

Abstract
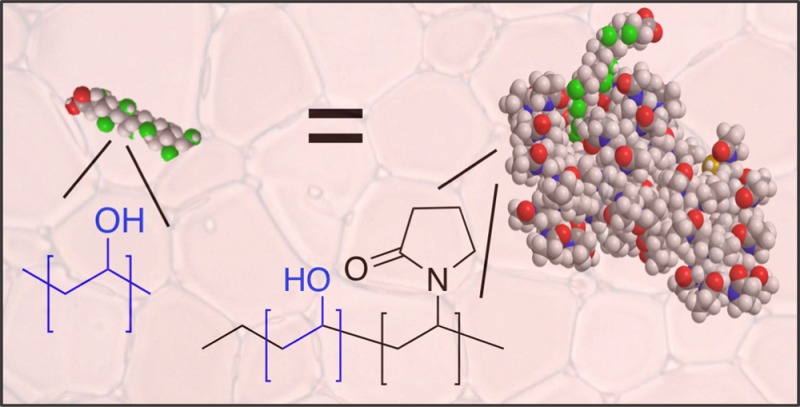
Antifreeze (glyco) proteins are produced by many cold-acclimatized species to enable them to survive subzero temperatures. These proteins have multiple macroscopic effects on ice crystal growth which makes them appealing for low-temperature applications—from cellular cryopreservation to food storage. Poly(vinyl alcohol) has remarkable ice recrystallization inhibition activity, but its mode of action is uncertain as is the extent at which it can be incorporated into other high-order structures. Here the synthesis and characterization of well-defined block copolymers containing poly(vinyl alcohol) and poly(vinylpyrrolidone) by RAFT/MADIX polymerization is reported, as new antifreeze protein mimetics. The effect of adding a large second hydrophilic block is studied across a range of compositions, and it is found to be a passive component in ice recrystallization inhibition assays, enabling retention of all activity. In the extreme case, a block copolymer with only 10% poly(vinyl alcohol) was found to retain all activity, where statistical copolymers of PVA lose all activity with very minor changes to composition. These findings present a new method to increase the complexity of antifreeze protein mimetic materials, while retaining activity, and also to help understand the underlying mechanisms of action.
Introduction
Antifreeze (glyco) proteins (AF(G)Ps) are produced by species which live in extreme cold environments (i.e., the Earth’s poles or at high altitude) as an evolutionary adaptation to control the formation and growth of ice crystals, which would otherwise lead to death.1 AF(G)Ps have three main effects: IRI (ice recrystallization inhibition), TH (thermal hysteresis; noncolligative depression of freezing point), and DIS (dynamic ice shaping) associated with the protein–ice interactions. IRI is a particularly interesting property for a range of biotechnological applications which involve cellular cryopreservation, as ice growth on thawing has been shown to be a key source of cell death. The ability to control ice growth would also be useful in industrial settings including aircraft wings, wind turbines, and food storage or in the space industry.2−5
AF(G)Ps themselves are not always suitable for application due to potential toxicity issues and the price of scale up, especially for AFGPs which cannot easily be expressed recombinantly in bacteria (due to the need for glycosylation).6 A particular problem with AF(G)Ps when applied to cryopreservation is the onset of DIS, which reduces cell recovery due to the formation of spicular ice which pierces cell membranes.7 Ben et al. have developed low molecular weight peptides and glyco-mimetics which show selective IRI activity and enhanced cryopreservation.8,9 Crucially, these simplified structures show that reductionist approaches to mimicking AF(G)P function are plausible and that new structures can selectivity show one AF(G)P “antifreeze” property rather than all three, with the implication that multiple molecular mechanisms may be in action.10 Native AFPs show different levels of TH and IRI with the two properties not always correlated.11,12
Inspired by this, we have shown that synthetic polymers can mimic AF(G)P’s IRI activity, even though they lack the precise secondary/tertiary structure which is essential for AF(G)P function.5 These have been applied to enhance the cryopreservation of blood13−15 and somatic cells16 but retain low cytotoxicity. Polymer mimetics have the advantage of being scalable and highly tunable in terms of composition and architecture. Additionally, native AFGPs show an increase in activity as the highly repetitive glyco-tripeptide motif is elongated,1 implying that synthetic polymers which can easily have their molecular weight tuned could have enhanced activity. The most potent polymeric AF(G)P mimetics identified thus far are those based on poly(vinyl alcohol), PVA, which inhibits all ice growth at sub mg·mL–1 concentrations. PVA also has well-known biocompatibility and is used in a range of medical or food applications.17 Polyampholytes (with mixed positive/negative side groups) have also been developed by Matsumura et al., which are potent cryoprotectants18−20 despite having less IRI,21 demonstrating the complex relationship between inhibiting ice growth and cellular cryopreservation.
Key questions still exist about the mode of action of PVA polymers, especially to understand if ice binding is occurring, or if they function at the quasi-liquid layer at the ice/water interface and slow down the exchange of water between ice grains. This latter mechanism does not require ice binding, and also explains the lack of (or very weak) TH and DIS activity observed with most PVAs. Budke and Koop suggested that the spacing of hydroxyl groups in PVA precisely matches a single crystal face of ice and have observed ice shaping.22 However, this study used high molecular weight PVA (27 kg mol–1) with large molecular weight dispersity, making it unclear as to which molecular weight fractions were responsible for the activity. We addressed this using reversible addition–fragmentation chain transfer (RAFT) polymerization23 to obtain well-defined PVAs from 800 to 20000 g·mol–1. Quantitative IRI studies showed that even DP 10 PVAs had activity but that a significant enhancement occurred at DP 20.24 For these shorter polymers, no TH or DIS was observed which suggests that ice binding is a molecular weight dependent effect. Addition of comonomers with PVA (or residual acetate groups from the synthesis) removed all activity from the PVA, suggesting that it is not tolerant of modifications and potentially supporting the ice binding hypothesis.24 To address this, we have investigated (supramolecular) branched PVAs and found that addition of a third arm to a PVA has no effect on IRI activity,25 as has been seen for branched AFPs.26 This is significant, as although the molecular weight was increased, there was no enhancement compared to increasing the molecular weight of a linear polymer. Brush-like PVA also shows identical activity to linear equivalents12 despite having far higher molecular weight.
The role of an additional (chemically different) block on the IRI activity of a polymer has not been studied in detail but could provide an alternative approach to increase the functionality such as increasing blood circulation lifetime or to enable incorporation into polymer composites or as protein conjugates.27 Interestingly, AFP–fusion proteins retain or even have enhanced activity28 and many antifreeze proteins are already tagged with, e.g., a His-tag to enable purification via nickel affinity chromatography, supporting this hypothesis. Mastai and Baruch studied branched poly(ethylene oxide)–poly(ethylenimine) block copolymers with polyglycidol side chains for ice interactions by DSC, observing some impact of the copolymer structure, but only DSC was used to probe this.29 Antonietti and co-workers found that phosphorylated poly(ethylene oxide)-block-poly[2-(2-hydroxyethyl) ethylene] had some IRI activity but only at high concentrations of 30 mg·mL–1.30 There has not been a detailed structure–activity study on block copolymer structure and ice growth inhibition, in part due to the challenges of polymerizing lesser activated monomers such as vinyl acetate (where poly(vinyl acetate) is the precursor to PVA) using RAFT CRP, although cobalt31 and vanadium mediators have been reported to give excellent control over Vac polymerizations.32
Controlled radical polymerization affords straightforward routes to preparing block copolymers as, due to the inherent living nature of the reaction, polymerizations are not irreversibly terminated. Using the RAFT/MADIX (reversible additional transfer fragmentation/macromolecular design via interchange of xanthates) methodology, vinyl acetate can be effectively polymerized, but there are few other monomers with which it undergoes effective (block) copolymerization. Stenzel and co-workers used MADIX to polymerize N-vinylpyrrolidone (NVP), examining chain extension using vinyl acetate, and accessing NVP star polymers.33 This has also been demonstrated by Okamoto and co-workers, using a benzyl and o-ethoxy functional MADIX agent in fluoroalcohols.34 Klumperman and co-workers also published several reports on the preparation of NVP-containing block copolymers35,36 demonstrating that PVA blocks can be accessed.
To address the questions around block copolymer architecture on IRI activity, this manuscript reports a detailed study on the effect of block copolymer architecture on PVA’s IRI activity compared to previous work where statistical copolymerization was used leading to a loss in activity.24 A library of vinylpyrrolidone block copolymers were synthesized, and the role of each block is evaluated by a quantitative IRI assay. Remarkably, PVA activity is shown to be tolerant to PVP inclusion, even at very high PVP weight percentages, which may support a mechanism not based entirely on ice binding.
Experimental Section
General
Phosphate-buffered saline (PBS) solutions were prepared using preformulated tablets (Sigma-Aldrich) in 200 mL of Milli-Q water (>18.2 Ω mean resistivity) to give [NaCl] = 0.138 M, [KCl] = 0.0027 M, and pH 7.4. Vinyl acetate and N-vinylpyrrolidone were purchased from Sigma-Aldrich and were filtered through a plug of basic alumina to remove inhibitors prior to their use. 4,4′-Azobis(4-cyanovaleric acid) was recrystallized from methanol and stored at −8 °C in the dark. Hydrazine hydrate solution (80%) was purchased from Sigma-Aldrich. All solvents were purchased from VWR or Sigma-Aldrich and used without further purification, except for 1,4-dioxane, which was filtered through a plug of alumina prior to use.
Physical and Analytical Methods
1H and 13C NMR spectra were recorded on Bruker DPX-300 and DPX-400 spectrometers using deuterated solvents obtained from Sigma-Aldrich. Chemical shifts are reported relative to residual nondeuterated solvent. Mass spectral analyses were obtained using Bruker MicroTOF or Bruker MaXis electrospray instruments using positive or negative electrospray mode. The molecular ion and mass fragments are quoted and assigned. Gel permeation chromatography (GPC) was used to determine the molecular weights and dispersity of the synthesized polymers. The THF GPC system was comprised of a Varian 390-LC-Multi detector suite fitted with differential refractive index (DRI), light scattering (LS), and ultraviolet (UV) detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 μm, 50 × 7.5 mm) and two mixed D columns of the same type. The mobile phase was THF with 5% trimethylamine (TEA) eluent at a flow of 1.0 mL/min, and samples were calibrated against Varian Polymer Laboratories Easi-Vials linear poly(styrene) and poly(methyl methacrylate) standards (162–2.4 × 105 g mol–1) using Cirrus v3.3. The DMF GPC system was comprised of a Varian 390-LC-Multi detector suite fitted with a differential refractive index (DRI) detector equipped with a guard column (Varian Polymer Laboratories PLGel 5 μm, 50 × 7.5 mm) and two mixed D columns of the same type. The mobile phase was DMF with 5 nM NH3BF4 eluent at a flow of 1.0 mL·min–1, and samples were calibrated against Varian Polymer Laboratories Easi-Vials poly(methyl methacrylate) standards (162–2.4 × 105 g mol–1) using Cirrus v3.3. Ice wafers were annealed on a Linkam Biological Cryostage BCS196 with T95-Linkpad system controller equipped with an LNP95-Liquid nitrogen cooling pump, using liquid nitrogen as the coolant (Linkam Scientific Instruments UK, Surrey, UK). An Olympus CX41 microscope equipped with a UIS-2 20×/0.45/∞/0–2/FN22 lens (Olympus Ltd., Southend on sea, UK) and a Canon EOS 500D SLR digital camera was used to obtain all images. Image processing was conducted using ImageJ, which is freely available from http://imagej.nih.gov/ij/.
Polymerization of N-Vinylpyrrolidone
As a representative example, N-vinylpyrrolidone (2.09 g, 18.78 mmol), CTA 1 (0.037 g, 0.17 mmol), and ACVA (0.005 g, 0.02 mmol) were dissolved in dioxane (4 mL) in a stoppered vial equipped with a stir bar. The reaction mixture was thoroughly degassed by bubbling N2 through the solution for 20 min, and the reaction mixture was allowed to polymerize at 70 °C for typically 3 h. The dark yellow solutions were then cooled to room temperature, and the block copolymer was recovered as white flakes by precipitation into cold diethyl ether held under ice. The bulk of the solvent was carefully decanted and the solid collected by centrifugation. The product was thoroughly dried under a vacuum at 40 °C for 24 h, forming a yellow solid. Representative characterization data for PVP55: 1H NMR (400 MHz, CDCl3) δ 3.74 (−CHN–, br, 1H), 3.19 (−NCH2CH2–, br, 2H), 2.38 (−NCOCH2–, br, 2H), 2.21(−NCH2CH2CH2–, br, 2H), 1.81–1.53 (−CH2–, br, 2H); MnSEC(THF) = 3350 Da, Mw/Mn = 1.42.
Chain Extension of Poly(vinylpyrrolidone) macroCTA with Vinyl Acetate
As a representative example, PVP macroCTA (0.158 g, 2.6 × 10–5 mol), vinyl acetate (1.12 g, 13 × 10–3 mol), dioxane (2.6 mL), and ACVA (4,4′-azobis(4-cyanovaleric acid); 0.001 g, 4.2 × 10–6 mol) were added to a stoppered vial. The solution was thoroughly degassed by bubbling N2 through the solution for 20 min, and the reaction mixture was then allowed to polymerize at 68 °C for 12 h. The yellow solutions were then cooled and opened to air. The block copolymer was then recovered as a yellow sticky solid after precipitation into diethyl ether. The diethyl ether was then decanted and the block copolymer was redissolved in THF, which was then concentrated in vacuo and thoroughly dried under a vacuum at 40 °C for 24 h, forming a yellow solid. Representative characterization data for PVAc18-b-PVP55: 1H NMR (400 MHz, CDCl3) δ 4.61 (−CHOAc–, br, 1H), δ 3.74 (−CHN–, br, 1H), 3.19 (−NCH2CH2–, br, 2H), 2.38 (−NCOCH2–, br, 2H), 2.21(−NCH2CH2CH2–, br, 2H), δ 2.00 (−CH3, t, 3H), 1.81–1.53 (−CH2–, br, 2H); MnSEC (DMF) = 9130 Da; Mw/Mn = 1.36.
Polymerization of Vinyl Acetate
As a representative example, CTA (5.2 mg, 2.6 × 10–5 mol), vinyl acetate (4.67 g, 2.6 × 10–3 mol), and ACVA (4,4′-azobis(4-cyanovaleric acid); 0.0013 g, 4.6 × 10–6 mol) were added to a stoppered vial. The reaction mixture was thoroughly degassed by bubbling N2 through the solution for 20 min, and the reaction mixture was then allowed to polymerize at 68 °C for 10–30 min, depending on the desired degree of conversion (at 1/100 [CTA]/[monomer] conversion = 1% /min). The yellow solutions were then quenched in liquid nitrogen and exposed to air. The polymer was then recovered as a yellow sticky solid after precipitation into hexane. The hexane was then decanted and the poly(vinyl acetate) redissolved in THF, which was then concentrated in vacuo and thoroughly dried under a vacuum at 40 °C for 24 h, forming a yellow solid. Representative characterization data for PVAc30: 1H NMR (400 MHz, CDCl3) δ 4.61 (−CHO–CH2, br, 1H), 1.74 (−CO–CH3, br, 3H), 1.53 (−CH2–, br, 2H); MnSEC(THF) = 2642 Da, Mw/Mn = 1.12.
Chain Extension of Poly(vinyl acetate) macroCTA with N-Vinylpyrrolidone
As a representative example, PVAc macroCTA (0.307 g, 1.16 mmol), N-vinylpyrrolidone (3 g, 28.2 mmol), dioxane (0.5 mL), and ACVA (4,4′-azobis(4-cyanovaleric acid); 0.028 g, 1 × 10–4 mol) were added to a stoppered vial. The reaction mixture was thoroughly degassed by bubbling N2 through the solution for 20 min, and the reaction mixture was then allowed to polymerize at 70 °C for 12 h. The yellow solutions were then cooled and opened to air. The block copolymer was then recovered as a yellow sticky solid after precipitation into diethyl ether. The diethyl ether was then decanted and the block copolymer redissolved in THF, which was then concentrated in vacuo and thoroughly dried under a vacuum for 24 h, forming a pale yellow solid. Representative characterization data for PVAc30-b-PVP24: 1H NMR (400 MHz, CDCl3) δ 4.61 (−CHOAc–, br, 1H), δ 3.74 (−CHN–, br, 1H), 3.19 (−NCH2CH2–, br, 2H), 2.38 (−NCOCH2–, br, 2H), 2.21(−NCH2CH2CH2–, br, 2H), δ 2.00 (−CH3, t, 3H), 1.81–1.53 (−CH2–, br, 2H); MnSEC (THF) = 5376 Da; Mw/Mn = 1.16.
Hydrolysis of PVAc-b-PVP Block Copolymers
As a representative example, PVAc30-b-PVP24 (0.3 g, 5376 Da, Mw/Mn = 1.16) was dissolved in a methanol (2 mL) and hydrazine hydrate solution (5 mL, 80% in water) in a stoppered vial. The reaction mixture was stirred at 60 °C for 5 h. The reaction mixture was then dialyzed using distilled water, and PVA30-b-PVP24 was recovered as a spongy white solid by freeze-drying the dialysis solution. Complete hydrolysis was confirmed by 1H NMR. Representative characterization data for PVA30-b-PVP24: 1H NMR (400 MHz, D2O): δ = 3.52 (−CHN–, br d, 1H), δ = 4.00 (−CHO–, br, 1H), δ = 3.12 (−NCH2CH2–, br d, 2H), δ = 2.49 (−NCOCH2–, br, 2H), δ = 2.13 (−NCH2CH2CH2–, br d, 2H), δ = 1.80–1.50 (−CH2–, br, 2H).
Results and Discussion
The key aim of this study was to investigate if block copolymers of PVA would retain or lose their potent IRI activity, as all other modifications to the PVA structure have been reported to result in a complete loss of activity.24 These prior observations raise questions about the mode of action and limit its translation into applications, but they also create the opportunity to use macromolecular architecture to modulate activity. To introduce a second water-soluble block, poly(vinylpyrrolidone) (PVP) was selected as it is compatible with the same xanthates required for PVA, which will allow reinitiation and hence block copolymerization (vide infra). The chain transfer agent (xanthate) was synthesized according to previous reports and is detailed in the Supporting Information. RAFT/MADIX was conducted in dioxane (Scheme 1), at various [monomer]:[CTA] ratios, isolated and characterized by 1H NMR and SEC, Table 1.
Scheme 1. Preparation of PVP macroCTA.

Table 1. PVP macroCTAs Prepared for This Study.
| macro CTA | [M]:[CTA]a | conv.b (%) | Mn,NMRc (g mol–1) | Mn,SECd (g mol–1) | Mw/Mnd | DPne | DPnf |
|---|---|---|---|---|---|---|---|
| PVP30 | 50 | 60 | 3350 | 1700 | 1.46 | 15 | 30 |
| PVP55 | 100 | 30 | 6110 | 3350 | 1.42 | 30 | 55 |
| PVP70 | 100 | 38 | 7780 | 3840 | 1.42 | 35 | 70 |
Monomer to RAFT agent ratio. [M] = 2 M in 1,4-dioxane.
Determined by 1H NMR spectroscopy.
Determined by 1H NMR of PVP after precipitation and drying.
Determined by SEC.
Number-average degree of polymerization from SEC.
Number-average degree of polymerization from 1H NMR.
As previously reported, the SEC Mn values obtained for PVP are underestimated, as DMF (or other standard SEC solvents) is not an ideal solvent for it, but dispersity values around 1.4 were observed, as would be expected for this monomer by RAFT/MADIX.37,381H NMR end-group analysis enabled estimation of the molecular weight by comparing the aromatic end-groups to the main chain, which were in agreement with that predicted from the conversion. From this point on, the NMR degree of polymerization will be the stated value. With these macro-CTAs on hand, reinitiation using vinyl acetate as the monomer was conducted (Scheme 2) and the following block copolymers were isolated and characterized, Table 2. Acceptable dispersity and monomodal distributions were obtained (Supporting Information), indicating quantitative reinitiation and no evidence of unreacted homopolymer. Hydrazinolysis was used to quantitatively remove the acetate protecting groups, confirmed by infrared spectroscopy and 1H NMR (see the Supporting Information).
Scheme 2. Preparation of PVA-b-PVP Block Copolymer.

Table 2. PVA-b-PVP Block Copolymers.
| PVAc-b-PVP | [M]/[mCTA]a | conv.b (%) | Mn,NMRc (g mol–1) | Mn,SECd (g mol–1) | Mw/Mnd | PVAce DP (−) |
|---|---|---|---|---|---|---|
| PVAc20-b-PVP30 | 20 | 96.8 | 5050 | 3400 | 1.47 | 20 |
| PVAc18-b-PVP55 | 500 | 15.6 | 7650 | 5400 | 1.35 | 18 |
| PVAc32-b-PVP70 | 1000 | 11.2 | 10520 | 9130 | 1.36 | 32 |
| PVAc37-b-PVP70 | 2000 | 6.1 | 10950 | 9650 | 1.38 | 37 |
[Total monomer]/[RAFT agent] ratio; polymerizations conducted in 2 M monomer solutions in dioxane.
Determined by 1H NMR spectroscopy. Theoretical Mn determined from the monomer to RAFT agent ratio.
Determined by 1H NMR of the copolymers after precipitation and drying.
Determined by SEC in DMF using PMMA standards.
Determined by 1H NMR spectroscopy of the PVP-b-PVAc block copolymers.
With this panel of polymers on hand, their IRI activity could be tested. This was done using the ‘splat’ assay. Briefly, 10 μL droplets of the polymers in phosphate buffered saline (PBS) were dropped onto a glass coverslip that sat on a chilled (−80 °C) aluminum plate. This generates a large number of <10 μm ice crystals by rapid nucleation. This is then transferred to a cold stage set at −8 °C and the ice crystal size measured after 30 min, enabling only growth (or inhibition of growth) to be measured. Data is reported relative to a PBS control, with smaller numbers indicating more activity. Control experiments confirmed that PVP homopolymers are nonactive. To enable a critical comparison of PVA activity, all data is reported as the PVA concentration in solution, not the total polymer concentration, to enable the effect of the second block to be probed and the results are shown in Figure 1.
Figure 1.
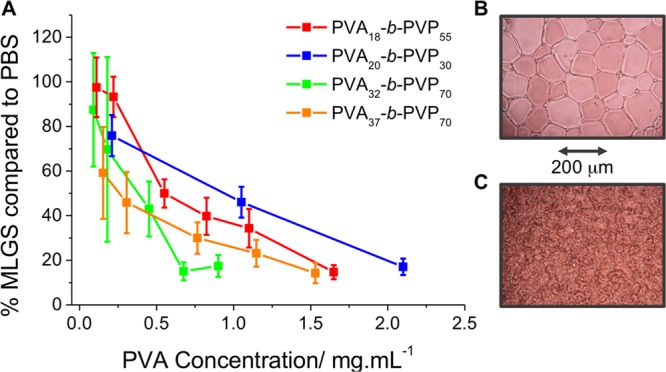
Ice recrystallization inhibition activity of PVA-b-PVP block copolymers. (A) Activity derived from splat test. (B) Example ice wafer of PBS alone. (C) Ice wafer of 1 mg·mL–1 of PVA32-b-PVP70. MLGS = mean largest grain size relative to a PBS control, expressed as %. Error bars represent the standard deviation from at least three measurements.
In line with our earlier work,24 the PVA containing polymers all showed remarkably strong IRI activity, comparable to that of native antifreeze proteins. In all cases, even with large PVP blocks, IRI activity was retained with the longer (DP 32) PVAs being more active than shorter ones (DP = 20), highlighting the crucial molecular weight dependence of this property. Considering the large size of the PVP component, it is somewhat surprising that activity was not affected. An additional series of experiments was conducted to compare block copolymer versus random copolymers to ascertain the role of sequence. A polymer was synthesized containing just 20 PVP and 70 PVA and tested for IRI, Figure 2. DP 70 PVA should be a very potent IRI, but the addition of the PVP units removed activity. In comparison, a DP 20 PVA as a homo or block copolymer retained all activity, again highlighting that the macromolecular architecture, not just the composition, is crucial for these polymers. One current hypothesis for the disruption of activity in copolymers is that unbroken sequences of hydroxyl groups are required for ice binding. This is discussed later in the manuscript.
Figure 2.
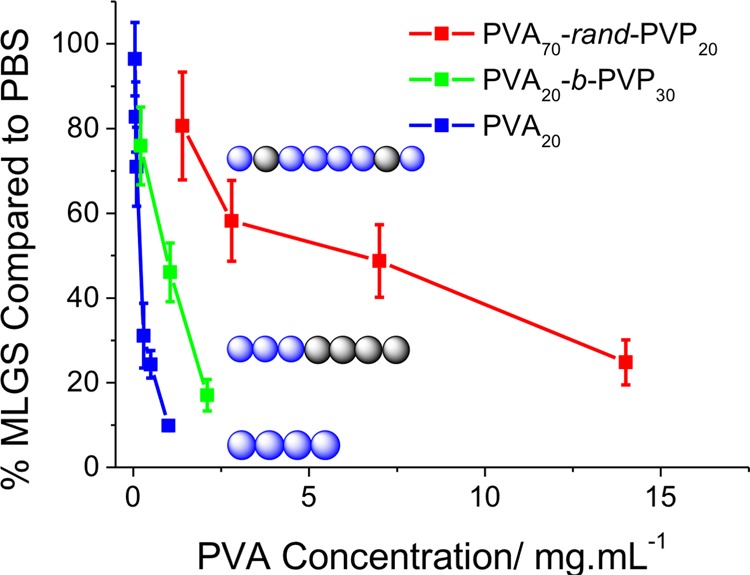
Comparison of sequence distribution on ice recrystallization inhibition activity. PVA70-rand-PVP20 random copolymer from previous work24 (Mn = 4320, Đ = 1.33, PVA = 48 wt %). Cartoons for PVA (blue) and PVP (black) are intended as a guide to structure only. MLGS = mean largest grain size relative to a PBS control, expressed as %. Error bars represent the standard deviation from at least three measurements.
The above data provided some insight into the role of a second block on PVA activity. However, the synthesis method of the PVP first meant that each PVA block was slightly different in length and dispersity which might conflict with the interpretation. As IRI activity is very sensitive to PVA block length in this regime, the synthetic strategy was revised, to synthesize the PVA blocks first, with predefined lengths, and to use these in a series of reinitiation experiments using PVP, Scheme 3. This ensured that the PVA component is identical across the series for a more systematic approach. Two PVAs were synthesized with a DP of 10 and 30, Table 3. These lengths were chosen, as the DP range of 10–30 is where the most significant changes in PVA’s IRI activity have been seen. The macro-CTAs were then used for subsequent chain extension with PVP, and the acetate groups removed by hydrazinolysis, Table 4. As with the homo-PVPs, the SEC molecular weights were lower than expected, and NMR values were used.
Scheme 3. Preparation of PVAc macroCTA.

Table 3. Initial PVAc Homopolymers Prepared for This Study.
| entry | [M]:[CTA]a | convb (%) | Mn,Theoc (g mol–1) | Mn,NMRd (g mol–1) | Mn,SECe (g mol–1) | Mw/Mne | DPnf |
|---|---|---|---|---|---|---|---|
| PVAc30 | 100 | 24.2 | 8610 | 2670 | 2640 | 1.12 | 30 |
| PVAc10 | 100 | 10 | 1056 | 780 | 920 | 1.32 | 9 |
Monomer to CTA ratio.
Determined by 1H NMR spectroscopy.
Theoretical Mn determined from the monomer to RAFT agent ratio.
Determined by 1H NMR spectroscopy.
Determined by SEC in THF using PMMA standards.
Number-average degree of polymerization.
Table 4. PVAc10-b-PVP Block Copolymers Prepared for This Study.
| PVAc-b-PVP | [M]/[CTA]a | NVP (mL) | conv.b (%) | Mn,NMRc (g mol–1) | Mn,DMFd (g mol–1) | Mn,THFe (g mol–1) | Mw/Mnd |
|---|---|---|---|---|---|---|---|
| PVA30-b-PVP60 | 240 | 3 | 18.3 | 4730 | 5380 | N/A | 1.16 |
| PVA10-b-PVP27 | 10 | 0.75 | 18.3 | 1970 | 3080 | 570 | 1.37 |
| PVA10-b-PVP90 | 30 | 1.90 | 100 | 4190 | 4600 | 740 | 1.59 |
| PVA10-b-PVP72 | 50 | 3.79 | 56.8 | 3640 | 7479 | 760 | 1.76 |
| PVA10-b-PVP85 | 75 | 5.78 | 43.2 | 3970 | 10060 | 690 | 1.99 |
[Total monomer]/[RAFT agent] ratio; polymerization conducted in 5 mL of 1,4-dioxane and varying concentrations of N-vinylpyrrolidone.
Determined by 1H NMR spectroscopy.
Determined by 1H NMR of the copolymers after precipitation and drying.
Determined by SEC in THF using PMMA standards.
Determined by 1H NMR spectroscopy of the PVAc-b-PVP block copolymers.
Figure 3 shows the results of the IRI testing of DP 30 PVA homopolymer and following two different chain extensions with PVP. The data shows remarkably good correlation with the PVA-weighted activity, being statistically identical in all cases, confirming that addition of a large, hydrophilic addition block does not disrupt function when using this length of PVA.
Figure 3.
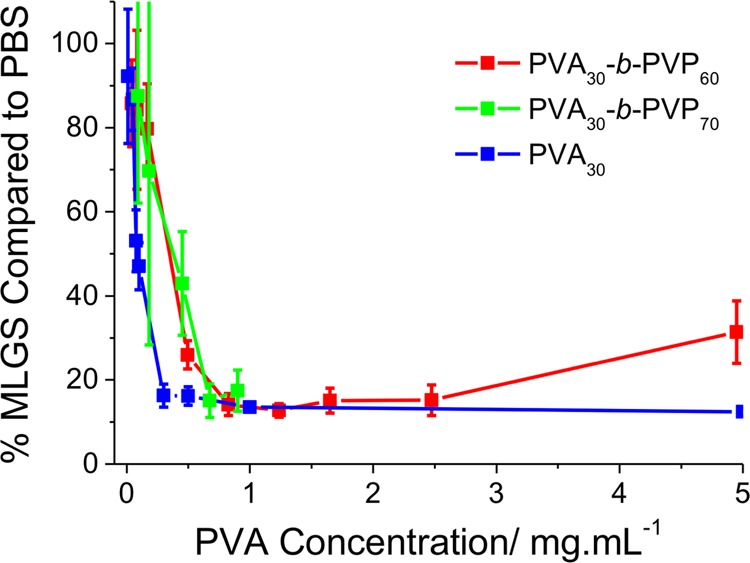
Effect of PVP block on ice recrystallization inhibition activity of PVA30. Concentration reported is correct to PVA concentration not total polymer concentration. MLGS = mean largest grain size relative to a PBS control, expressed as %. Error bars represent the standard deviation from at least three measurements.
The shorter DP 10 polymers were also tested. DP 10 PVA has significantly lower IRI activity (but still far more than other synthetic IRIs) than those with DP > 20, making it a more stringent test for the impact of chain extension. Four different PVP block lengths were tested and compared to DP 10 PVA and reported as a function of PVA concentration. In line with the data above, activity was retained in all cases with there being no statistically significant differences between any of the individual block copolymers compared to the homopolymer. The only exception to this was PVA10 and PVA10-b-PVP72. The block copolymer appears to have slightly higher activity but only in a rather narrow concentration range which we are hesitant to explain as an enhancement as the activity plateaus to a similar level.
The data presented above demonstrates that although main chain modifications to IRI active polymers are not tolerated additional functionality can be easily included via block copolymerization. We have recently observed that with supramolecular star-branched PVAs where addition of a third arm does not provide additional activity, relative to a pseudo-two-armed equivalent (a linear polymer).25 This is in agreement with these findings, which can enable some insight into the potential mechanisms of action. Native antifreeze proteins appear to function by direct interaction with specific faces of growing ice crystals, leading to thermal hysteresis and dynamic ice shaping. For these polymers, we see no evidence of ice shaping. Ben et al. have studied several small molecule IRI active compounds and found IRI without any TH or DIS and using solid state NMR have found evidence that disruption of the quasi liquid layer interface may be the crucial mechanism of action to obtain IRI without ice binding.39 There is also speculation that PVA’s activity is due to its specific hydroxyl group spacing interfacing with the ice crystals, and hence disruption of the PVA primary sequence (as a copolymer, for example) reduces activity. In the case of PVA10-b-PVP90, over 90% of the polymer is PVP, shown in a space filling model in Figure 4B. Such a large modification would be expected to dramatically affect the polymer’s ability to bind ice if that was the dominant mechanism of action. Davies et al. have shown that conjugation of a 7 kDa AFP to a much larger maltose-binding protein (42 kDa) is not only tolerated but can enhance TH activity and suggests AFP–AFP contact (cooperativity) is not crucial for TH activity.28 The difference here is that these short PVAs do not show TH nor ice shaping and hence major structural changes maybe have different effects. If the PVA is acting on bulk water or the ice/water interface, the PVP “tail” would simply be dissolved in the liquid water fraction, perhaps explaining why it has no effect on IRI activity. The passive nature of the PVP block also rules out viscosity as being crucial for IRI; for any polymer, increasing the molecular weight dramatically increases their solution viscosity, which could slow the rate of water exchange. However, the long PVP chains did not increase activity, despite these polymers having higher viscosity relative to PVA homopolymers or shorter block copolymers. Recent studies on native AFPs have revealed an ice-like hydration layer on the antifreeze protein surfaces, which directs the interaction with ice; such a mechanism cannot be ruled out here.40 While not conclusive evidence, these findings could support the hypothesis that ice growth inhibiting polymers (or molecules) may not have the same molecular-level mechanism as antifreeze proteins, even though they give rise to similar macroscopic effects. The data presented here on relatively short PVAs also may not scale with high molecular weight PVAs (which are extremely challenging to access using controlled radical polymerization), where ice shaping and thermal hysteresis is reported22 and the key dependence on unbroken hydroxyl units also suggests some specific interactions, which will form the basis of future studies.
Figure 4.
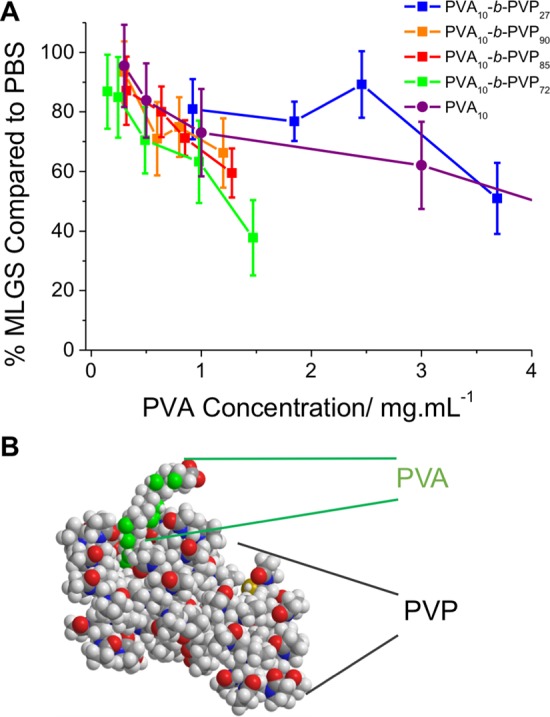
(A) PVA weight % dependent IRI activity of PVA10-b-PVP block copolymers compared to PVA10 (Mn = 440, Đ = 1.18). MLGS = mean largest grain size relative to a PBS control, expressed as %. Error bars represent the standard deviation from at least three measurements. (B) Space-filling model highlighting the small amount of PVA present in the block copolymer. Generated using Chem3D 15.1, this cartoon is for illustrative purposes only.
Conclusions
Antifreeze proteins and glycoproteins are complex macromolecules which enable life to survive at extreme low temperatures, but their mechanism of action, and the mechanism of action of their synthetic mimics, is not understood. Here a detailed study in the influence of block copolymer structure on the ice recrystallization inhibition activity is presented. Using RAFT/MADIX polymerization, a library of well-defined polymers were obtained, and for the first time, chain extension of vinylpyrrolidone from a poly(vinyl acetate) macromonomer was demonstrated. Full characterization of the polymers confirmed monomodal distributions and full reinitiation of the first block. Quantitative ice recrystallization inhibition analysis on this library revealed that addition of the PVP block was essentially passive; it did not increase or decrease activity. Polymers synthesized with the extreme composition of only 10% PVA and 90% PVP retained activity statistically identical to that of PVA homopolymers. These observations are important, as all previous reports on structural modifications of ice recrystallization inhibition active polymers resulted in a loss of activity. The observations with block copolymers show that macromolecular architecture could be a powerful tool to incorporate additional functionality into these polymers, for example, to increase circulation times in vivo, or to enable them to be conjugated to other macromolecules to create hybrid materials. The lack of influence of the PVP blocks also gives some insight into the mechanism of action of PVA and its interaction with ice, or the ice/water interface. The additional steric bulk of a PVP block would be expected to influence the interaction with ice (either positively or negatively), but the passive nature might confirm that PVAs (at least at the low molecular range being used here) tend to act at the interface rather than on the ice crystal surface, but further detailed studies are needed to confirm any of these mechanisms. In summary, this work represents the first quantitative and systematic study into block copolymer structure on IRI activity and will have implications both in polymer mimetics and also in the rational design of de novo antifreeze peptides/proteins. These results not only raise hope that ice inhibiting polymer can be incorporated into more complex structures for advanced application but also gives some insight into the mechanisms of ice growth.
Acknowledgments
The authors acknowledge the Leverhulme trust (RPG 144) for providing a studentship for T.R.C. M.I.G. holds an ERC starting grant (CRYOMAT 638661). R.N. is a Royal Society University research fellow. The Royal Society are also thanked for funding the cryo-microscopes used in this study.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.6b00915.
Ice recrystallization inhibition (splat) assay, synthesis of methyl (ethoxycarbonothioyl) sulfanyl benzene (CTA), SEC traces of polymers (S1–S6), 1H NMR spectroscopic analysis of polymers (S7 and S8), and IRI activity of commercial PVP (S9) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Harding M. M.; Anderberg P. I.; Haymet A. D. J. Eur. J. Biochem. 2003, 270 (7), 1381–1392. 10.1046/j.1432-1033.2003.03488.x. [DOI] [PubMed] [Google Scholar]
- Ustun N. S.; Turhan S. J. Food Process. Preserv. 2015, 39 (6), 3189–3197. 10.1111/jfpp.12476. [DOI] [Google Scholar]
- Esser-Kahn A. P.; Trang V.; Francis M. B. J. Am. Chem. Soc. 2010, 132 (38), 13264–13269. 10.1021/ja103038p. [DOI] [PubMed] [Google Scholar]
- Balcerzak A. K.; Capicciotti C. J.; Briard J. G.; Ben R. N. RSC Adv. 2014, 4 (80), 42682–42696. 10.1039/C4RA06893A. [DOI] [Google Scholar]
- Gibson M. I. Polym. Chem. 2010, 1 (8), 1141–1152. 10.1039/c0py00089b. [DOI] [Google Scholar]
- Liu S.; Wang W.; von Moos E.; Jackman J.; Mealing G.; Monette R.; Ben R. N. Biomacromolecules 2007, 8 (5), 1456–1462. 10.1021/bm061044o. [DOI] [PubMed] [Google Scholar]
- Chao H.; Davies P. L.; Carpenter J. F. J. Exp. Biol. 1996, 199 (Pt 9), 2071–2076. [DOI] [PubMed] [Google Scholar]
- Capicciotti C. J.; Kurach J. D. R.; Turner T. R.; Mancini R. S.; Acker J. P.; Ben R. N. Sci. Rep. 2015, 5, 9692. 10.1038/srep09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czechura P.; Tam R. Y.; Dimitrijevic E.; Murphy A. V.; Ben R. N. J. Am. Chem. Soc. 2008, 130 (10), 2928–2929. 10.1021/ja7103262. [DOI] [PubMed] [Google Scholar]
- Tam R. Y.; Rowley C. N.; Petrov I.; Zhang T.; Afagh N. A.; Woo T. K.; Ben R. N. J. Am. Chem. Soc. 2009, 131, 15745–15753. 10.1021/ja904169a. [DOI] [PubMed] [Google Scholar]
- Yu S. O.; Brown A.; Middleton A. J.; Tomczak M. M.; Walker V. K.; Davies P. L. Cryobiology 2010, 61 (3), 327–334. 10.1016/j.cryobiol.2010.10.158. [DOI] [PubMed] [Google Scholar]
- Olijve L. L. C.; Hendrix M. M. R. M.; Voets I. K. Macromol. Chem. Phys. 2016, 217 (8), 951–958. 10.1002/macp.201500497. [DOI] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. Nat. Commun. 2014, 5, 3244. 10.1038/ncomms4244. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. ACS Biomater. Sci. Eng. 2015, 1 (9), 789–794. 10.1021/acsbiomaterials.5b00162. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Lovett J. R.; Armes S. P.; Gibson M. I. Angew. Chem., Int. Ed. 2016, 55 (8), 2801–2804. 10.1002/anie.201511454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller R. C.; Pessin J. E.; Vatish M.; Mitchell D. A.; Gibson M. I. Biomater. Sci. 2016, 47, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMerlis C.; Schoneker D. Food Chem. Toxicol. 2003, 41 (3), 319–326. 10.1016/S0278-6915(02)00258-2. [DOI] [PubMed] [Google Scholar]
- Rajan R.; Hayashi F.; Nagashima T.; Matsumura K. Biomacromolecules 2016, 17 (5), 1882–1893. 10.1021/acs.biomac.6b00343. [DOI] [PubMed] [Google Scholar]
- Matsumura K.; Hyon S.-H. Biomaterials 2009, 30 (27), 4842–4849. 10.1016/j.biomaterials.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Matsumura K.; Bae J. Y.; Kim H. H.; Hyon S. H. Cryobiology 2011, 63 (2), 76–83. 10.1016/j.cryobiol.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Cameron N. R.; Gibson M. I. Chem. Commun. 2015, 51 (65), 12977–12980. 10.1039/C5CC04647E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budke C.; Koop T. ChemPhysChem 2006, 7 (12), 2601–2606. 10.1002/cphc.200600533. [DOI] [PubMed] [Google Scholar]
- Congdon T.; Shaw P.; Gibson M. I. Polym. Chem. 2015, 6 (26), 4749–4757. 10.1039/C5PY00775E. [DOI] [Google Scholar]
- Congdon T.; Notman R.; Gibson M. I. Biomacromolecules 2013, 14 (5), 1578–1586. 10.1021/bm400217j. [DOI] [PubMed] [Google Scholar]
- Phillips D. J.; Congdon T. R.; Gibson M. I. Polym. Chem. 2016, 7 (9), 1701–1704. 10.1039/C5PY01948F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens C. A.; Drori R.; Zalis S.; Braslavsky I.; Davies P. L. Bioconjugate Chem. 2015, 26 (9), 1908–1915. 10.1021/acs.bioconjchem.5b00290. [DOI] [PubMed] [Google Scholar]
- Mancini R. J.; Lee J.; Maynard H. D. J. Am. Chem. Soc. 2012, 134 (20), 8474–8479. 10.1021/ja2120234. [DOI] [PubMed] [Google Scholar]
- DeLuca C. I.; Comley R.; Davies P. L. Biophys. J. 1998, 74 (3), 1502–1508. 10.1016/S0006-3495(98)77862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch E.; Mastai Y. Macromol. Rapid Commun. 2007, 28 (23), 2256–2261. 10.1002/marc.200700501. [DOI] [Google Scholar]
- Mastai Y.; Rudloff J.; Cölfen H.; Antonietti M. ChemPhysChem 2002, 3 (1), 119–123. . [DOI] [PubMed] [Google Scholar]
- Debuigne A.; Willet N.; Jérôme R.; Detrembleur C. Macromolecules 2007, 40 (20), 7111–7118. 10.1021/ma0712908. [DOI] [Google Scholar]
- Shaver M. P.; Hanhan M. E.; Jones M. R. Chem. Commun. 2010, 46 (12), 2127–2129. 10.1039/b922202b. [DOI] [PubMed] [Google Scholar]
- Nguyen T. L. U.; Eagles K.; Davis T. P.; Barner-Kowollik C.; Stenzel M. H. J. Polym. Sci., Part A: Polym. Chem. 2006, 44 (15), 4372–4383. 10.1002/pola.21518. [DOI] [Google Scholar]
- Wan D.; Satoh K.; Kamigaito M.; Okamoto Y. Macromolecules 2005, 38 (25), 10397–10405. 10.1021/ma0515230. [DOI] [Google Scholar]
- Bailly N.; Thomas M.; Klumperman B. Biomacromolecules 2012, 13 (12), 4109–4117. 10.1021/bm301410d. [DOI] [PubMed] [Google Scholar]
- Bailly N.; Pound-Lana G.; Klumperman B. Aust. J. Chem. 2012, 65 (8), 1124–1131. 10.1071/CH12185. [DOI] [Google Scholar]
- Ieong N. S.; Brebis K.; Daniel L. E.; O’Reilly R. K.; Gibson M. I. Chem. Commun. 2011, 47 (42), 11627–11629. 10.1039/c1cc15171a. [DOI] [PubMed] [Google Scholar]
- Ieong N. S.; Hasan M.; Phillips D. J.; Saaka Y.; O’Reilly R. K.; Gibson M. I. Polym. Chem. 2012, 3 (3), 794–799. 10.1039/c2py00604a. [DOI] [Google Scholar]
- Capicciotti C. J.; Leclere M.; Perras F. A.; Bryce D. L.; Paulin H.; Harden J.; Liu Y.; Ben R. N. Chem. Sci. 2012, 3 (5), 1408–1416. 10.1039/c2sc00885h. [DOI] [Google Scholar]
- Meister K.; Strazdaite S.; DeVries A. L.; Lotze S.; Olijve L. L. C.; Voets I. K.; Bakker H. J. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (50), 17732–17736. 10.1073/pnas.1414188111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.