

Abstract
Next-generation sequencing has revolutionized cancer genetics, but accurately detecting mutations in repetitive DNA sequences, especially mononucleotide runs, remains a challenge. This is a particular concern for tumors with defective mismatch repair (MMR) that accumulate strand-slippage mutations. We developed MonoSeq to improve indel mutation detection in mononucleotide runs, and used MonoSeq to investigate strand-slippage mutations in endometrial cancers, a tumor type that has frequent loss of MMR. We performed extensive Sanger sequencing to validate both clonal and sub-clonal MonoSeq mutation calls. Eighty-one regions containing mononucleotide runs were sequenced in 542 primary endometrial cancers (223 with defective MMR). Our analyses revealed that the overall mutation rate in MMR-deficient tumors was 20–30-fold higher than in MMR normal tumors. MonoSeq analysis identified several previously unreported mutations, including a novel hotspot in an A7 run in the terminal exon of ARID5B.The ARID5B indel mutations were seen in both MMR-deficient and MMR normal tumors, suggesting biologic selection. Analysis of tumor mRNAs revealed the presence of mutant transcripts that could result in translation of neopeptides. Improved detection of mononucleotide run strand-slippage mutations has clear implications for comprehensive mutation detection in tumors with defective MMR. Indel frameshift mutations and the resultant antigenic peptides could help guide immunotherapy strategies.
Keywords: mononucleotide run, next-generation sequencing, microsatellite instability, endometrial cancer
INTRODUCTION
Accurate and comprehensive mutation detection has become increasingly important in cancer therapeutics with the growing emphasis on personalized medicine. Enhancing the ability to detect mutations present in previously “unanalyzable” genomic sequences could improve therapy, either through discovery of new pathways or molecules for targeted agents, or by identification of novel neoantigens for cancer vaccines. DNA mismatch repair (MMR)-defective tumors in particular have shown promising responses to immunotherapies predicated on immune cell recognition of tumor neoantigens (Le, et al., 2015), and response to immune-based therapies has been directly correlated with neoantigen load (Rizvi, et al., 2015; Snyder, et al., 2014).
Massively parallel deep sequencing has dramatically improved the ability to detect mutations. Sequencing low complexity DNA, however, remains a challenge (Bragg, et al., 2013; Huse, et al., 2007; Minoche, et al., 2011). Pyrosequencing (454 Life Sciences) and semiconductor sequencing (Ion Torrent) approaches have well-documented limitations for mononucleotide run analysis, attributable to their respective sequencing chemistries (Balzer, et al., 2011; Van den Hoecke, et al., 2015). Reversible terminator sequencing (RTS, used by Illumina platforms) in principle reduces homopolymer sequencing errors by the addition of only a single base at a time. However, in practice accurate sequencing of longer homopolymers using RTS remains problematic (Erlich, et al., 2008). In fact, the Illumina MiSeq Reporter™ default variant caller filters any variants occurring in mononucleotide runs >8bp, thereby excluding a large number of loci. The inability to reliably analyze mononucleotide repeats limits our understanding of the mutational burden in the ~15% of colorectal cancer, ~30% of endometrial cancer (EC), and other tumors types with defective MMR. Recent estimates of strand-slippage mutations associated with defective MMR indicate that mononucleotides are far more mutable than di- and tri-nucleotide repeats (Kim, et al., 2013). Given that mononucleotides are the most frequently mutated class of repeats and that reliable detection of mutations in mononucleotide runs is suboptimal, it is logical to assume the mutational burden in tumors with defective MMR has been underestimated.
We undertook studies to investigate and improve methods of sequencing mononucleotide runs. Here we show MiSeq®-based RTS has a highly consistent and quantifiable homopolymer sequencing error rate. We designed our MonoSeq variant caller to account for this intrinsic error rate, and validated its performance by comparison to Sanger sequencing. Our preliminary estimates from sequencing 223 ECs with defective MMR as indicated by microsatellite instability (MSI) provide evidence that a large number of strand-slippage mutations in this tumor type have not yet been discovered. Using MonoSeq we were able to identify a novel hotspot mutation in ARID5B (MIM# 608538) that is highly likely to be under cancer specific selection.
MATERIALS AND METHODS
DNA samples
The normal germline DNA samples used in this study were provided with informed consent (Washington University Human Research Protection Office and The Ohio State University IRB protocols 201106361 [legacy conversion 04–1009] and 2012C0097, respectively). The EC patient population and tumor specimens investigated have been previously described (Billingsley, et al., 2015; Walker, et al., 2015; Zighelboim, et al., 2007; Zighelboim, et al., 2014). All 542 subjects studied were treated at Washington University School of Medicine, St. Louis, MO, and this study is covered under the Washington University Human Research Protection Office (IRB protocols 91–507 and 93–0828) and The Ohio State University IRB protocol 2012C0116. All tumor DNAs were prepared from high neoplastic cellularity (>66%) flash-frozen tissues. Results for tumor microsatellite instability (MSI) testing were previously reported (Novetsky, et al., 2013; Zighelboim, et al., 2007). Nine MSI-low tumors were included with the MSS group.
Targeted deep sequencing
Targeted deep sequencing was performed using TruSeq Custom Amplicon Assays (Illumina, San Diego, CA). The germline panel consisted of 456 amplicons targeting approximately 162kb (average 355bp/amplicon), and contained 80 homopolymers 7–10bp long. The EC panel contained 521 amplicons targeting approximately 217kb (average 417bp/amplicon), with 178 homopolymers 7–11bp long. Nineteen amplicons targeted the coding exons of ARID5B. Libraries were prepared separately for 798 germline DNAs and 542 EEC DNAs, using TruSeq Custom Amplicon Kits v1.5 (Illumina, San Diego, CA). Library pools were made using equimolar amounts of bar-coded and amplified DNA from 71 patient samples per pool. Multiplexed sequencing reactions were performed with 250-base paired-end reads on an Illumina MiSeq®, using MiSeq reagent kits v2 (Illumina San Diego, CA). Variants were called using VarScan v2.3.7 (Koboldt, et al., 2009), and compiled using MuCor software (Kroll, et al., 2016). All variants detected in this paper were submitted to COSMIC public database (http://cancer.sanger.ac.uk/cosmic) and variants are reported using HGVS guidelines (http://www.hgvs.org/varnomen).
Sanger sequencing
Sanger sequencing was performed using 14 different sets of homopolymer-spanning primers to amplify DNA with Phusion high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA). PCR products were treated with ExoSAP-IT (Affymetrix, Santa Clara, CA) and sequenced with ABI Prism BigDye Terminator Cycle Sequencing Kit version 3.1 (Applied Biosystems, Waltham, MA) in the Genomics Shared Resource at the Ohio State University. Mutant peak height in chromatograms was quantified using Mutation Surveyor software (Softgenetics, State College, PA).
Read quantification
To quantify the homopolymer length in each read at each locus, a custom hidden Markov model allowing up to 300bp upstream flanking sequence, an arbitrary number of the repeated nucleotide, and up to 300bp downstream flanking sequence was constructed for every locus. All reads were matched to these models over their entire length and accepted as relevant to a locus if they contained at most one mismatch in each of the 5bp regions immediately flanking the homopolymer run and at most 10 mismatches total (ignoring bases with quality scores <20). The length of the homopolymer in each accepted read was recorded directly from the resulting alignment.
Allelic distributions in non-mutant samples
The homopolymer length data from all germline samples were pooled for all monomorphic 7–10bp homopolymer loci to obtain apparent allelic distributions at each locus. Because the germline panel did not target any monomorphic 11N homopolymers, five monomorphic 11N homopolymers from MSS EEC samples were treated in the same way to obtain the apparent allelic distribution in 11N homopolymers. Finally, the data for all loci of the same length k were pooled to obtain the reference apparent allele distributions Pi(k) that quantify the probability to see a homopolymer of length i at a locus with true alleles of length k.
MonoSeq maximum likelihood model
MonoSeq variant caller is available at https://github.com/rbundschuh/MonoSeq/. For a given locus we define vk to be the (unknown) true fraction of alleles with k-mers. Then, the probability distribution at the locus can be calculated by
If we observe N reads at such a locus, they should be distributed according to a multinomial distribution, i.e.,
The log-likelihood of this is
The first term of this does not depend on the unknown parameters vk and can thus be dropped, which leaves
to be minimized over the vk. We then minimize this numerically over all pairs of non-zero vk that add up to one, i.e., under the assumption that at most two alleles coexist. The VAF is then 1-νn where n is the wild type length of the homopolymer locus. When applying the model, we selected the 81 loci for analysis based on having only a single homopolymer in the amplicon.
RESULTS
MonoSeq: a variant caller that accounts for RTS homopolymer errors
To explore the homopolymer error rate in RTS, we analyzed data from two different sequencing experiments focusing on monomorphic (not polymorphic in our study populations) mononucleotide repeats of 7–10bp: 798 normal DNA samples and 317 microsatellite stable (MSS) endometrioid endometrial cancers (EECs) tested were expected to have very few if any strand-slippage mutations. Large numbers of variants were, however, detected by VarScan variant caller in both sample sets, reflecting the high false positive call rate in mononucleotide runs (Fig. 1A). A small number of variant calls in the MSS EEC samples had much higher variant allele fractions (VAFs) and we confirmed that some of these were true positives (Fig. 1A). When variant calls were stratified by reference allele homopolymer length, we observed a step-wise increase in false positive call VAF, which was remarkably similar in the two data sets (Fig. 1A). This suggests that for a given repeat length the intrinsic homopolymer error rate is nearly constant. The error rate was indeed shown to be constant by examining the erroneous reads in every locus (Fig. 1B and Supp. Table S1). For example, for 8N homopolymers, 94% of reads contained the 8bp run, whereas 4% and 2% of reads contained an erroneous 7N or 9N homopolymer, respectively (Fig. 1B, second row, and Supp. Table S1).
Figure 1. MiSeq®-based amplicon sequencing has an intrinsic mononucleotide run indel error rate.
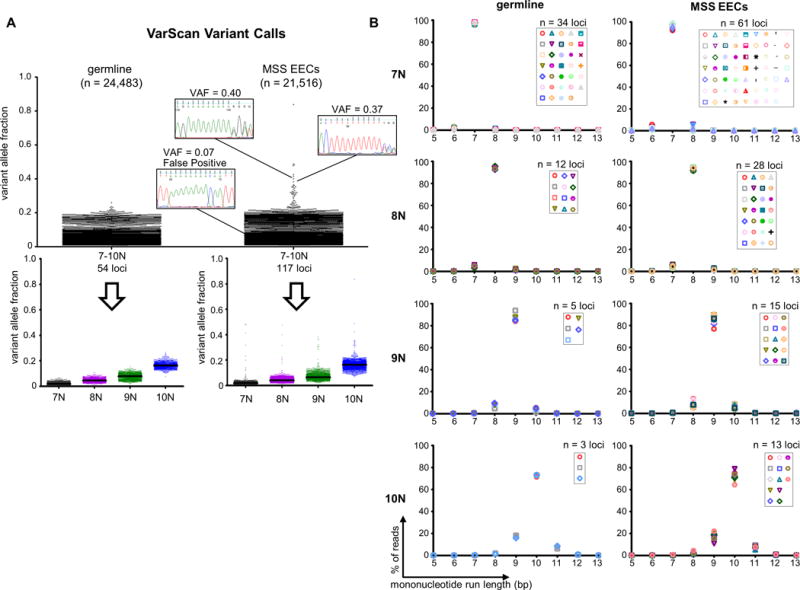
(A) Dot plots show the VarScan variant allele fractions (VAFs) for mononucleotide run indel calls in germline DNA samples and microsatellite stable endometrioid endometrial cancers (MSS EECs). Total number of calls (n) is given in parenthesis. Sanger sequencing using high fidelity Phusion polymerase for representative true and false positive calls is shown. Because the mononucleotide repeats analyzed are monomorphic, the vast majority of variants are presumed to be false positives. Bottom panels show the calls stratified by reference sequence mononucleotide run length, and illustrate increasing VAF (sequencing noise) with increasing run length. Black lines indicate medians. (B) Distributions for the repeat length for the combined reads from all germline or MSS EECs samples. Each marker represents a different repeat. Graphs illustrate that allele distributions are similar for mononucleotide runs of the same length. See also Supp. Table S1.
We devised a variant caller that accounts for the RTS homopolymer error rate by incorporating the homopolymer allelic distributions from the germline DNA samples (Fig. 1B and Supp. Table S1) into a maximum likelihood model (see ‘Materials and Methods’ section). We used our caller, MonoSeq, to identify mononucleotide run strand-slippage mutations (indel variants) in a series of MSI+ EECs (n=223), and similarly applied MonoSeq to the 317 MSS EECs investigated. To test MonoSeq’s accuracy we performed a total of 458 Sanger sequencing validations for 14 different loci, using strand-slippage mutation calls with a wide range of VAFs. MonoSeq’s variant calls were highly concordant with Sanger sequencing traces (Fig. 2). All putative clonal mutations (VAF > 0.30) tested were validated, and the MonoSeq VAFs showed a strong correlation with the minor peak heights of the Sanger traces. MonoSeq correctly assigned very low VAFs to wild-type samples (no evidence of mutation in Sanger traces) whereas the corresponding VarScan calls had comparatively higher VAFs, indicating that MonoSeq filtered false positive reads and improved the ability to detect sub-clonal mutations, especially in longer homopolymers (Fig. 2).
Figure 2. Comparison of MonoSeq and VarScan variant calls relative to Sanger sequencing validation.
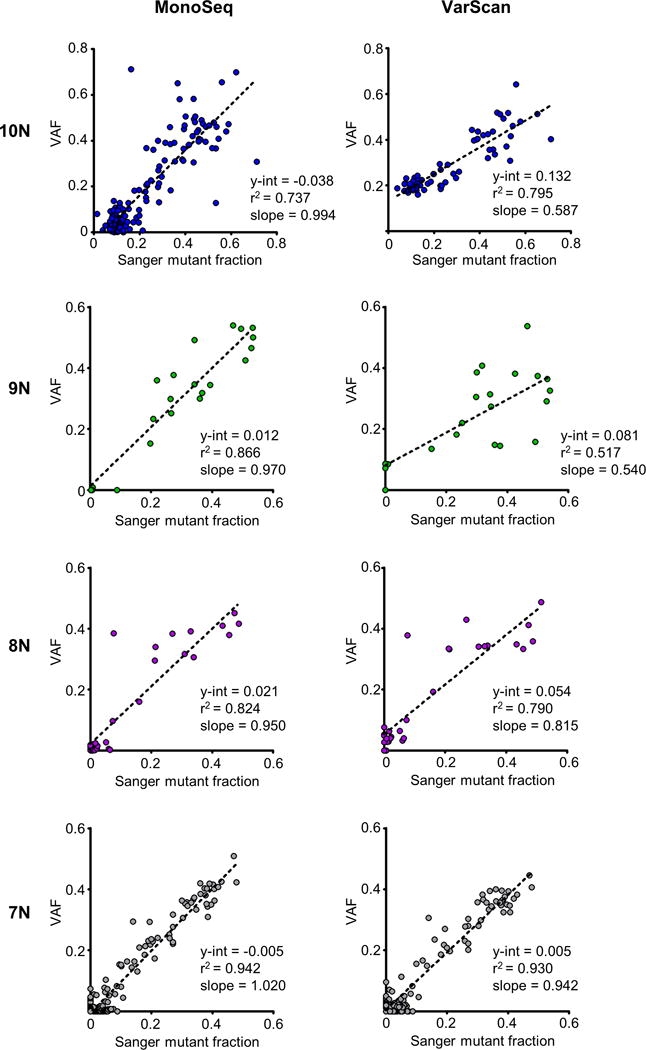
Sanger sequencing validation was performed for 458 total indel calls in 14 different mononucleotide runs. Plots show correlation between mutant peak heights in Sanger traces and variant allele fractions (VAFs) from MonoSeq (left) and VarScan VAFs (right). The y-intercept is a measurement of the false positive next-generation sequencing reads in samples with no detectable mutation by Sanger sequencing. MonoSeq corrects for false positive reads as indicated by a reduction of the y-intercept in linear regression lines and linear regression line slopes approximately equal to one. Sanger mutant fraction was determined by Mutation Surveyor software (Softgenetics, State College, PA).
MSI+ tumors have a 20–30 fold increase in mononucleotide run strand-slippage mutations
To gain insight into the relative increase in strand-slippage mutations in MSI+ compared to MSS tumors, we examined the somatic mutations detected by MonoSeq in 71 well-covered (>100x) non-coding homopolymers. A total of 586 mutations were detected in the 223 MSI+ samples (average 2.63 mutations/sample), and 40 mutations in the 317 MSS samples (average 0.125 mutations/sample) (Fig. 3A). The mutations detected had a range of VAFs indicative of both clonal and sub-clonal mutations. There was a non-linear increase in the mutation rate with increasing mononucleotide run length in both the MSS and MSI+ samples (Fig. 3B). Of note, the increase in mutations in the MSI+ samples was approximately 20–30-fold for all length mononucleotide runs examined (Fig. 3C).
Figure 3. Estimation of the increased mononucleotide repeat mutational load in tumors with microsatellite instability (MSI).
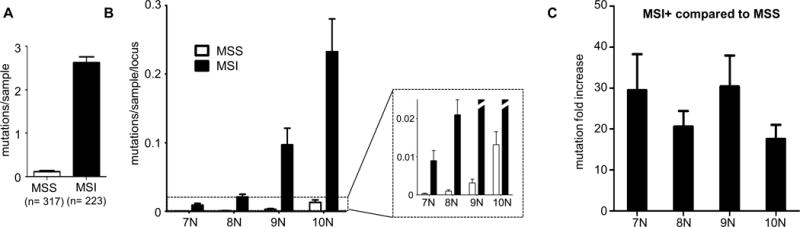
(A) Average mutations per tumor sample in 71 non-coding mononucleotide runs for microsatellite stable (MSS) and MSI+ EECs. Error bars are s.e.m. (B) Distribution of non-coding mutations normalized to the number of samples and number of mononucleotide repeat loci sequenced shows an increase in mutability with increasing mononucleotide run length. Error bars are s.e.m. (C) Fold increase in non-coding strand-slippage mutations for MSI+ compared to MSS tumors. Error bars are s.e.m.
A novel hotspot strand-slippage mutation in ARID5B
Our target panel included 10 mononucleotide runs in the coding sequences of six different cancer-relevant genes, five of which were identified by The Cancer Genome Atlas (TCGA) as significantly mutated genes in EEC (The Cancer Genome Atlas Research Network, 2013). Three of these genes contain mononucleotide runs that were previously reported to harbor strand-slippage mutations in EECs (CASP5 [MIM# 602665] (Vassileva, et al., 2004), CTCF [MIM# 604167] (Zighelboim, et al., 2014) and ZFHX3 [MIM# 104155] (Walker, et al., 2015)), whereas the other three (ARID5B, LIMCH1 and CSMD3 [MIM# 608399]) contain homopolymers not reported to be mutated. We used MonoSeq to test for repeat mutations in the 540 EEC specimens at these loci, and found strand-slippage mutations in 9 of the 10 coding sequence mononucleotide runs (five of the six genes) (Table 1).
TABLE 1.
Coding homopolymer indels stratified by MSI status
| gene | TCGA for EC SMG status | Mononucleotide Run | variant | no. observations | ||
|---|---|---|---|---|---|---|
| protein | transcript | MSI+MSS | ||||
| ARID5B | MSI+ tumors | A7 | p.I497* | NM_032199.1:c.1489delA | 10 (4%) | 1 (0.3%) |
| p.I497fs*31 | NM_032199.1:c.1489dupA | 6 (3%) | 5 (2%) | |||
|
| ||||||
| CASP5a | no | A10 | p.R23fs*21 | NM_001136112.1:c.67delA | 80 (36%) | 3 (1%) |
| A10 | p.T81fs*26 | NM_001136112.1:c.241delA | 46 (21%) | 1(0.3%)b | ||
| p.T81fs*3 | NM_001136112.1:c.241dupA | 11 (5%) | 5 (2%) | |||
| p.T81fs*27 | NM_001136112.1:c.240_241dupAA | 1 (0.4%) | 1 (0.3%)b | |||
| A8 | p.N191fs*8 | NM_001136112.1:c.572delA | 4 (2%) | 0 | ||
|
| ||||||
| CSMD3 | MSS tumors | A7 | p.E1051fs*14 | NM_198123.1:c.3151dupA | 1 (0.4%) | 0 |
| T7 | p.F3640fs*61 | NM_198123.1:c.10920delT | 7 (3%) | 0 | ||
|
| ||||||
| CTCF | MSI+ and MSS tumors | A7 | p.T204fs*26 | NM_006565.3:c.610dupA | 50 (22%) | 1 (0.3%) |
| p.T204fs*18 | NM_006565.3:c.610delA | 16 (7%) | 0 | |||
| p.T204fs*19 | NM_006565.3:c.609_610dupAA | 1 (0.4%) | 0 | |||
| A7 | p.E691fs*30 | NM_006565.3:c.2070delA | 2 (1%) | 0 | ||
|
| ||||||
| LIMCH1 | MSI+ tumors | A7 | n/a | n/a | 0 | 0 |
|
| ||||||
| ZFHX3 | MSI+ tumors | G7 | p.E763fs*61 | NM_006885.3:c.2287delG | 6 (3%) | 0 |
| p.E763fs*26 | NM_006885.3:c.2287dupG | 2 (1%) | 0 | |||
A10 repeats have VAF cutoff of >.15, which may exclude mutations present in small subclones
sample MSS1946 harbors both NM_001136112.1:c.240_241dupAA and NM_001136112.1:c.240_241delA, with both variants present in germline DNA
MSI: microsatellite instability; TCGA for EC: The Cancer Genome Atlas for endometrial cancer; SMG: significantly mutated gene
All variants detected in this paper were submitted to COSMIC public database (http://cancer.sanger.ac.uk/cosmic).
An A7 repeat in the terminal exon of ARID5B was mutated in 7.2% of MSI+ samples, and was also mutated in a smaller fraction of MSS tumors (1.9%), which might indicate it is under biologic selection (Table 1). Both insertion and deletion mutations were seen. Single base insertions were observed in 11 tumors (6 MSI+ and 5 MSS), and another 11 tumors harbored single base deletions (10 MSI+ and one MSS). Both clonal and sub-clonal mutations were detected, and MonoSeq was able to identify sub-clonal mutations that were almost undetectable by Sanger sequencing (Fig. 4A). A single-base insertion with VAF = 0.103 was detected in tumor sample MSI-1712T by both Sanger sequencing and MonoSeq (Fig. 4A). To confirm that the minor peaks present in the Sanger sequencing chromatogram are indicative of a true sub-clonal mutation rather than sequencing noise, we re-amplified the region from both tumor and paired normal DNA from patient 1712, then TA cloned the PCR products. One of the 19 clones tested from the tumor sample had the insertion mutation (consistent with the predicted variant fraction), whereas none of the 20 clones from the normal DNA had mutations (Fig. 4A). Other Sanger sequencing validations highlighted the correlation between the minor peak height of Sanger trances and MonoSeq VAF (Fig. 4A).
Figure 4. A novel mutational hotspot in ARID5B.
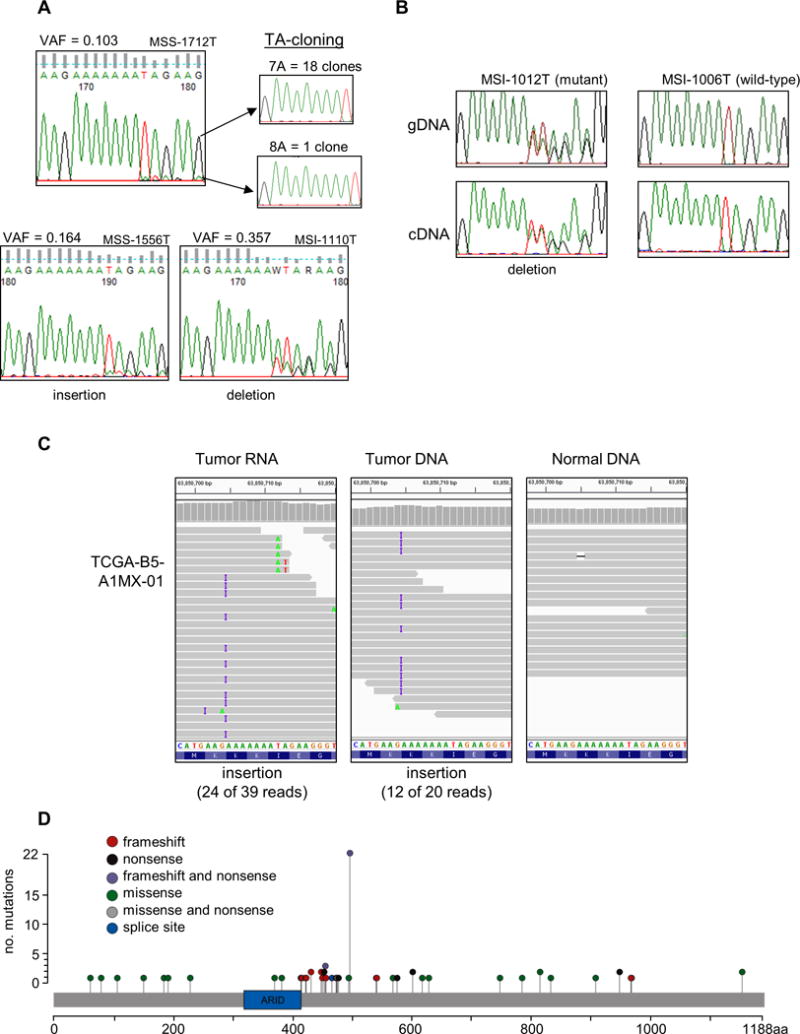
(A) Sanger sequencing chromatograms validate sub-clonal mutations called using MonoSeq. The MonoSeq variant allele fraction (VAF) parallels Sanger sequencing minor peak height. Inserts are chromatograms of cloned and sequenced PCR products from sample MSS-1712T, which confirms the tumor harbors a sub-clonal mutation. (B) Transcripts harboring the frameshift mutation were detectable in cDNA from sample MSI-1012T. Chromatogram for sample MSI-1006T is shown as a representative wild-type. (C) The ARID5B hotspot mutation is in TCGA-B5-A1MX-01. Twenty-four of 39 RNAseq reads displayed the insertion, and 12 of 20 DNA reads had the same mutation. In normal DNA from the same patient, only one of 18 reads displayed an A track mutation, which we attribute to sequencing error. (D) All ARID5B coding exons were sequenced for 540 endometrioid endometrial cancer specimens. Lollipop plot shows 72 mutational events (47 different mutations in 67 samples). Variants were called using VarScan and MonoSeq. ARID domain is represented by green box. Data displayed using Integrated Genomics Viewer software (Broad Institute, Cambridge, MA). All variants detected in this paper were submitted to COSMIC public database (http://cancer.sanger.ac.uk/cosmic).
Because these ARID5B strand-slippage mutations occur in the terminal exon, nonsense mediated decay (NMD) of the mutant transcripts would not be expected. Tumor RNA was available for three samples carrying the ARID5B mutation. RT-PCR and Sanger sequencing revealed that mutated transcripts were present in two of the three tumors (Fig. 4B). We further validated the existence of the ARID5B hotspot mutations and escape from NMD by using RNAseq and exome sequencing data from TCGA for EC (The Cancer Genome Atlas Research Network, 2013). RNAseq data for 546 endometrial cancers was downloaded from Cancer Genomics Hub (Kent, et al., 2002) to screen for the hotspot mutation at the transcript level. Although most of the ARID5B transcript had high read counts indicating it was expressed in many tumors, the ~200bp region containing the A7 mononucleotide run was poorly covered by RNAseq, with only 17 tumors sequenced to a depth ≥15X. However, one of these 17 tumors, TCGA-B5-A1MX-01 (an MSI+ endometrioid tumor), clearly harbors a single base insertion at this hotspot, present in 62% of RNAseq reads (Fig. 4C). A single base insertion was present in 60% of reads from the tumor DNA, whereas the paired normal DNA did not harbor the insertion, consistent with somatic origin (Fig. 3C). This mutation was not reported by TCGA, which emphasizes the need for robust mononucleotide strand-slippage repeat variant calling (Cerami, et al., 2012; Gao, et al., 2013).
To better understand how frequent the ARID5B indel mutation is relative to other mutations, we sequenced all ARID5B coding exons in the 540 EEC tumors samples. A total of 47 different mutations occurred in 69 samples, with five samples that harbored two mutations each (Fig. 4D). Most mutations were truncating—28% were nonsense, 35% were frameshifts and 1% were splice site mutations. There was a cluster of frameshift mutations found in a 124bp stretch in exon 9 consisting of 12 samples that harbored 9 different frameshifts, suggesting a possible fragile site. Expectedly, the overall mutation rate was higher in MSI+ tumors (19.3%) compared to MSS (8.2%). The mutational spectrum of ARID5B indicates clearly that there is selection for genetic inactivation in endometrial cancers and that the A7 indel mutations are among the most common seen in MSI+ endometrial cancers.
Enhanced mutation detection in previously characterized MSI targets
Our custom amplicon panel included two A10 repeats in the CASP5 coding sequence (exons 2 and 3) and an A8 repeat in exon 4. Mutation of the exon 3 A10 homopolymer in EC was first reported more than a decade ago (Vassileva, et al., 2004). Using MonoSeq we revisited this locus and found that 26% of MSI+ tumors had mutations, compared to only a 5% mutation rate described in the original report (Vassileva, et al., 2004) (Table 1). We speculate this difference is due to MonoSeq’s improved detection of sub-clonal mutations (VAFs < 0.30), and our much larger sample size. The mutation rates of the exon 2 A10 repeat and exon 4 A8 repeat were 36% and 2%, respectively, with 52% of MSI+ tumors harboring at least one frameshift mutation in a CASP5 coding sequence mononucleotide run (Table 1). There was no evidence for mutual exclusivity or co-occurrence of mutations at multiple CASP5 mononucleotide runs (P-value > 0.1 determined by Fisher’s exact test). There were 9 MSS tumors with indel variants in either the exon 2 or exon 3 A10 tracks (Table 1). Sanger sequencing using matched normal DNA proved that these patients harbored germline single base insertions, and one had a germline two base insertion as well as a germline one base deletion in trans. The discovery of the compound heterozygote (no functional copy of CASP5 in the patient’s germline DNA) speaks to the redundancy of the caspase pathway, and suggests selection for CASP5 mutation in endometrial cancers is unlikely.
We used MonoSeq to re-assess mutations in a CTCF A7 hotspot we identified in a previous Sanger-based sequencing study using this EC cohort (Zighelboim, et al., 2014). MonoSeq detected sub-clonal mutations that were not evident in our previous study. In addition to the ~20% of MSI+ tumors that contained fully clonal mutations, we also identified ~10% of MSI+ tumors with sub-clonal indel mutations, many of which we attributed to low level sequencing noise in our initial report (Supp. Fig. S1A). Our original report estimated that this hotspot mutation occurs in 25% of MSI+ tumors, but we now estimate 29% of MSI+ tumors are mutated (Zighelboim, et al., 2014) (Supp. Fig. S1B). We also found a single instance of a sub-clonal mutation in an MSI-low tumor (sample MSI-L1378T), which we confirmed via Sanger sequencing with proofreading polymerase (Supp. Fig. S1B), furthering supporting the selection for mutation of this locus in tumors with defective MMR.
DISCUSSION
Although there have been significant advances in improving the accuracy of sequencing repeats, including di- and tri-nucleotides useful for genotyping and tumor MSI typing (Albers, et al., 2011; Cantarella and D’Agostino, 2015; Gan, et al., 2015; Gymrek, et al., 2012; Highnam, et al., 2013; Narzisi, et al., 2014; Salipante, et al., 2014), MonoSeq specifically addresses the need for improved sequencing of the lowest complexity DNA sequences (mononucleotide runs). Although MonoSeq was trained on the Illumina MiSeq®’s intrinsic homopolymer error rate (Fig. 1), it can easily be adapted to other RTS sequencing platforms (e.g. Illumina HiSeq® and NextSeq®), and potentially for platforms with other sequencing chemistries.
Genes preferentially mutated in MSI+ tumors and specifically those known to be important in tumor biology (i.e. selected mutations) were first identified in colorectal cancers more than two decades ago (Duval and Hamelin, 2002; Markowitz, et al., 1995; Rampino, et al., 1997; Salahshor, et al., 1999). Although defective MMR is more common in endometrial that colorectal cancers, the spectrum of MSI mutations and genes for which there is strong selection for mutation is less well studied, with only a few confirmed MSI targets (Bertoni, et al., 1999; Giannakis, et al., 2014; Novetsky, et al., 2013; Schwartz, et al., 1999; Vassileva, et al., 2002; Zighelboim, et al., 2014). A recent reanalysis of TCGA exome sequencing data for colorectal and endometrial cancers with MMR deficiency highlighted the fact that many mutations associated with MSI have not been fully appreciated, that they are difficult to detect, and that different genes and repeats are mutated in the two tumor types (Kim, et al., 2013). Our work to develop MonoSeq focused on endometrial cancers and primarily on noncoding repeats that are unlikely to be subject to strong selection, but we did evaluate a small number of coding sequence mononucleotide repeats and discovered multiple novel non-synonymous mutations in EECs (Table 1).
We identified a novel hotspot in ARID5B that is mutated at modest frequency in both MSI+ and MSS tumors (4% overall). Although ARID5B was reported as a “significantly mutated gene” by TCGA, and the spectrum of mutations seen in the EECs was similar to that seen by TCGA, the A7 repeat mutations made up a third of all ARID5B mutations identified using custom amplicon sequencing combined with MonoSeq. Furthermore, Kim et al. did not report on the A7 mutation, suggesting that the lower coverage for the exome capture sequencing used by TCGA many not be sufficient to reliably reveal some mononucleotide repeat variants (Kim, et al., 2013). The ARID5B frameshift we identified could be one example of a potentially large number of mutations that give rise to novel peptides because the mutation is in the last exon and as such is not subject to NMD (Nagy and Maquat, 1998). Although we were able to show the frameshift mutation was present in mRNA from the tumors we studied and a TCGA specimen, we were unable to test for the truncated frameshift ARID5B protein in our primary endometrial cancers due to lack of a suitable antibody.
MonoSeq combined with other sequencing efforts may afford opportunities for comprehensive/genome-wide discovery of other potential neoantigen-producing mutations. There are over 4,000 coding mononucleotide repeats ≥ 7bp in the human exome, and for each cancer and cell type a subset of these will be mutated and expressed. Frameshift peptides may prove to be important in designing personalized immunotherapies or in predicting response to therapies based on immune checkpoint blockade. To date, most cancer peptide therapies have primarily targeted tumor associated antigens or neoantigens derived from missense mutations (Baurain, et al., 2000; Hacohen, et al., 2013; Mandelboim, et al., 1994; Schumacher and Schreiber, 2015). When combined with HLA presentation algorithms (Karosiene, et al., 2012; Lundegaard, et al., 2008; Nielsen, et al., 2003; Peters and Sette, 2005; Sidney, et al., 2008) computational tools such as MonoSeq could uncover mutations suitable for directing immunotherapies that would otherwise be undetectable. In a recent study testing the effectiveness of a vaccine created from 11 different peptides, patients who showed an immune response to more than one peptide had better overall response rates, but only 8 of 27 evaluable patients achieved this outcome (Walter, et al., 2012). This finding illustrates how challenging designing multiple peptide vaccines can be due to the lack of common neoantigen-producing mutations between even highly mutated tumors (Karasaki, et al., 2015; Schumacher and Schreiber, 2015). Comprehensive identification of all mutations (including mononucleotide run indels) present in a large fraction of tumors will afford the maximum power to discover the best candidates for vaccination design.
PD-L1/PD-1 immune checkpoint inhibitors have emerged as a particularly promising approach for the treatment of MSI+ and other highly mutated tumor types tumors (Hamid, et al., 2013; Rizvi, et al., 2015; Van den Hoecke, et al., 2015). There are data supporting increased effectiveness for immune checkpoint inhibitors in tumors with higher mutational burdens, and neoantigen load (Rizvi, et al., 2015; Snyder, et al., 2014), however specific mutations contributing to response have not been elucidated. Better sequencing data might serve to identify predictive markers for MSI+ tumors and potentially define groups of MSS patients that might also respond based on shared mutations.
At present, detection of sub-clonal mutations in repetitive regions remains a barrier to reaching the objective of comprehensive mutation analysis, especially for those efforts focused on tumors with defective MMR and other highly mutable cancers. Although the mutational landscapes of endometrial and colorectal cancers described by TCGA excluded mutation calls in most homopolymers >7bp (The Cancer Genome Atlas Research Network, 2013; The Cancer Genome Atlas Research Network, 2012), further analysis of the sequencing data has pointed to many additional frameshift mutations (Kim, et al., 2013). We believe that the methods reported here afford greater sensitivity for detection of clonal and sub-clonal mutations in repeats and that a subset of these will contribute to tumor phenotypes. Moreover, the ability to detect sub-clonal mutations is critical for inferring information about tumor evolution, and is often necessary for separating driver and passenger mutations, and for identifying therapy resistant clones.
Other groups have estimated the increased mutational burden in MSI+ tumors both using global mutational analysis and by determining the relative mutability of selected MSI targets (Ji and King, 2001; The Cancer Genome Atlas Research Network, 2013). In our analysis of noncoding repeats (intronic, intergenic and UTR) we saw a 20–30-fold increase in homopolymer indel mutations (Figure 3C). We also observed a relative increase in the mutability of MSS samples with increasing homopolymer length (Figure 3B), and a constant fold change in the number of mutations between MSI+ and MSS tumors regardless of homopolymer length (Figure 3C).
Approaches such as the one we described here will afford opportunities to better characterize the overall mutational burden in tumors with defective MMR and may point to novel genes important in tumorigenesis. We acknowledge the fact that multiple analysis methods are required for mutation profiling efforts, with MSI+ tumors presenting particular challenges both in terms of the numbers and classes of mutations they carry. The ability to detect all classes of somatic mutations and sub-clonal mutations has important implications for precision medicine approaches to genetically targeted cancer treatments.
Supplementary Material
Acknowledgments
We would like to thank Alexis Chassen for manuscript editing and data curation. We would like to acknowledge Pearlly Yan and the Ohio State University Genomics Core Facility, and Tea Meulia and the Ohio State University Molecular and Cellular Imagine Center, a CFAES/OARDX core facility, in Wooster, OH. We would like to acknowledge the Ohio Supercomputer Center, which was used for data processing. We are very grateful to all of the patients who contributed specimens to this study and all of the attending physicians and staff at the Washington University School of Medicine Division of Gynecologic Oncology, and The Ohio State University College of Medicine Division of Gynecologic Oncology. This work was supported by The National Institutes of Health (R21 CA155674 and R01 CA151853 to P.J.G.), the Pelotonia Fellowship Program (C.J.W., C.L.M. and A.K.E.), and the National Cancer Institute (P30 CA016058 to the Genomics and Biostatistics shared resources at the Ohio State University Comprehensive Cancer Center).
Footnotes
All authors declare no conflict of interest.
References
- Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21(6):961–73. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzer S, Malde K, Jonassen I. Systematic exploration of error sources in pyrosequencing flowgram data. Bioinformatics. 2011;27(13):i304–9. doi: 10.1093/bioinformatics/btr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baurain JF, Colau D, van Baren N, Landry C, Martelange V, Vikkula M, Boon T, Coulie PG. High frequency of autologous anti-melanoma CTL directed against an antigen generated by a point mutation in a new helicase gene. J Immunol. 2000;164(11):6057–66. doi: 10.4049/jimmunol.164.11.6057. [DOI] [PubMed] [Google Scholar]
- Bertoni F, Codegoni AM, Furlan D, Tibiletti MG, Capella C, Broggini M. CHK1 frameshift mutations in genetically unstable colorectal and endometrial cancers. Genes Chromosomes Cancer. 1999;26(2):176–80. [PubMed] [Google Scholar]
- Billingsley CC, Cohn DE, Mutch DG, Stephens JA, Suarez AA, Goodfellow PJ. Polymerase varepsilon (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer. 2015;121(3):386–94. doi: 10.1002/cncr.29046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg LM, Stone G, Butler MK, Hugenholtz P, Tyson GW. Shining a light on dark sequencing: characterising errors in Ion Torrent PGM data. PLoS Comput Biol. 2013;9(4):e1003031. doi: 10.1371/journal.pcbi.1003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarella C, D’Agostino N. PSR: polymorphic SSR retrieval. BMC Res Notes. 2015;8:525. doi: 10.1186/s13104-015-1474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62(9):2447–54. [PubMed] [Google Scholar]
- Erlich Y, Mitra PP, delaBastide M, McCombie WR, Hannon GJ. Alta-Cyclic: a self-optimizing base caller for next-generation sequencing. Nat Methods. 2008;5(8):679–82. doi: 10.1038/nmeth.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan C, Love C, Beshay V, Macrae F, Fox S, Waring P, Taylor G. Applicability of next generation sequencing technology in microsatellite instability testing. Genes (Basel) 2015;6(1):46–59. doi: 10.3390/genes6010046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46(12):1264–6. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymrek M, Golan D, Rosset S, Erlich Y. lobSTR: A short tandem repeat profiler for personal genomes. Genome Res. 2012;22(6):1154–62. doi: 10.1101/gr.135780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1(1):11–5. doi: 10.1158/2326-6066.CIR-13-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highnam G, Franck C, Martin A, Stephens C, Puthige A, Mittelman D. Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res. 2013;41(1):e32. doi: 10.1093/nar/gks981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8(7):R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji HP, King MC. A functional assay for mutations in tumor suppressor genes caused by mismatch repair deficiency. Hum Mol Genet. 2001;10(24):2737–43. doi: 10.1093/hmg/10.24.2737. [DOI] [PubMed] [Google Scholar]
- Karasaki T, Nagayama K, Kawashima M, Hiyama N, Murayama T, Kuwano H, Nitadori JI, Anraku M, Sato M, Miyai M, et al. Identification of Individual Cancer-Specific Somatic Mutations for Neoantigen-Based Immunotherapy of Lung Cancer. J Thorac Oncol. 2015 doi: 10.1016/j.jtho.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64(3):177–86. doi: 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Laird PW, Park PJ. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell. 2013;155(4):858–68. doi: 10.1016/j.cell.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–5. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll KW, Eisfeld AK, Lozanski G, Bloomfield CD, Byrd JC, Blachly JS. MuCor: Mutation Aggregation and Correlation. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–12. doi: 10.1093/nar/gkn202. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelboim O, Berke G, Fridkin M, Feldman M, Eisenstein M, Eisenbach L. CTL induction by a tumour-associated antigen octapeptide derived from a murine lung carcinoma. Nature. 1994;369(6475):67–71. doi: 10.1038/369067a0. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268(5215):1336–8. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- Minoche AE, Dohm JC, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011;12(11):R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23(6):198–9. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- Narzisi G, O’Rawe JA, Iossifov I, Fang H, Lee YH, Wang Z, Wu Y, Lyon GJ, Wigler M, Schatz MC. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods. 2014;11(10):1033–6. doi: 10.1038/nmeth.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–17. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novetsky AP, Zighelboim I, Thompson DM, Jr, Powell MA, Mutch DG, Goodfellow PJ. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol Oncol. 2013;128(3):470–4. doi: 10.1016/j.ygyno.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275(5302):967–9. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahshor S, Kressner U, Pahlman L, Glimelius B, Lindmark G, Lindblom A. Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosomes Cancer. 1999;26(3):247–52. [PubMed] [Google Scholar]
- Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014;60(9):1192–9. doi: 10.1373/clinchem.2014.223677. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Jr, Yamamoto H, Navarro M, Maestro M, Reventos J, Perucho M. Frameshift mutations at mononucleotide repeats in caspase-5 and other target genes in endometrial and gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res. 1999;59(12):2995–3002. [PubMed] [Google Scholar]
- Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008;4:2. doi: 10.1186/1745-7580-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Hoecke S, Verhelst J, Vuylsteke M, Saelens X. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC Genomics. 2015;16:79. doi: 10.1186/s12864-015-1284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002;62(14):4095–9. [PubMed] [Google Scholar]
- Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Apoptotic and growth regulatory genes as mutational targets in mismatch repair deficient endometrioid adenocarcinomas of young patients. Oncol Rep. 2004;11(4):931–7. [PubMed] [Google Scholar]
- Walker CJ, Miranda MA, O’Hern MJ, McElroy JP, Coombes KR, Bundschuh R, Cohn DE, Mutch DG, Goodfellow PJ. Patterns of CTCF and ZFHX3 Mutation and Associated Outcomes in Endometrial Cancer. J Natl Cancer Inst. 2015;107(11) doi: 10.1093/jnci/djv249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18(8):1254–61. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- Zighelboim I, Goodfellow PJ, Gao F, Gibb RK, Powell MA, Rader JS, Mutch DG. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J Clin Oncol. 2007;25(15):2042–8. doi: 10.1200/JCO.2006.08.2107. [DOI] [PubMed] [Google Scholar]
- Zighelboim I, Mutch DG, Knapp A, Ding L, Xie M, Cohn DE, Goodfellow PJ. High frequency strand slippage mutations in CTCF in MSI-positive endometrial cancers. Hum Mutat. 2014;35(1):63–5. doi: 10.1002/humu.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.