AA. Curr. Med. Chem. 2010;17:1–9. doi: 10.2174/092986710789957832. [DOI] [PubMed] [Google Scholar]
- 111.Janosi L, Li Z, Hancock JF, Gorfe AA. Proc. Natl. Acad. Sci. U. S. A. 2012;109:8097–8102. doi: 10.1073/pnas.1200773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. J. Am. Chem. Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 113.Bayburt TH, Grinkova YV, Sligar SG. Nano Lett. 2002;2:853–856. [Google Scholar]
- 114.Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R. Biochem. J. 2016 doi: 10.1042/BCJ20160031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Prakash P, Zhou Y, Liang H, Hancock JF, Gorfe AA. Biophys. J. 2016;110:1125–1138. doi: 10.1016/j.bpj.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Muratcioglu S, Chavan TS, Freed BC, Jang H, Khavrutskii L, Freed RN, Dyba MA, Stefanisko K, Tarasov SG, Gursoy A, Keskin O, Tarasova NI, Gaponenko V, Nussinov R. Structure. 2015;23:1325–1335. doi: 10.1016/j.str.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Güldenhaupt J, Rudack T, Bachler P, Mann D, Triola G, Waldmann H, Kötting C, Gerwert K. Biophys. J. 2012;103:1585–1593. doi: 10.1016/j.bpj.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lin W-C, Iversen L, Tu H-L, Rhodes C, Christensen SM, Iwig JS, Hansen SD, Huang WYC, Groves JT. Proc. Natl. Acad. Sci. U. S. A. 2014;111:2996–3001. doi: 10.1073/pnas.1321155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li Z, Cao S, Buck M. Biophys. J. 2016;110:1033–1035. doi: 10.1016/j.bpj.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gorfe AA, Hanzal-bayer M, Abankwa D, Hancock JF, Mccammon JA. J. Med. Chem. 2007;50:674–684. doi: 10.1021/jm061053f. [DOI] [PubMed] [Google Scholar]
- 121.Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, Mattos C. Structure. 2015;23:505–516. doi: 10.1016/j.str.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL. Cell. 2000;103:931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 123.Lu S, Li S, Zhang J. Med. Res. Rev. 2014;34:1242–1285. doi: 10.1002/med.21317. [DOI] [PubMed] [Google Scholar]
- 124.Nussinov R, Tsai C-J. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 125.Nussinov R, Tsai C, Liu J. J. Am. Chem. Soc. 2014;136:17692–176701. doi: 10.1021/ja510028c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang W, Wang G, Shen Q, Liu X, Lu S, Geng L, Huang Z, Zhang J. Bioinformatics. 2015;31:2598–2600. doi: 10.1093/bioinformatics/btv169. [DOI] [PubMed] [Google Scholar]
- 127.Ribeiro AAST, Ortiz V. Chem. Rev. 2016;116:6488–6502. doi: 10.1021/acs.chemrev.5b00543. [DOI] [PubMed] [Google Scholar]
- 128.Nussinov R, Tsai C. Curr. Pharm. Des. 2012;18:1311–1316. doi: 10.2174/138161212799436377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, Zhang J. Chem. Rev. 2016;116:6607–6665. doi: 10.1021/acs.chemrev.5b00542. [DOI] [PubMed] [Google Scholar]
- 130.McCarthy M, Prakash P, Gorfe AA. Acta Biochim. Biophys. Sin. (Shanghai) 2016;48:3–10. doi: 10.1093/abbs/gmv100. [DOI] [PubMed] [Google Scholar]
- 131.Prakash P, Sayyed-Ahmad A, Gorfe AA. PLoS Comput. Biol. 2015;11:e1004469. doi: 10.1371/journal.pcbi.1004469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aramini JM, Vorobiev SM, Tuberty LM, Janjua H, Campbell ET, Seetharaman J, Su M, Huang YJ, Acton TB, Xiao R, Tong L, Montelione GT. Structure. 2015;23:1382–1393. doi: 10.1016/j.str.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ting PY, Johnson CW, Fang C, Cao X, Graeber TG, Mattos C, Colicelli J. FASEB J. 2015;29:3750–3761. doi: 10.1096/fj.15-271510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Iversen L, Tu H-L, Lin W-C, Christensen SM, Abel SM, Iwig J, Wu H-J, Gureasko J, Rhodes C, Petit RS, Hansen SD, Thill P, Yu C-H, Stamou D, Chakraborty AK, Kuriyan J, Groves JT. Science. 2014;345:50–54. doi: 10.1126/science.1250373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Buhrman G, O’Connor C, Zerbe B, Kearney BM, Napoleon R, Kovrigina EA, Vajda S, Kozakov D, Kovrigin EL, Mattos C. J. Mol. Biol. 2011;413:773–789. doi: 10.1016/j.jmb.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hubbard PA, Moody CL, Murali R. Front. Physiol. 2014;5:478. doi: 10.3389/fphys.2014.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sun Q, Phan J, Friberg AR, Camper DV, Olejniczak ET, Fesik SW. J. Biomol. NMR. 2014;60:11–14. doi: 10.1007/s10858-014-9849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lu S, Huang W, Zhang J. Drug Discov. Today. 2014;19:1595–1600. doi: 10.1016/j.drudis.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 139.Shen Q, Wang G, Li S, Liu X, Lu S, Chen Z, Song K, Yan J, Geng L, Huang Z, Huang W, Chen G, Zhang J. Nucleic Acids Res. 2016;44:D527–D535. doi: 10.1093/nar/gkv902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Buhrman G, Wink G, Mattos C. Structure. 2007;15:1618–1629. doi: 10.1016/j.str.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Buhrman G, Holzapfel G, Fetics S, Mattos C. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4931–4936. doi: 10.1073/pnas.0912226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Buhrman G, Kumar VSS, Cirit M, Haugh JM, Mattos C. J. Biol. Chem. 2011;286:3323–3331. doi: 10.1074/jbc.M110.193854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Marcus K, Mattos C. Clin. Cancer Res. 2015;21:1810–1818. doi: 10.1158/1078-0432.CCR-14-2148. [DOI] [PubMed] [Google Scholar]
- 144.Holzapfel G, Buhrman G, Mattos C. Biochemistry. 2012;51:6114–6126. doi: 10.1021/bi300509j. [DOI] [PubMed] [Google Scholar]
- 145.Gorfe AA, Grant BJ, McCammon JA. Structure. 2008;16:885–896. doi: 10.1016/j.str.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hall BE, Bar-sagi D, Nassar N. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12138–12142. doi: 10.1073/pnas.192453199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ma J, Karplus M. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11905–11910. doi: 10.1073/pnas.94.22.11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Raimondi F, Portella G, Orozco M, Fanelli F. PLoS Comput. Biol. 2011;7:e1001098. doi: 10.1371/journal.pcbi.1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lukman S, Grant BJ, Gorfe AA, Grant GH, McCammon JA. PLoS Comput. Biol. 2010;6:e1000922. doi: 10.1371/journal.pcbi.1000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Grant BJ, Gorfe AA, McCammon JA. PLoS Comput. Biol. 2009;5:e1000325. doi: 10.1371/journal.pcbi.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grant BJ, Lukman S, Hocker HJ, Sayyah J, Brown JH, McCammon JA, Gorfe AA. PLoS One. 2011;6:e25711. doi: 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lu S, Jang H, Nussinov R, Zhang J. Sci. Rep. 2016;6:21949. doi: 10.1038/srep21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Spoerner M, Nuehs A, Herrmann C, Steiner G, Kalbitzer HR. J. Biol. Chem. 2010;285:39768–39778. doi: 10.1074/jbc.M110.145235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Meierhofer T, Rosnizeck IC, Graf T, Reiss K, König B, Kalbitzer HR, Spoerner M. J. Am. Chem. Soc. 2011;133:2048–2051. doi: 10.1021/ja108779j. [DOI] [PubMed] [Google Scholar]
- 155.Rosnizeck IC, Filchtinski D, Lopes RP, Kieninger B, Herrmann C, Kalbitzer HR, Spoerner M. Biochemistry. 2014;53:3867–3878. doi: 10.1021/bi401689w. [DOI] [PubMed] [Google Scholar]
- 156.Cherfils J, Zeghouf M. Physiol. Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 157.Scheffzek K, Ahnadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 158.Shipman L. Nat. Rev. Drug Discov. 2016 doi: 10.1038/nrd.2016.40. [DOI] [PubMed] [Google Scholar]
- 159.Prakash P, Sayyed-Ahmad A, Gorfe AA. PLoS Comput. Biol. 2012;8:e1002394. doi: 10.1371/journal.pcbi.1002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hocker HJ, Cho K-J, Chen C-YK, Rambahal N, Sagineedu SR, Shaari K, Stanslas J, Hancock JF, Gorfe AA. Proc. Natl. Acad. Sci. U. S. A. 2013;110:10201–10206. doi: 10.1073/pnas.1300016110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nan X, Tamgüney TM, Collisson EA, Lin L-J, Pitt C, Galeas J, Lewis S, Gray JW, McCormick F, Chu S. Proc. Natl. Acad. Sci. U. S. A. 2015;112:7996–8001. doi: 10.1073/pnas.1509123112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen M, Peters A, Huang T, Nan X. Mini Rev. Med. Chem. 2016;16:391–403. doi: 10.2174/1389557515666151001152212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chung JK, Lee YK, Lam HYM, Groves JT. J. Am. Chem. Soc. 2016;138:1800–1803. doi: 10.1021/jacs.5b12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Philips MR, Der CJ. Proc. Natl. Acad. Sci. U. S. A. 2015;112:9793–9794. doi: 10.1073/pnas.1511805112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sayyed-Ahmed A, Cho KJ, Hancock JF, Gorfe AA. J. Phys. Chem. B. 2016 doi: 10.1021/acs.jpcb.6b02403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jambrina PG, Rauch N, Pilkington R, Rybakova K, Nguyen LK, Kholodenko BN, Buchete N-V, Kolch W, Rosta E. Angew. Chem. Int. Ed. Engl. 2016;55:983–986. doi: 10.1002/anie.201509272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jambrina PG, Bohuszewicz O, Buchete N-V, Kolch W, Rosta E. Biochem. Soc. Trans. 2014;42:784–790. doi: 10.1042/BST20140025. [DOI] [PubMed] [Google Scholar]
- 168.Inouye K, Mizutani S, Koide H, Kaziro Y. J. Biol. Chem. 2000;275:3737–3740. doi: 10.1074/jbc.275.6.3737. [DOI] [PubMed] [Google Scholar]
- 169.Kolch W. Biochem. J. 2000;351:289–305. [PMC free article] [PubMed] [Google Scholar]
- 170.Nussinov R. Phys. Biol. 2013;10:045004. doi: 10.1088/1478-3975/10/4/045004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marshall J. Curr. Opin. Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 172.Koide H, Satoh T, Nakafuku M, Kaziro Y. Proc. Natl. Acad. Sci. U. S. A. 1993;90:8683–8686. doi: 10.1073/pnas.90.18.8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, Wittinghofer A. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4944–4949. doi: 10.1073/pnas.081441398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Huang L, Hofer F, Martin GS, Kim SH. Nat. Struct. Biol. 1998;5:422–426. doi: 10.1038/nsb0698-422. [DOI] [PubMed] [Google Scholar]
- 175.Stieglitz B, Bee C, Schwarz D, Yildiz O, Moshnikova A, Khokhlatchev A, Herrmann C. EMBO J. 2008;27:1995–2005. doi: 10.1038/emboj.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bunney TD, Harris R, Gandarillas NL, Josephs MB, Roe SM, Sorli SC, Paterson HF, Rodrigues-Lima F, Esposito D, Ponting CP, Gierschik P, Pearl LH, Driscoll PC, Katan M. Mol. Cell. 2006;21:495–507. doi: 10.1016/j.molcel.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 177.Qamra R, Hubbard SR. PLoS One. 2013;8:e72473. doi: 10.1371/journal.pone.0072473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Scheffzek K, Grünewald P, Wohlgemuth S, Kabsch W, Tu H, Wigler M, Wittinghofer A, Herrmann C. Structure. 2001;9:1043–1050. doi: 10.1016/s0969-2126(01)00674-8. [DOI] [PubMed] [Google Scholar]
- 179.Keskin O, Gursoy A, Ma B, Nussinov R. Chem. Rev. 2008;108:1225–1244. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 180.Lu S, Jang H, Zhang J, Nussinov R. Chem Med Chem. 2016;11:814–821. doi: 10.1002/cmdc.201500481. [DOI] [PubMed] [Google Scholar]
- 181.Milroy L, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Chem. Rev. 2014;114:4695–4748. doi: 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- 182.Sheng C, Dong G, Miao Z, Zhang W, Wang W. Chem. Soc. Rev. 2015;44:8238–8259. doi: 10.1039/c5cs00252d. [DOI] [PubMed] [Google Scholar]
- 183.Wilson AJ. Chem. Soc. Rev. 2009;38:3289–3300. doi: 10.1039/b807197g. [DOI] [PubMed] [Google Scholar]
- 184.Waldmann H, Karaguni I-M, Carpintero M, Gourzoulidou E, Herrmann C, Brockmann C, Oschkinat H, Müller O. Angew. Chemie Int. Ed. 2004;43:454–458. doi: 10.1002/anie.200353089. [DOI] [PubMed] [Google Scholar]
- 185.Lu S, Deng R, Jiang H, Song H, Li S, Shen Q, Huang W, Nussinov R, Yu J, Zhang J. Structure. 2015;23:1725–1734. doi: 10.1016/j.str.2015.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, Pei D. Angew. Chem. Int. Ed. Engl. 2015;54:7602–7606. doi: 10.1002/anie.201502763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Upadhyaya P, Qian Z, Habir NAA, Pei D. Tetrahedron. 2014;70:7714–7720. doi: 10.1016/j.tet.2014.05.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Spiegel J, Cromm PM, Itzen A, Goody RS, Grossmann TN, Waldmann H. Angew. Chem. Int. Ed. Engl. 2014;53:2498–2503. doi: 10.1002/anie.201308568. [DOI] [PubMed] [Google Scholar]
- 189.Wu X, Upadhyaya P, Villalona-Calero MA, Briesewitz R, Pei D. Med chem comm. 2013;4:378–382. doi: 10.1039/C2MD20329D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Trinh TB, Upadhyaya P, Qian Z, Pei D. ACS Comb. Sci. 2016;18:75–85. doi: 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Reddy MVR, Venkatapuram P, Mallireddigari MR, Pallela VR, Cosenza SC, Robell KA, Akula B, Ho BS, Reddy EP. J. Med. Chem. 2011;54:6254–6276. doi: 10.1021/jm200570p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lopez-Alcalá C, Alvarez-Moya B, Villalonga P, Calvo M, Bachs O, Agell N. J. Biol. Chem. 2008;283:10621–10631. doi: 10.1074/jbc.M706238200. [DOI] [PubMed] [Google Scholar]
- 193.Nussinov R, Muratcioglu S, Tsai C-J, Jang H, Gursoy A, Keskin O. Expert Opin. Ther. Targets. 2016 doi: 10.1517/14728222.2016.1135131. [DOI] [PubMed] [Google Scholar]
- 194.Yuan TL, Fellmann C, Lee C-S, Ritchie CD, Thapar V, Lee LC, Hsu DJ, Grace D, Carver JO, Zuber J, Luo J, McCormick F, Lowe SW. Cancer Discov. 2014;4:1182–1197. doi: 10.1158/2159-8290.CD-13-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, Bhattacharya R, Maharaj A, Azam S, Rodriguez-Aguayo C, Nagaraja AS, Morelli MP, Gharpure KM, Waugh TA, Gonzalez-Villasana V, Zand B, Dalton HJ, Kopetz S, Lopez-Berestein G, Ellis LM, Sood AK. Mol. Cancer Ther. 2014;13:2876–2885. doi: 10.1158/1535-7163.MCT-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhu H, Liang ZY, Ren XY, Li TH. Cancer Biol. Ther. 2014;5:1693–1698. doi: 10.4161/cbt.5.12.3466. [DOI] [PubMed] [Google Scholar]
- 197.Xue W, Dahlman JE, Tammela T, Khan OF, Sood S, Dave A, Cai W, Chirino LM, Yang GR, Bronson R, Crowley DG, Sahay G, Schroeder A, Langer R, Anderson DG, Jacks T. Proc. Natl. Acad. Sci. 2014;111:E3553–E3561. doi: 10.1073/pnas.1412686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lin G, Hu R, Law W-C, Chen C-K, Wang Y, Li Chin H, Nguyen QT, Lai CK, Yoon HS, Wang X, Xu G, Ye L, Cheng C, Yong K-T. Small. 2013;9:2757–2763. doi: 10.1002/smll.201201716. [DOI] [PubMed] [Google Scholar]
- 199.Zorde Khvalevsky E, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, Shemi A, Golan T, Domb AJ, Yavin E, Giladi H, Rivkin L, Simerzin A, Eliakim R, Khalaileh A, Hubert A, Lahav M, Kopelman Y, Goldin E, Dancour A, Hants Y, Arbel-Alon S, Abramovitch R, Shemi A, Galun E. Proc. Natl. Acad. Sci. U. S. A. 2013;110:20723–20728. doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bäumer S, Bäumer N, Appel N, Terheyden L, Fremerey J, Schelhaas S, Wardelmann E, Buchholz F, Berdel WE, Müller-Tidow C. Clin. Cancer Res. 2015;21:1383–1394. doi: 10.1158/1078-0432.CCR-13-2017. [DOI] [PubMed] [Google Scholar]
- 201.Coussens LM, Tinkle CL, Hanahan D, Werb Z. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 203.Gottesman MM, Fojo T, Bates SE. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 204.Borst P, Elferink RO. Annu. Rev. Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 205.Fojo T, Bates S. Oncogene. 2003;22:7512–7523. doi: 10.1038/sj.onc.1206951. [DOI] [PubMed] [Google Scholar]
- 206.Todaro M, Francipane MG, Medema JP, Stassi G. Gastroenterology. 2010;138:2151–2162. doi: 10.1053/j.gastro.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 207.Bouwman P, Jonkers J. Nat. Rev. Cancer. 2012;12:587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 208.Maier S, Dahlstroem C, Haefliger C, Plum A, Piepenbrock C. Am. J. pharmacogenomics. 2005;5:223–232. doi: 10.2165/00129785-200505040-00003. [DOI] [PubMed] [Google Scholar]
- 209.Taylor ST, Hickman JA, Dive C. J. Natl. Cancer Inst. 2000;92:18–23. doi: 10.1093/jnci/92.1.18. [DOI] [PubMed] [Google Scholar]
- 210.Swanton C. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Nat. Rev. Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 212.Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, V Sharma S, Brannigan BW, Mohapatra G, Settleman J, Haber DA. Nat. Genet. 2005;37:1315–1316. doi: 10.1038/ng1671. [DOI] [PubMed] [Google Scholar]
- 213.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 214.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Science. 2004;305:399–402. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 216.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Longley DB, Johnston PG. J. Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 218.Nussinov R, Tsai C-J, Mattos C. Trends Mol. Med. 2013;19:695–704. doi: 10.1016/j.molmed.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 219.Garraway LA, Jänne PA. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 220.Nussinov R, Tsai C-J, Jang H, Korcsmáros T, Csermely P. Semin. Cell Dev. Biol. 2016 doi: 10.1016/j.semcdb.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 221.Cope CL, Gilley R, Balmanno K, Sale MJ, Howarth KD, Hampson M, Smith PD, Guichard SM, Cook SJ. J. Cell Sci. 2014;127:788–800. doi: 10.1242/jcs.137588. [DOI] [PubMed] [Google Scholar]
- 222.Ilic N, Utermark T, Widlund HR, Roberts TM. Proc. Natl. Acad. Sci. U. S. A. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SMB. Nat. Chem. Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, Mateus C, Routier E, Kamsu-Kom N, Agoussi S, Eggermont AM, Désaubry L, Robert C, Vagner S. Nature. 2014;513:105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 225.Wei G-H, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, Yan J, Talukder S, Turunen M, Taipale M, Stunnenberg HG, Ukkonen E, Hughes TR, Bulyk ML, Taipale J. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nguyen LT, Tretiakova MS, Silvis MR, Lucas J, Klezovitch O, Coleman I, Bolouri H, Kutyavin VI, Morrissey C, True LD, Nelson PS, Vasioukhin V. Cancer Cell. 2015;27:797–808. doi: 10.1016/j.ccell.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Arqués O, Chicote I, Puig I, Tenbaum SP, Argilés G, Dienstmann R, Fernández N, Caratù G, Matito J, Silberschmidt D, Rodon J, Landolfi S, Prat A, Espín E, Charco R, Nuciforo P, Vivancos A, Shao W, Tabernero J, Palmer HG. Clin. Cancer Res. 2016;22:644–656. doi: 10.1158/1078-0432.CCR-14-3081. [DOI] [PubMed] [Google Scholar]
- 228.Mologni L, Brussolo S, Ceccon M, Gambacorti-Passerini C. PLoS One. 2012;7:e51449. doi: 10.1371/journal.pone.0051449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tenbaum SP, Ordóñez-Morán P, Puig I, Chicote I, Arqués O, Landolfi S, Fernández Y, Herance JR, Gispert JD, Mendizabal L, Aguilar S, Ramón y Cajal S, Schwartz S, Vivancos A, Espín E, Rojas S, Baselga J, Tabernero J, Muñoz A, Palmer HG. Nat. Med. 2012;18:892–901. doi: 10.1038/nm.2772. [DOI] [PubMed] [Google Scholar]
- 230.Fearon ER, Wicha MS. J. Natl. Cancer Inst. 2014;106:djt444. doi: 10.1093/jnci/djt444. [DOI] [PubMed] [Google Scholar]
- 231.Moon B-S, Jeong W-J, Park J, Il Kim T, Min DS, Choi K-Y. J. Natl. Cancer Inst. 2014;106:djt373. doi: 10.1093/jnci/djt373. [DOI] [PubMed] [Google Scholar]
- 232.Lu S, Huang W, Wang Q, Shen Q, Li S, Nussinov R, Zhang J. PLoS Comput. Biol. 2014;10:e1003831. doi: 10.1371/journal.pcbi.1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Nussinov R, Tsai C-J, Ma B. Annu. Rev. Biophys. 2013;42:169–189. doi: 10.1146/annurev-biophys-083012-130257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Aeluri M, Chamakuri S, Dasari B, Guduru SKR, Jimmidi R, Jogula S, Arya P. Chem. Rev. 2014;114:4640–4694. doi: 10.1021/cr4004049. [DOI] [PubMed] [Google Scholar]
- 235.Keskin O, Ma B, Rogale K, Gunasekaran K, Nussinov R. Phys. Biol. 2005;2:S24–S35. doi: 10.1088/1478-3975/2/2/S03. [DOI] [PubMed] [Google Scholar]
- 236.Pawson T, Nash P. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- 237.Khoo KH, Hoe KK, Verma CS, Lane DP. Nat. Rev. Drug Discov. 2014;13:217–236. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 238.Lee K-B, Hwang JM, Choi IS, Rho J, Choi J-S, Kim G-H, Il Kim S, Kim S, Lee Z-W. Angew. Chem. Int. Ed. Engl. 2011;50:1314–1317. doi: 10.1002/anie.201005333. [DOI] [PubMed] [Google Scholar]
- 239.Lee JH, Zhang Q, Jo S, Chai SC, Oh M, Im W, Lu H, Lim H. J. Am. Chem. Soc. 2011;133:676–679. doi: 10.1021/ja108230s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Salter JD, Morales GA, Smith HC. Trends Biochem. Sci. 2014;39:373–380. doi: 10.1016/j.tibs.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park C-M, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 242.Arkin MR, Tang Y, Wells JA. Chem. Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]