

Abstract
Introduction
Aberrant RON (Recepteur d'Origine Nantais) tyrosine kinase activation causes the epithelial cell to evade normal growth pathways, resulting in unregulated cell proliferation, increased cell motility and decreased apoptosis. Wildtype (wt) RON has been shown to play a role in metastasis of epithelial malignancies. It presents an important potential therapeutic target for colorectal, breast, gastric and pancreatic cancer. Little is known about functional differences amongst RON isoforms RON155, RON160 and RON165. The purpose of this study was to determine the effect of various RON kinase isoforms on cell motility.
Methods
Cell lines with stable expression of wtRON were generated by inserting the coding region of RON in pTagRFP (tagged red fluorescence protein plasmid). The expression constructs of RON variants (RON155, RON160 and RON165) were generated by creating a mutagenesis-based wtRON-pTag RFP plasmid and stably transfected into HEK 293 cells. The wound closure scratch assay was used to investigate the effect on cell migratory capacity of wild type RON and its variants.
Results
RON transfected cells demonstrated increased cell motility compared to HEK293 control cells. RON165 cell motility was significantly increased compared to RON160 (mean percentage of wound covered 37.37% vs. 32.40%; p = 0.03).
Conclusions
RON tyrosine kinase isoforms have variable cell motility. This may reflect a difference in the behavior of malignant epithelial cells and their capacity for metastasis.
Keywords: Biological sciences, Biochemistry, Cell biology
1. Introduction
Aberrant RON (Recepteur d' Origine Nantais) is a receptor tyrosine kinase member of the MET proto-oncogene family with known involvement in both tumor progression and metastasis [1, 2, 3, 4, 5, 6, 7]. In normal cells, RON is transiently activated through binding with its ligand, macrophage-stimulating protein (MSP), which initiates receptor autophosphorylation and upregulation of kinase activity [8, 9, 10]. However, RON is likely abnormally activated through overexpression of receptor proteins or point mutations in the kinase domain, leading to dimerization [11]. RON is not only directly implicated in tumorigenesis and invasiveness, but also interacts downstream with oncogenic pathways phosphatidyl inositol-3 kinase (PI3 K)/Akt and Ras/Raf/mitogen-activated protein kinase (MAPK) [12, 13, 14]. RON also participates in cross-talk with other oncogenic receptor tyrosine kinases (RTKs) such as the epidermal growth factor receptor (EGFR) and Met receptor [3, 7]. Constitutive activation of the RON receptor causes the epithelial cell to evade normal growth pathways, resulting in unregulated cell proliferation, increased cell motility and decreased apoptosis [12, 15]. Both in vitro and in vivo experiments have shown RON to be capable of inducing invasive phenotypes and distant tumor metastasis [14, 16, 17].
RON receptor splice variants are increasingly implicated in mechanisms underlying oncogenesis, metastasis and the creation of drug-resistant phenotypes [18, 19, 20]. While generally the creation of isoforms is thought to positively contribute to protein diversity, normal expression of splice variants can be significantly altered in cancer [21, 22]. In fact, variants are estimated to be abnormal in over half of cancers studied in the literature, particularly those with high cellular proliferation such as epithelial cancers [23]. Wildtype RON is a 180-kDa heterodimer containing 20 exons and 19 introns, composed of an extracelullar 40-kDa alpha chain and a 150-kDa transmembrane beta chain containing an intracellular kinase [6]. Deletion or truncation of RON receptor mRNA transcripts may result in splice variants that demonstrate constitutive autophosphorylation and increased kinase activity [24].
At least 8 RON isoforms have been identified to date: 55 (also known as short-form RON), 85, 110, 155, 160, 160E2/E3, and 170 [6, 15, 18, 24, 25, 26]. Three commonly identified splice variants 155, 160 and 165 are generated by various exon deletions in different regions of the B-chain of the RON receptor, resulting from abnormal mRNA splicing [20]. The major known characteristics of these isoforms are summarized in Table 1. Both RON155 and RON 160 induce tumor formation in vivo and exhibit cell migration abilities in a ligand-independent manner [20, 26]. RON 165, discovered in 1996 in gastric carcinoma cell line KATO-III [24], is capable of inducing cell motility in transfected cells, though does not demonstrate transformative or tumorigenic potential.
Table 1.
Characteristics of wtRON and RON isoforms.
| wtRON | RONΔ155 | RONΔ160 | RONΔ165 | |
|---|---|---|---|---|
| Deletion/Insertion | None | Exon 5,6,11 | Exon 5,6 | Exon 11 |
| Molecular Weight(kDa) | 180 | 155 | 160 | 165 |
| Structure | α-chain, mature β-chain | Single chain | 125 kDa mature β-chain | Single chain |
| Cellular location | Membrane | Cytoplasm | Membrane | Cytoplasm |
| Autophosphorylation? | No | Yes | Yes | Yes |
| MSP Stimulation | Yes | Yes | Yes | Yes |
RON plays a role in the metastasis of epithelial malignancies and presents an important potential therapeutic target for colorectal, ovarian, breast, gastric, lung and pancreatic cancer [19, 27, 28, 29]. While advances in control of local disease have been made in recent years, breast cancer remains the second leading cause of death in women and colorectal cancer the third leading cause of cancer deaths in both sexes in the United States [30]. The sequelae of tumor metastasis accounts for the majority of cancer deaths. Approximately 20% of patients with colorectal cancer are found to have distant disease at the time of diagnosis [31]. While the development of monoclonal antibodies to wtRON is underway, little is known about the functional differences amongst RON isoforms RON155, RON160 and RON165, particularly differences in metastatic potential. The purpose of this study was to determine the effect of various RON kinase isoforms on cell motility. We hypothesized that RON isoforms will demonstrate varying cell motility.
2. Methods and materials
2.1. Cell culture
HEK 293 cells were obtained as a gift from Dr. Diane Lidke (University of New Mexico). The cells were cultured and maintained in MEM supplemented with 10% (v/v) fetal bovine serum at 37 °C in a humidified atmosphere of 95% air and 5% CO2.
2.2. Creation of RON and RON variant constructs
Plasmid (pDONR223-MST1R) containing the full cDNA coding region of wtRON (Addgene, Cambridge, MA, USA) was obtained. PCR primers (forward 5′-TCAAGCTTCGAATTCAT GGAGCTCCTCCCGCCGCTGC-3′, reverse 5′-TCGACTGCAGAATTCTAGTGGGCCGAGGAGG-3′) were designed to amplify the fragment from start codon to stop codon by PCR reaction (Sigma Labs, Santa Fe, NM, USA). Amplified PCR fragments were isolated and inserted into expression plasmid pTagRFP (red fluorescence protein). The wtRON plasmid construct was used to generate RON isoforms RON155 (by deletion of exons 5, 6, and 11), RON160 (by deletion of exons 5 and 6), and RON165 (by deletion of exon 11) by target deletion with InFusion HD Cloning Plus Kit (Clontech Laboratories, Mountain View, CA, USA). All the expression plasmids were confirmed by DNA sequencing.
2.3. Plasmid transfection and immunoblotting analysis
Plasmid constructs for wtRON, RON155, RON160 and RON165 were transfected into HEK293 cells using Xfect Transfection Reagent (Clontech). We selected four individual clones for each isoform. The culture medium plus 800 μg/ml of G418 was used to select the clones. The expression of wtRON and RON variant was confirmed by immunoblotting assay using an anti-RON antibody. Endogenous RON expression in HEK293 cells was compared with expression in five common colon cancer cell lines to confirm the utility of employing this cell line for wtRON and RON isoform transfection. To compare the effect of RON isoforms on downstream signaling of the PI3 K pathway, we performed immunoblotting with the indicated antibodies and BioRad Image Lab™ software to calculate relative quantity analysis.
2.4. Wound scratch assay
HEK293 cells, which were individually transfected with empty plasmid vector (pTagRFP as a control) or plasmids containing wtRON, RON155, RON160 and RON165 respectively were cultured in a 60-mm dish coated with collagen and grew until confluence. A uniform wound was created manually in the confluent monolayer of cells with a pipette tip. Wound closure was monitored at 0, 12 and 24 hours using a Nikon light microscope at 10x magnification. The percentage of wound coverage was measured using Image-J software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA).
2.5. Statistical analysis
All experiments were repeated independently and in triplicate. Variance in cell motility values, if significant, was detected using first an F-test, followed by a Student’s T-test to compare mean values. Statistical significance was considered for p values ≤ 0.05.
3. Results
3.1. Low to undetectable levels of endogenous RON found in HEK293 cells
We found near undetectable endogenous RON expression in HEK cells compared to five commonly used colon cancer cell lines (Fig. 1a). This confirmed the utility of employing HEK293 cells for wtRON and RON isoform transfection and subsequent motility analysis.
Fig. 1.

(a) Endogenous expression of RON. HEK293 cells demonstrate nearly undetectable levels of RON expression compared to five colon cancer cell lines. Please refer to Supplementary Material Fig. 1a for full immunoblotting image. (b) Immunoblotting assay to confirm the expression of wtRON and RON isoforms in stably transfected HEK cells. Prominent bands were confirmed for each of RON isoforms (155, 160 and 165 kDa respectively). Please refer to Supplementary Material Fig. 1b for full immunoblotting image.
3.2. WtRON and RON isoforms demonstrated increased cell motility compared to control cells
To determine differences in cell motility between RON and RON isoforms 155, 160 and 165 and control cells, a wound scratch assay was performed (Fig. 2). Migration was monitored at 12 and 24 hours. All RON cell lines were 100% confluent at 24 hours. Cell motility was measured by the mean change in percentage of wound coverage at 12 hours. WtRON demonstrated the fastest migration of all 5 cell lines. WtRON (36.85% +/−5.83; p < 0.002), RON 155 (34.77% +/5.27; p < 0.01) and 165 (37.37% +/− 3.66; p < 0.001) demonstrated significantly increased motility compared to control (28.14% +/− 5.23) (Fig. 3). RON 160 showed increased motility compared to control (mean 32.4% +/− 1.44 vs. 28.15%, +/− 1.40; p = 0.09), but did not obtain statistical significance.
Fig. 2.

In vitro wound scratch assay. All lines except for pTagRFP-HEK control cells had complete confluence by 24 hours. Wound closure at 12 hours was used to compare cell motility.
Fig. 3.

Effect of RON and RON isoforms on cell motility, pTagRFP-wtRON and −RON isoforms, as measured by the mean percent change in wound coverage at 12 hours. The error bars demonstrate the SD of each cell line, ranging from 3.65 to 5.83.
3.3. RON 165 demonstrated increased cell motility compared to RON 160
The wound scratch assay analysis was also employed to compare differences between wtRON and the three RON isoforms. RON 165 was found to have significantly increased motility compared to RON 160 (37.37% vs. 32.40%; p = 0.03) (Fig. 2).
3.4. WtRON and RON isoforms have variable affects on PI3 K pathway downstream signaling
Relative quantity analysis demonstrated RON160 had increased levels of Akt, pGSK3β and FOXO3a protein expression compared to control, wtRON, RON 155 and165 (Fig. 4). WtRON and RON 160 had increased levels of GSK3β compared to control, RON 155 and 165. PFOXO3a protein expression was increased in RON160 and 165 compared to control, wtRON and RON 155. RON and all isoforms were found to have decreased pAkt levels compared to control.
Fig. 4.
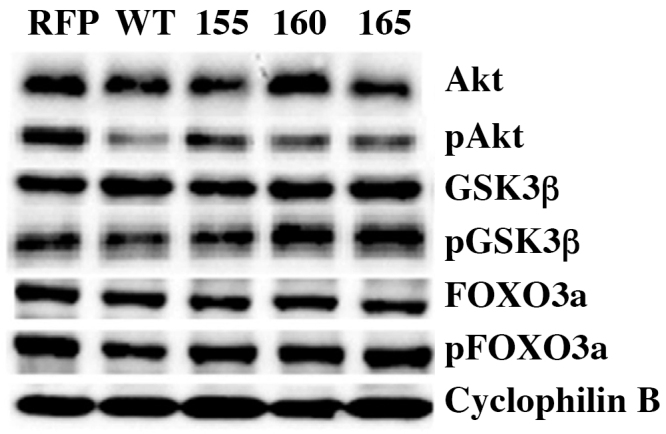
The effect of wt RON and RON isoforms on downstream signaling of the PI3 K pathway. GSK3β, FOXO3a and its phosphorylated proteins were increased in RON 160 while the remaining isoforms showed minimal variation. PAkt was decreased in wt RON and its isoforms. Please refer to Supplementary Materials Fig. 2a–g for full immunoblotting images.
4. Discussion
RON tyrosine kinase is involved in tumorigenesis and metastasis in a variety of epithelial cancers. The RON receptor interacts with the extracellular environment, promoting cell adhesion and epithelial-mesenchymal transition, avoids apoptosis, transforms normal cells into invasive phenotypes and activates downstream oncogenic pathways such as PI3 K/Akt and Ras/Raf/MAPK. Various RON splice variants also demonstrate tumorigenic and metastatic potential, though the literature has only begun to elucidate the functional differences between them. We have shown that HEK293 cells transfected with RON isoforms RON155, RON160 and RON165 demonstrate variable cell motility, specifically RON 165 which exhibited increased motility compared to RON 160.
Consistent with previous literature, wtRON showed increased cell motility compared to control cells [4, 6] and demonstrated the fastest cell migration of all the cell lines studied. This is an interesting finding considering that wtRON has ligand-dependent activation while RON isoforms are constitutively activated. We suspect if MSP ligand was added there may be an even larger difference in cell motility. There continues a discussion as to whether wtRON is tumorigenic or promotes metastasis. Initial studies suggested that human wtRON was neither tumorigenic nor metastatic [4] However, further studies [11] showed wtRON to have transformative properties in NIH3T3 cells including long cytoplasmic extensions and loss of contact inhibition, even in the absence of ligand stimulation. Zhou et al. confirmed wtRON to be incapable of tumorigenesis [20]. Eyob et al. also found knocking out RON had no effect on tumor development or growth rates, though eliminated lung metastasis [32]. Current knowledge suggests that while human wtRON may not have transformative properties, it is undoubtedly linked to the ability of tumors to metastasize.
Interestingly, RON 165 exhibited the fastest migration of the three RON isoforms. RON 165 is found in normal colorectal mucosa cells and is thought to contribute to metastatic spread and invasive phenotypes, though not tumorigenesis [20, 24]. Conversely, RON160 is able to induce transformation, cell scattering and tumorigenesis in athymic nude mice. RON155, first reported by Zhou et al., was also found to induce cell scattering, transformation and tumor growth in vivo, though less efficiently than RON160. RON160 and 155 were found to cause multiple foci in transfected NIH3T3 cells, induce tumor formation and metastatic spread. However, in this study the metastatic potential of RON165 was not studied in vivo, as it was not tumorigenic in nude mice. Our study suggests that RON165 significantly increases cell motility. Overexpression of RON165 was noted in some types of breast and colon cancer, which may explain these cancers’ extraordinary capacity for metastasis. Further experiments in an orthotopic model are needed to examine the true functional differences in metastatic potential of wtRON and RON isoforms, particularly RON165.
Understanding the ability of tumors to metastasize is of utmost clinical importance. The invasion of metastatic disease to distant organs is the main cause of death in most cancer patients [33]. A hallmark of colorectal cancer is the ability to invade adjacent organs and metastasize to remote locations such as the liver or lung [34]. In normal colon epithelial cells, RON is barely expressed, though is highly expressed and constitutively activated in many colorectal cancer cell lines [33]. However, treatment of colorectal cancer is difficult as a significant degree of genetic heterogeneity leads to highly variable phenotypes [35]. The existence of splice variation adds yet another level of complexity to an individual patient’s tumor. RON isoforms are related to deregulation of mRNA splicing machinery and not genetic mutations [15]. Research into splicing factors is currently developing, specifically examining the functions of a silencer and enhancer on exon 12 that may regulate aberrant splicing in epithelial cancers [18] or studying the SRSF2 gene which promotes inclusion of exon 11 [36, 37]. In addition, clarifying the functional differences between isoforms important as RON variants display differing cellular locations, with RON160 being entirely intracellular, while RON 155 and 165 span the cellular membrane. As RON and other RTKs represent a developing target for monoclonal antibodies [38, 39] RON isoforms may require a different therapeutic approach.
In addition to comparing cell motility, we performed a preliminary analysis of wtRON and RON isoform effects on downstream signaling in the PI3 K pathway. Most of the current literature regarding PI3 K/Akt pathways in cancer cells supports the hypothesis that increased cell motility would correlate with increased downstream protein expression, signifying activation of RON and PI3 K pathways. We were surprised to find that RON 160 increased the expression of several proteins compared to wtRON, RON 155 and 165 and that pAkt was decreased in all cell lines compared to control. Recent literature has revealed that protein signaling in the PI3 K/Akt signally pathway is anything but linear, with evidence that in some cases PI3 K and Akt may operate independently of each other in cancer cells [40]. Our preliminary data shows that there may be variation in downstream protein expression, though the clinical significance of these findings are not yet clear. Further study is needed to explore the effects of RON and its splice variants on the downstream pathways such as PI3 K/Akt and Ras/Raf/MAPK.
This study includes a few notable limitations. The three RON isoforms studied demonstrated increased cell motility compared to control cells as expected. In our study, RON 160 was increased compared to control, though did not reach statistical significance, which we attribute to a smaller sample size of available images. Our plasmid constructs were transfected into normal epithelial cells as opposed to a colon cancer cell lines. This is because many colon cancer cell lines have wtRON and RON splice variant expression [15, 33]. This HEK293 cell model, having near undetectable levels of endogenous RON expression, allows for preliminary analysis of the impact of isolated splice variants on cell motility. Thus, we cannot yet comment on RON isoforms cell motility in tumor cells or how the differences in cell motility will translate in an in vivo animal model.
In this study we demonstrate that RON tyrosine kinase splice variants demonstrate variable cell motility in normal cells. Variation in cell motility between RON isoforms may represent a difference in metastatic potential. Future studies aimed at exploring the functional differences of RON isoforms in in vivo pre-clinical models will help to determine their role as potential therapeutic targets in both localized cancers and metastatic disease.
Declarations
Author contribution statement
Alissa Greenbaum: Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Ashwani Rajput: Conceived and designed the experiments; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Guanghua Wan: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data.
Funding statement
This work was supported by University of New Mexico Department of Surgery and UNM Comprehensive Cancer Center P30CA 118110. The sponsors had no involvement in study design, collection analysis or interpretation of data, or in writing the article.
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
Dr. Diane Lidke, PhD, University of New Mexico for her generous donation of HEK293 cells.
Appendix A. Supplementary data
References
- 1.Camp E.R., Liu W., Fan F., Yang A., Somcio R., Ellis L.M. RON, a tyrosine kinase receptor involved in tumor progression and metastasis. Ann. Surg. Oncol. 2005;12(4) doi: 10.1245/ASO.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y., Yao H.P., Wang M.H. Multiple variants of the RON receptor tyrosine kinase: Biochemical properties, tumorigenic activities, and potential drug targets. Cancer Letters. 2007;257(2):157–164. doi: 10.1016/j.canlet.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Peace B.E., Hill K.J., Degen S.J.F., Waltz S.E. Cross-talk between the receptor tyrosine kinases Ron and epidermal growth factor receptor. Exp. Cell Res. 2003;289:317–325. doi: 10.1016/s0014-4827(03)00280-5. [DOI] [PubMed] [Google Scholar]
- 4.Santoro M.M., Collesi C., Grisendi S., Gaudino G., Comoglio P.M. Constitutive activation of the RON gene promotes invasive growth but not transformation. Mol. Cell. Biol. 1996;16(12):7072–7083. doi: 10.1128/mcb.16.12.7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santoro M.M., Penengo L., Minetto M., Orecchia S., Cilli M., Gaudino G. Point mutations in the tyrosine kinase domain release the oncogenic and metastatic potential of the Ron receptor. Oncogene. 1998;17(6):741–749. doi: 10.1038/sj.onc.1201994. [DOI] [PubMed] [Google Scholar]
- 6.Wang M.H., Wang D., Chen Y.Q. Oncogenic and invasive potentials of human macrophage-stimulating protein receptor, the RON receptor tyrosine kinase. Carcinogenesis. 2003;24(8):1291–1300. doi: 10.1093/carcin/bgg089. [DOI] [PubMed] [Google Scholar]
- 7.Follenzi A., Bakovic S., Gual P., Stella M.C., Longati P., Comoglio P.M. Cross-talk between the proto-oncogenes Met and Ron. Oncogene. 2000;19(27):3041–3049. doi: 10.1038/sj.onc.1203620. [DOI] [PubMed] [Google Scholar]
- 8.Wang M.H., Ronsin C., Gesnel M.C., Coupey L., Skeel A., Leonard E.J., Breathnach R. Identification of the ron gene product as the receptor for the human macrophage stimulating protein. Science. 1994;266(5182):117–119. doi: 10.1126/science.7939629. [DOI] [PubMed] [Google Scholar]
- 9.Danilkovitch-Miagkova A. Oncogenic signaling pathways activated by RON receptor tyrosine kinase. Curr. Cancer Drug Targets. 2003;3(1):31–40. doi: 10.2174/1568009033333745. [DOI] [PubMed] [Google Scholar]
- 10.Gaudino G., Follenzi A., Naldini L., Collesi C., Santoro M., Gallo K., Comoglio P.M. RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J. 1994;13(15):3524–3532. doi: 10.1002/j.1460-2075.1994.tb06659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peace B.E., Hughes M.J., Degen S.J., Waltz S.E. Point mutations and overexpression of Ron induce transformation, tumor formation, and metastasis. Oncogene. 2001;20(43):6142–6151. doi: 10.1038/sj.onc.1204836. [DOI] [PubMed] [Google Scholar]
- 12.Danilkovitch A., Skeel A., Leonard E.J. Macrophage stimulating protein-induced epithelial cell adhesion is mediated by a PI3-K-dependent, but FAK-independent mechanism. Exp. Cell Res. 1999;248(2):575–582. doi: 10.1006/excr.1999.4429. [DOI] [PubMed] [Google Scholar]
- 13.Liu X., Zhao L., Derose Y.S., Lin Y.-C., Bieniasz M., Eyob H. Short-Form Ron Promotes Spontaneous Breast Cancer Metastasis through Interaction with Phosphoinositide 3-Kinase. Genes Cancer. 2011;2(7):753–762. doi: 10.1177/1947601911421924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J., Rajput A., Kan J.L.C., Rose R., Liu X.-Q., Kuropatwinski K. Knockdown of Ron kinase inhibits mutant phosphatidylinositol 3-kinase and reduces metastasis in human colon carcinoma. J. Biol. Chem. 2009;284(16):10912–10922. doi: 10.1074/jbc.M809551200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y.Q., Zhou Y.Q., Angeloni D., Kurtz A.L., Qiang X.Z., Wang M.H. Overexpression and activation of the RON receptor tyrosine kinase in a panel of human colorectal carcinoma cell lines. Exp. Cell Res. 2000;261(1):229–238. doi: 10.1006/excr.2000.5012. [DOI] [PubMed] [Google Scholar]
- 16.Maggiora P., Marchio S., Stella M.C., Giai M., Belfiore A., De Bortoli M. Overexpression of the RON gene in human breast carcinoma. Oncogene. 1998;16(22):2927–2933. doi: 10.1038/sj.onc.1201812. [DOI] [PubMed] [Google Scholar]
- 17.Wang D., Shen Q., Chen Y.-Q., Wang M.-H. Collaborative activities of macrophage-stimulating protein and transforming growth factor-beta1 in induction of epithelial to mesenchymal transition: roles of the RON receptor tyrosine kinase. Oncogene. 2004;23(9):1668–1680. doi: 10.1038/sj.onc.1207282. [DOI] [PubMed] [Google Scholar]
- 18.Ghigna C., De Toledo M., Bonomi S., Valacca C., Gallo S., Apicella M. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: Therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol. 2014;7(4):495–503. doi: 10.4161/rna.7.4.12744. [DOI] [PubMed] [Google Scholar]
- 19.Liu X., Zhao L., Derose Y.S., Lin Y.-C., Bieniasz M., Eyob H. Short-Form Ron Promotes Spontaneous Breast Cancer Metastasis through Interaction with Phosphoinositide 3-Kinase. Genes Cancer. 2011;2(7):753–762. doi: 10.1177/1947601911421924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y.Q., He C., Chen Y.Q., Wang D., Wang M.H. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene. 2003;22(2):186–197. doi: 10.1038/sj.onc.1206075. [DOI] [PubMed] [Google Scholar]
- 21.Pajares M.J., Ezponda T., Catena R., Calvo A., Pio R., Montuenga L.M. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol. 2007;8(4):349–357. doi: 10.1016/S1470-2045(07)70104-3. [DOI] [PubMed] [Google Scholar]
- 22.Skotheim R.I., Nees M. Alternative splicing in cancer: Noise, functional, or systematic? Int. J. Biochem. Cell Biol. 2007;39(7-8):1432–1449. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Ritchie W., Granjeaud S., Puthier D., Gautheret D. Entropy measures quantify global splicing disorders in cancer. PLoS Comput. Biol. 2008;4(3):1–9. doi: 10.1371/journal.pcbi.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collesi C., Santoro M.M., Gaudino G., Comoglio P.M. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol. Cell. Biol. 1996;16(10):5518–5526. doi: 10.1128/mcb.16.10.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Q., Zhang K., Yao H.-P., Zhou Y.-Q., Padhye S., Wang M.H. Inhibition of MSP-RON signaling pathway in cancer cells by a novel soluable form of RON comprising the entire sema sequence. Int. J. Oncol. 2010;36:1551–1561. doi: 10.3892/ijo_00000642. [DOI] [PubMed] [Google Scholar]
- 26.Wang M.H., Kurtz A.L., Chen Y. Identification of a novel splicing product of the RON receptor tyrosine kinase in human colorectal carcinoma cells. Carcinogenesis. 2000;21(8):1507–1512. [PubMed] [Google Scholar]
- 27.Angeloni D., Danilkovitch-Miagkova A., Ivanov S.V., Breathnach R., Johnson B.E., Leonard E.J., Lerman M.I. Gene structure of the human receptor tyrosine kinase RON and mutation analysis in lung cancer samples. Genes Chromosomes Cancer. 2000;29:147–156. [PubMed] [Google Scholar]
- 28.Thomas R.M., Toney K., Fenoglio-Preiser C., Revelo-Penafiel M.P., Hingorani S.R., Tuveson D.A. The RON receptor tyrosine kinase mediates oncogenic phenotypes in pancreatic cancer cells and is increasingly expressed during pancreatic cancer progression. Cancer Res. 2007;67:6075–6082. doi: 10.1158/0008-5472.CAN-06-4128. [DOI] [PubMed] [Google Scholar]
- 29.Zou Y., Howell G.M., Humphrey L.E., Wang J., Brattain M.G. Ron Knockdown and Ron Monoclonal Antibody IMC-RON8 Sensitize Pancreatic Cancer to Histone Deacetylase Inhibitors (HDACi) PLoS One. 2013;8(7):1–10. doi: 10.1371/journal.pone.0069992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siegel R.L., Miller K.D., Jemal A. Cancer statistics. CA Cancer J. Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 31.Calonge N., Fisher N.L., Berg A.O. Recommendations from the EGAPP Working Group: can testing of tumor tissue for mutations in EGFR pathway downstream effector genes in patients with metastatic colorectal cancer improve health outcomes by guiding decisions regarding anti-EGFR therapy? Genet. Med. 2013;15:517–527. doi: 10.1038/gim.2012.184. [DOI] [PubMed] [Google Scholar]
- 32.Eyob H., Ekiz H.A., DeRose Y.S., Waltz S.E., Williams M.A., Welm A.L. Inhibition of Ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013;3(7):751–760. doi: 10.1158/2159-8290.CD-12-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C.-T., Chow N.-H., Su P.-F., Lin S.-C., Lin P.-C., Lee J.-C. The Prognostic Significance of RON and MET Receptor Coexpression in Patients with Colorectal Cancer. Dis. Colon Rectum. 2008;51(8):1268–1274. doi: 10.1007/s10350-008-9297-1. [DOI] [PubMed] [Google Scholar]
- 34.Yokota J. Tumor progression and metastasis. Carcinogenesis. 2000;21(3):497–503. doi: 10.1093/carcin/21.3.497. [DOI] [PubMed] [Google Scholar]
- 35.Chowdhury S., Ongchin M., Sharratt E., Dominguez I., Wang J., Brattain M.G., Rajput A. Intra-tumoral heterogeneity in metastatic potential and survival signaling between iso-clonal HCT116 and HCT116b human colon carcinoma cell lines. PloS One. 2013;8(4) doi: 10.1371/journal.pone.0060299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon H., Cho S., Loh T.-J., Oh H.-K., Jang H.-N., Zhou J., Kwon Y.S., Liao D.J., Jun Y., Eom S., Ghigna C., Biamonti G., Green M.R., Zheng X., Shen H. SRSF2 promotes splicing and transcription of exon 11 included isoform in Ron proto-oncogene. Biochim. Biophys. Acta. 2014;1839(11):1132–1140. doi: 10.1016/j.bbagrm.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghigna C., Giordano S., Shen H., Benvenuto F., Castiglioni F., Comoglio P.M., Green M.R., Riva S., Biamonti G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell. 2005;20:881–890. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 38.Wang M.-H., Yao H.-P., Zhou Y.-Q. Oncogenesis of RON receptor tyrosine kinase: a molecular target for malignant epithelial cancers. Acta Pharmacol. Sin. 2006;27(6):641–650. doi: 10.1111/j.1745-7254.2006.00361.x. [DOI] [PubMed] [Google Scholar]
- 39.Yao H.-P., Zhou Y.-Q., Ma Q., Guin S., Padhye S.S., Zhang R.-W., Wang M.-H. The monoclonal antibody Zt/f2 targeting RON receptor tyrosine kinase as potential therapeutics against tumor growth-mediated by colon cancer cells. Mol. Cancer. 2011;10:82. doi: 10.1186/1476-4598-10-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faes S., Dormond O. PI3 K and AKT: unfaithful partners in cancer. Int. J. Mol. Sci. 2015;16(9):21138–21152. doi: 10.3390/ijms160921138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.