

Abstract
Cancer vaccines based on mRNA are extensively studied. The fragile nature of mRNA has instigated research into carriers that can protect it from ribonucleases and as such enable its systemic use. However, carrier-mediated delivery of mRNA has been linked to production of type I interferon (IFN) that was reported to compromise the effectiveness of mRNA vaccines. In this study, we evaluated a cationic lipid for encapsulation of mRNA. The nanometer-sized, negatively charged lipid mRNA particles (LMPs) efficiently transfected dendritic cells and macrophages in vitro. Furthermore, i.v. delivery of LMPs resulted in rapid expression of the mRNA-encoded protein in spleen and liver, predominantly in CD11c+ cells and to a minor extent in CD11b+ cells. Intravenous immunization of mice with LMPs containing ovalbumin, human papilloma virus E7, and tyrosinase-related protein-2 mRNA, either combined or separately, elicited strong antigen-specific T-cell responses. We further showed the production of type I IFNs upon i.v. LMP delivery. Although this decreased the expression of the mRNA-encoded protein, it supported the induction of antigen-specific T-cell responses. These data question the current notion that type I IFNs hamper particle-mediated mRNA vaccines.
Keywords: cytotoxic T lymphocyte, immunotherapy, liposome, mRNA, particle, type I interferon
Introduction
Nucleic acids like DNA and mRNA have been used to develop vaccines for priming of tumor-specific cytotoxic T lymphocytes (CTLs). Compared with other antigen forms like proteins, nucleic acids are easy to produce and can be engineered to target antigen-derived epitopes to both the major histocompatibility complex I (MHC I) and MHC II antigen-presenting pathways. As such nucleic acid vaccines enable activation of strong antitumor T-cell responses.1,2 In contrast to DNA, the use of mRNA does not require nuclear translocation or transcription to mediate protein expression. Moreover, the use of mRNA is not faced with the risk of genomic integration of the delivered sequence. Consequently, tumor antigen encoding mRNA has been studied as an off-the-shelf cancer vaccine.2,3,4 The use of mRNA for cancer vaccination brings forth the challenge of its delivery to antigen-presenting cells (APCs). In this regard, the existence of multiple APC types, their different location, the mode of mRNA uptake, and finally their activation by mRNA should be considered.
APCs are a heterogeneous group of cells that have the ability to capture, process, and present antigens to T cells. Dendritic cells (DCs) are considered professional APCs, as they can initiate primary T-cell responses. Therefore, several strategies have been devised to target DCs.5,6,7,8 However, a recent study underscored that selective targeting of antigens to DCs does not result in strong antigen-specific T-cell responses. Strong T-cell stimulation was only observed when the antigen was also targeted to macrophages, another important APC subset.7 These data suggest that mRNA vaccines should be delivered to both DCs and macrophages to function optimally.
Furthermore, it was shown that the location of mRNA delivery determines the strength of the ensuing immune response. Antigen mRNA formulated in lactated Ringer's solution has been delivered to CD11c+ cells within lymph node and skin. Intranodal delivery of mRNA resulted in stronger immune responses than its delivery in skin, most likely because intranodal DCs are intimately associated with T cells.9,10 However, intranodal delivery of vaccines in mice is invasive, requiring an incision to expose the lymph node. In humans, intranodal delivery of mRNA is performed under ultrasound guidance and although well tolerated, it is challenging. Therefore, alternative routes of delivering mRNA like intrasplenic and i.v. injection have been evaluated.11,12,13 These approaches envisage mRNA transfection of splenic APCs that are also intimately associated with T cells and were shown to efficiently activate antigen-specific CTLs. Importantly, intrasplenic and i.v. delivery of mRNA requires its encapsulation to protect the mRNA from degradation by ribonucleases.14,15 Nonviral mRNA encapsulation is often performed using lipids, polymers, and combinations thereof.2 The resulting nanoparticles and (sometimes) microparticles have been tested in vivo with varying success using i.v., s.c, i.d., i.p., i.m., and recently i.n. injections.16,17 An issue that arose during this preclinical work is the immunogenicity of particulate mRNA. Its delivery to APCs has been linked with the induction of strong type I interferon (IFN) responses.18,19,20,21,22,23 This innate immune response is elicited through the interaction of the mRNA with pattern-recognition receptors like toll-like receptors (TLRs) and retinoic acid-inducible protein I.18,21,24 In the context of mRNA vaccines, it was suggested that type I IFNs compromise transgene expression and as such induction of antigen-specific immunity.19,20 These data imply that an ideal anticancer mRNA vaccine should enable systemic delivery of tumor antigen mRNA to DCs and macrophages with minimal induction of type I IFNs in order to initiate strong CTL responses. We evaluated the encapsulation of mRNA in Lipofectamine RNAiMAX, a proprietary lipid formulation, as lipids are biocompatible and biodegradable. Moreover, lipid particles are highly versatile as their characteristics can be altered for instance through PEGylation or inclusion of targeting moieties. We addressed the best suited mRNA:lipid ratio for packaging and subsequent in vitro transfection of DCs and macrophages. We further examined the kinetics and location of the expression of the mRNA-encoded antigen after systemic delivery of lipid mRNA particles (LMPs) and whether APCs were transfected in vivo. Induction of antigen-specific CTLs was evaluated, addressing whether multiple antigens could be delivered simultaneously without affecting the strength of the CTL response. Finally, we examined the induction of type I IFNs and how these impacted on the antigen expression and CTL response.
Results
Efficient in vitro transfection of DCs and macrophages with LMPs
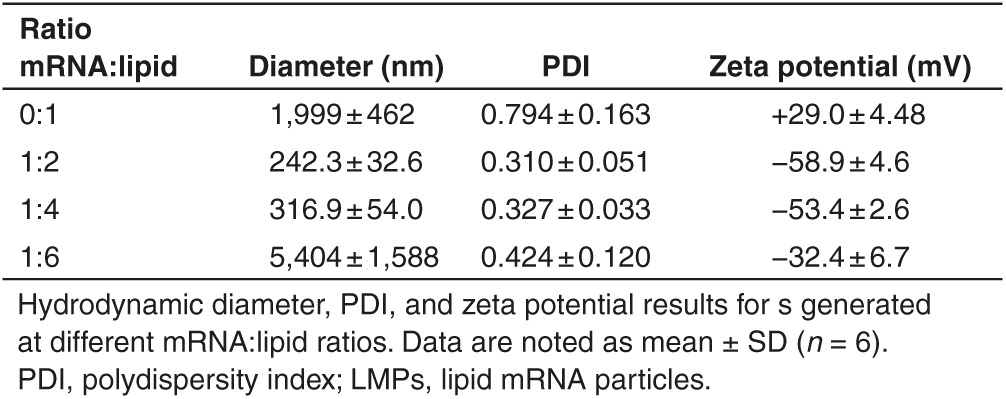
To establish whether Lipofectamine RNAiMAX, a cationic lipid, can package and protect mRNA, we admixed mRNA encoding truncated nerve growth factor receptor or Firefly luciferase (Fluc) at different mass (RNA in µg) per volume (lipid in µl) ratios with this lipid. The electrophoretic mobility shift assay showed that mRNA was completely packaged in LMPs at an mRNA:lipid ratio of 1:6, as the mRNA did not migrate in the agarose gel, whereas at a 1:2 and 1:4 ratio, free mRNA was detectable (Figure 1a). This free mRNA represented 50% and 25% of the total mRNA (data not shown), respectively, and was degraded upon exposure to serum (Figure 1b). The LMPs obtained through mRNA packaging at an mRNA:lipid ratio of 1:6, exhibited >5 µm diameter with a high polydispersity index and a negative surface charge, indicating the formation of large aggregates when all the mRNA is complexed with the lipid. At an mRNA:lipid ratio of 1:2 and 1:4, nanometer-sized particles with a lower polydispersity index were formed. Due to the relative excess of mRNA over lipid, these LMPs exhibited a more negative surface charge (Table 1). Flow cytometry measurements showed efficient in vitro transfection of human embryonal kidney (HEK) 293T cells in OptiMem with LMPs containing mRNA encoding enhanced green fluorescent protein (eGFP). Importantly, both the cell viability and transfection efficiency decreased when more lipids were used to package the same amount of mRNA (Figure 1c–e). Therefore, subsequent experiments were performed with LMPs generated at a 1:2 mRNA:lipid ratio. As the LMPs were intended for i.v. immunization, we also assessed the transfection efficiency of bone marrow-derived DCs and RAW246.7 macrophages. The results on CD11c+ DCs and CD11b+ macrophages showed high transfection efficiency in OptiMem as well as serum-containing medium (Figure 1f,g). Importantly, in the presence of serum, the level of protein expression was significantly higher compared with the situation in serum-free medium (Figure 1f,g), underpinning the suitability of the presented particles for the intended in vivo applications.
Figure 1.

Efficient in vitro transfection of dendritic cells (DCs) and macrophages with lipid mRNA particles (LMPs). (a,b) Electric mobility shift assay of mRNA packaged at different mRNA:lipid ratios. mRNA formulated in 0.8 lactated Ringer's solution served as a control. In a the samples were not exposed to serum, whereas in b the samples were incubated with 10% fetal clone I serum for 30 minutes. The photographs show representative pictures of the three independent experiments. Lanes 1 to 5 represent: (1) ladder; (2) mRNA in 0.8 lactated Ringer's solution; LMPs at a 1:2 (ref. 25); (4) 1:4, and (5) 1:6 ratio. (c) Representative histogram of 5 × 105 human embryonal kidney 293T cells transfected with 2.5 µg enhanced green fluorescent protein (eGFP) mRNA packaged at different mRNA:lipid ratios. Transfection was performed in OptiMem. Twenty-four hours later, cells were harvested, counted by trypan blue staining to determine the percentage of live/dead cells, and evaluated in flow cytometry for the expression of eGFP. The graph in d depicts the percentage of eGFP+ cells after transfection (n = 5). The graph in e depicts the cell viability after transfection (n = 5). (f) Five hundred thousand DCs were transfected with 2.5 µg eGFP mRNA, complexed at a 1:2 mRNA:lipid ratio in the absence or presence of serum. The graph depicts the percentage and the mean fluorescence intensity (MFI) of eGFP+ cells (n = 3). (g) Five hundred thousand RAW246.7 macrophages were transfected with 2.5 µg eGFP mRNA complexed at a 1:2 mRNA:lipid ratio in the absence or presence of serum. Twenty-four hours later, cells were harvested and evaluated in flow cytometry for the expression of eGFP. The graph depicts the percentage and the MFI of eGFP+ cells (n = 4). The graphs in d–g summarize the results of the indicated number of experiments as mean ± SEM. ns, not significant.
Table 1. Characterization of LMPs.
Intravenous but not s.c. or i.m. delivery of LMPs results in potent antigen-specific T-cell responses
To address whether LMPs are promising as a vaccine candidate, we first evaluated the expression of Fluc after i.v., s.c., and i.m. delivery of LMPs containing 10 µg Fluc mRNA. Expression of the Fluc protein was evaluated 24 hours later using in vivo bioluminescence imaging. Expression of Fluc was detectable after i.v. and i.m. delivery of the LMPs, while it was not detectable after their s.c. delivery (Figure 2a,b). Next, we evaluated the stimulation of ovalbumin (OVA)-specific T cells after i.v., s.c., and i.m. delivery of LMPs containing 5 µg OVA mRNA. The expansion of OVA-specific CD8+ T cells was evaluated in flow cytometry using MHC Dextramers for detection of H-2Kb/SIINFEKL (OVA)-specific T cells, while their cytolytic activity was evaluated using an in vivo cytotoxicity assay. Intravenous delivery of OVA mRNA containing LMPs resulted in the strongest expansion of OVA-specific CD8+ T cells (Figure 2c). In accordance with this result, we observed the strongest lyses of OVA-presenting target cells after i.v. delivery of the LMPs (Figure 2d). Based on these results, we continued with i.v. delivery of LMPs in the following experiments.
Figure 2.

Intravenous but not s.c. or i.m. delivery of lipid mRNA particles (LMPs) results in potent ovalbumin (OVA)-specific T-cell responses. (a) Representative in vivo bioluminescence images after s.c., i.m., or i.v. delivery of 10 µg Fluc mRNA (n = 4). (b) The graph summarizes the results shown in a as mean ± SEM (n = 4). (c,d) C57BL/6 mice received a s.c., i.m., or i.v. injection with LMPs containing 5 µg OVA mRNA. Five days later, we performed c a major histocompatibility complex (MHC) Dextramer staining to detect H-2Kb/SIINFEKL (OVA)-specific T cells and d an in vivo cytotoxicity assay to detect lyses of OVA-presenting target cells (n = 4). SC, subcutaneous; IM, intramuscular; IV, intravenous; ns, not significant.
Intravenous delivery of LMPs results in protein expression in spleen and liver
To evaluate the in vivo mRNA transfection kinetics after systemic delivery of LMPs, we packaged 10 µg Fluc mRNA and injected it intravenously in C57BL/6 mice. Expression of the Fluc protein was evaluated using in vivo bioluminescence imaging. We showed Fluc expression as soon as 15 minutes after injection and could detect Fluc expression up to 4 days after injection (Figure 3a,b). To determine which organs exhibited Fluc expression, we isolated lymph nodes, spleen, liver, lung, intestines, heart, kidneys, and stomach 4 hours after LMP delivery, showing positivity in the spleen and liver (Figure 3c).
Figure 3.

Kinetics of the in vivo expression of the mRNA-encoded protein after i.v. delivery of lipid mRNA particles (LMPs). (a) Representative in vivo bioluminescence images of the same mouse at different time points after i.v. injection of 10 μg Fluc mRNA packaged at a mRNA:lipid ratio of 1:2 (n = 4). (b) The graph depicts the expression of Fluc at the indicated time points, summarizing the results as mean ± SEM (n = 4). (c) Representative bioluminescence image of organs that were excised 4 hours after i.v. delivery of 10 µg Fluc mRNA to confirm the Fluc expression in the spleen and liver (n = 7).
Protein expression after i.v. delivery of LMPs is detected in CD11c+ and CD11b+ cells
Next, we addressed whether APCs, in particular CD11c+ DCs, express the mRNA-encoded protein upon i.v. delivery of LMPs, since this is critical for the subsequent activation of antigen-specific immunity. Therefore, we delivered Fluc LMPs (10 µg mRNA) intravenously in wild-type C57BL/6 and CD11c diphtheria toxin receptor transgenic mice. The latter were pretreated with diphtheria toxin, resulting in a depletion of CD11c+ cells. Expression of Fluc protein was assessed 4 hours after LMP delivery using in vivo bioluminescence imaging. Although we observed a significant decrease in luminescent signal in mice depleted for CD11c+ cells, we could still detect the expression of Fluc protein in the liver and spleen (Figure 4a,b). This indicates that CD11c+ cells are mainly, but not exclusively, responsible for the uptake and translation of mRNA delivered as LMPs. To confirm these findings and evaluate whether CD11b+ cells are coresponsible for the mRNA uptake, we next delivered LMPs containing Thy1.1 mRNA intravenously to Thy1.2 C57BL/6 mice. To ensure a detectable percentage of Thy1.1+ (mRNA transfected) cells, we delivered 20 µg mRNA. Four hours later, we performed flow cytometry measurements on spleen cells, showing that only a small fraction of spleen cells were Thy1.1+ (Figure 4c). These Thy1.1+ cells mainly consisted of CD11c+ cells, whereas a minor fraction was CD11b+ (Figure 4d).
Figure 4.

Intravenous delivery of lipid mRNA particles (LMPs) results in protein expression in CD11c+ cells. (a) Representative in vivo bioluminescence images out of two individual experiments of CD11c-diphteria toxin receptor mice (CD11c−) or littermates (CD11c+) that were treated with dihphteria toxin 24 hours prior to i.v. injection of 10 μg Fluc mRNA packaged at a mRNA:lipid ratio of 1:2. Mice with an intact CD11c+ cell population that received an i.v. injection of 10 μg truncated nerve growth factor receptor (tNGFR) mRNA packaged at a mRNA:lipid ratio of 1:2 served as a negative control (n = 8–10). (b) The graph summarizes the results of the Fluc expression as shown in a as mean ± SEM (n = 8–10). (c) Representative flow cytometry plots showing the expression of Thy1.1 mRNA in splenic cells from a mouse injected with 20 μg tNGFR mRNA (control) or Thy1.1 mRNA (n = 3). (d) Representative flow cytometry plot showing the percentage CD11c+ and CD11b+ cells in the Thy 1.1+ population after i.v. injection of Thy 1.1 mRNA (n = 3).
Intravenous delivery of LMPs induces antigen-specific CTLs
To further study whether i.v. delivery of LMPs induces effector cells that can specifically lyse target, we performed an in vivo cytotoxicity assay. We delivered LMPs formulated at an mRNA:lipid ratio of 1:2 that contained different amounts of mRNA encoding OVA. We showed that as little as 1 µg of OVA mRNA was sufficient to induce strong lyses of OVA-presenting target cells (>60%; Figure 5a). Dose-dependent induction of CTLs specific for the human papilloma virus (HPV) E7 antigen was also observed (Figure 5b). Importantly, CTL responses against HPV E7 could be significantly enhanced when LMPs (1 µg HPV E7 mRNA) were delivered 3 times at a 5-day interval (Figure 5c).
Figure 5.

Delivery of lipid mRNA particles (LMPs) results in the induction of antigen-specific lysis. (a) C57BL/6 mice were injected intravenously with LMPs containing varying amounts of ovalbumin (OVA) mRNA (plus truncated nerve growth factor receptor (tNGFR) mRNA). Five days later, an in vivo cytotoxicity assay was performed (n = 6–13). (b) Mice were injected intravenously with 5 μg or 1 μg E7 mRNA (plus tNGFR mRNA; n = 4). (c) Comparison of a single versus three injections at a 5-day interval of 1 μg E7 mRNA (n = 4). The graphs in (a–c) depict the percentage specific lysis as mean ± SEM. (d–f) Intravenous injection of multiple (5 μg per antigen) versus one antigen mRNA molecule (5 μg antigen mRNA plus tNGFR mRNA). The graphs in d, e, and f depict the percentage specific lysis of target cells presenting OVA, human pappiloma virus E7, or tyrosinase-related protein-2 (Trp2), respectively (n = 3–5). ns, not significant.
It has been shown that the differential MHC affinity of peptides results in the so-called CTL immunodominance.26 Therefore, APCs are often only loaded with one antigen to avoid competition between peptides for binding to MHC molecules and as such avoid skewing of the immune response. We further addressed whether delivery of multiple mRNA molecules encoding distinct antigens would result in similar target cell lysis as delivery of each antigen mRNA separately. Therefore, we injected LMPs containing three different antigen mRNA molecules (5 µg/mRNA) encoding OVA, HPV E7, or tyrosinase-related protein-2 and compared the induction of CTLs to delivery of LMPs containing mRNA for one of the aforementioned antigens supplemented with mRNA encoding truncated nerve growth factor receptor (control) to ensure delivery of equal amounts of mRNA. We showed similar induction of OVA-, HPV E7-, and tyrosinase-related protein-2-specific CTLs in mice immunized with LMPs containing only one type or multiple types of antigen mRNA molecules (Figure 5d–f).
Type I IFNs induced upon i.v. delivery of lipid:RNA particles support CTL activation
In order to evaluate the induction of a type I IFN response after systemic delivery of LMPs, we first determined the expression of Mx1 in splenocytes using quantitative real-time polymerase chain reaction, as the transcription of this gene is upregulated upon stimulation with type I IFNs. Therefore, splenocytes were isolated from nontreated mice (control) and mice that were injected intravenously with LMPs (10 µg mRNA) 24 hours prior to splenectomy. We showed that i.v. delivery of LMPs resulted in increased levels of Mx1, indicating the production of type I IFN (Figure 6a). The induction of type I IFNs was confirmed in enzyme-linked immunosorbent assay on sera that were collected 2, 6, and 24 hours after i.v. delivery of LMPs, showing a peak in IFN-α concentration at 6 hours (Figure 6b). We next compared the expression of Fluc protein upon systemic delivery of Fluc LMPs (10 µg) in wild-type C57BL/6 and interferon-alpha receptor (IFNAR) knockout mice (IFNAR−/−) to assess whether the protein expression is reduced due to type I IFN production. We observed that expression of Fluc was significantly enhanced in IFNAR−/− mice (Figure 6c). Finally, we determined whether the reduced expression of the mRNA-encoded protein due to type I IFNs hampered the induction of CTLs. Therefore, an in vivo cytotoxicity assay was performed in wild-type C57BL/6 and IFNAR−/− mice to measure the induction of OVA-specific CTLs. Surprisingly, we showed a reduction in specific lysis in IFNAR−/− mice, despite the aforementioned higher bioavailability of the mRNA-encoded protein (Figure 6d). These results suggest that induction of CTLs upon systemic delivery of LMPs is supported by type I IFN production rather than counteracted.
Figure 6.

Type I interferon (IFN) induced by i.v. delivery of lipid mRNA particles (LMPs) enhances antigen-specific immune responses. (a) Mx1 quantitative real-time polymerace chain reaction on mRNA extracted from the spleen 6 hours after i.v. delivery of LMPs. The graph depicts the 2−ΔCt values as mean ± SEM (n = 4). (b) IFN-α ELISA of blood samples collected 2, 6 and 24 hours after i.v. injection of LMPs. The graph depicts the concentration of IFN-α (ng/ml) as mean ± SEM. (c) Wild type or IFNAR−/− mice received an i.v. injection of 10 μg Fluc mRNA. Four hours later, we performed in vivo bioluminescence imaging. The graph depicts the Fluc expression as mean ± SEM (n = 8–10). (d) Wild type or IFNAR−/− mice received an i.v. injection with LMPs containing the 5 µg OVA mRNA. Five days later, we performed the in vivo cytotoxicity assays. The percentage-specific lysis is shown in the graph as mean ± SEM (n = 6–7).
Discussion
In this report, we show that Lipofectamine RNAiMAX, a cationic lipid that has been extensively explored for delivery of small interfering RNA, can be used for systemic delivery of mRNA for cancer immunotherapy purposes.
Since the physicochemical characteristics of particles critically influence their uptake by phagocytic cells, we first evaluated the charge and size of LMPs generated at different mRNA:lipid ratios. We showed that nanometer-sized particles with a negative charge were obtained at a 1:2 and 1:4 ratio, whereas micrometer-sized particles that also have a negative charge were obtained at a 1:6 ratio. The negative charge of the LMPs at a 1:2 and 1:4 ratio can in part be explained by the presence of noncomplexed mRNA as shown in the electrophoretic mobility shift assay. However, as the LMPs prepared at a 1:6 ratio also showed a negative charge without evidence of noncomplexed mRNA, the negative zeta potential of these LMPs can likely be attributed to a structural configuration where the majority of the mRNA is attached to the exterior of the particle, rather than encapsulated within the liposomes as previously reported for DNA-based lipoplexes. Overall, such a negative surface charge could be beneficial, as it has been described for DNA-based lipoplexes that a negative charge enables uptake of nanoparticles via phagocytosis.27,28 Size is another key factor that determines the uptake efficiency of nanoparticles.29,30,31 It was shown that nanometer-sized particles are internalized by various cell types, including fibroblasts, macrophages, and DCs.32,33 Therefore, it is no surprise that delivery of mRNA to HEK293T cells was most efficacious when using LMPs generated at a 1:2 ratio (242.3 ± 32.6 nm; −58.9 ± 4.6 mV). Since we further observed that particles generated with increasing amounts of Lipofectamine RNAiMAX negatively affected the cell viability, we proceeded with LMPs prepared at a 1:2 mRNA:lipid ratio to evaluate transfection of APCs.
In vitro transfection of bone marrow-derived DCs and RAW246.7 macrophages was performed in the absence as well as presence of serum. In this way, we indirectly evaluated the uptake of noncomplexed mRNA, which is still observed at a 1:2 LMP formulation and moreover gained information on the potential of the mRNA–lipoplexes for systemic delivery. It is generally accepted that the presence of serum can lead to particle aggregation,34,35,36,37 resulting in changes in physicochemical characteristics and as such different particle uptake and sometimes even premature release of its content.38 However, we observed the opposite, a higher percentage of DCs and macrophages expressed the mRNA-encoded protein and at higher levels when LMPs were delivered in serum-containing cultures. These observations exclude that noncomplexed mRNA is engulfed as we showed in the electric mobility shift assay that free mRNA is degraded upon contact with serum. Moreover, these data suggest that the studied nanoparticles are suited for the intended systemic delivery of mRNA. Also Marchini et al.39 observed enhanced transfection efficiency of murine fibroblasts using lipid:DNA complexes in the presence of serum. This was linked to an increase in particle stability and particle size,39 which in turn has been linked to enhanced uptake and trafficking to the cytosol.40 As the relation between the structural properties of mRNA–lipoplexes has not yet been as extensively studies as for their DNA-based counterparts, one could consider a plethora of factors that might explain the difference in transfection efficiency in serum-free versus serum-containing media. In addition to the particle's size and surface charge, one such factor could be the adsorption of serum proteins to the surface of LMPs. It has been described that adsorption of proteins like alpha-2-human serum glycoprotein and the opsonin immunoglobulin G to different microparticles promotes phagocytosis by immune cells.41 Another factor is the behavior of phagocytic cells in the presence of serum. It is known that serum in medium has marked effects on several properties of DCs, including their transfectability.42 Moreover, it has been described that the phagocytic capacity of mononuclear cells increases when cultured in increasing amounts of fetal bovine serum.42,43,44
Since LMPs generated at a mRNA:lipid ratio of 1:2 efficiently transfected APCs in vitro, we evaluated them in vivo. Using in vivo bioluminescence imaging and LMPs containing Fluc mRNA, we evaluated the timing and location of Fluc expression. We observed Fluc in the spleen as soon as 15 minutes after systemic injection of LMPs. The signal in the spleen accumulated over a period of 4 hours and afterward decreased over a period of 4 days. We furthermore observed expression of Fluc in the liver 4 hours after i.v. LMP delivery. It has been described that interaction of cationic particles and serum proteins after in vivo delivery can cause aggregation. The observed mRNA delivery to cells in liver and spleen using the LMPs prepared with Lipofectamine RNAiMAX argues against the formation of large aggregates in vivo. However, it cannot be entirely excluded that a fraction of the LMPs does not reach target organs. Therefore, it would be of interest in future studies to follow the distribution of the LMPs for instance using fluorescence or radioactive labeling and evaluate whether the location of the LMPs completely overlaps with the location of protein expression.
It has been described that after i.v. delivery, lipid-based nanoparticles first enter the spleens' marginal zone that contains DCs and macrophages, which in turn are characterized by their ability to engulf particulates.43 Therefore, it is not surprising that we detected the expression of the mRNA-encoded protein in CD11c+ and CD11b+ cells after i.v. delivery of LMPs. Importantly, flow cytometry showed that mainly CD11c+ cells are responsible for the mRNA uptake. Nonetheless, the study by Ciavarra et al.44 on the role of splenic DCs and macrophages in generating antiviral immunity suggests that CD11b+ cells should not be disregarded when it comes to their contribution to induce antigen-specific immunity.
We could show that i.v. administration of LMPs containing antigen mRNA results in strong antigen-specific immune responses. Importantly, these responses could be induced with as little as 1 µg antigen mRNA and could be significantly enhanced through repeated vaccination. Moreover, we addressed whether i.v. delivery of LMPs containing a mixture of mRNA molecules encoding three different antigens would evoke antigen-specific responses that are equally potent as the antigen-specific immune response elicited upon delivery of LMPs containing only one antigen mRNA. The latter was performed as most often DCs for cancer vaccination purposes are loaded with only one antigen to avoid competition between antigen-derived peptides for binding to MHC molecules and as such avoid a bias of the immune response. However, the ability to package and deliver multiple antigen mRNA molecules in one LMP would make the product commercially more attractive, since it takes the heterogeneity of tumors into account. Similar to the observations made by Schaft et al.,45 who electroporated human DCs with mRNA encoding three different antigens, we did not observe a negative influence on the presentation of the antigen and in our setting the subsequent in vivo elimination of antigen-presenting target cells.
Finally, we evaluated whether i.v. delivery of LMPs results in the induction of type I IFNs and is so, how these impact on vaccine efficacy. The latter is important as data from Pollard et al.20 suggest that type I IFN in the context of mRNA vaccination is detrimental for the ensuing immune response. Nonetheless, in several non-mRNA studies, type I IFN has been linked to protective CD8+ T-cell responses.46,47,48 Moreover, we previously performed a study in which mRNA encoding a fusokine consisting of IFN-β fused to the ectodomain of the transforming growth factor -beta receptor II (TGF-βIIR) was used to stimulate antitumor immunity.49 These studies question the notion that type I IFN hampers the efficacy of mRNA-based vaccines and warrant further investigation into this topic. Since we observed transient production of type I IFN after i.v. delivery of LMPs, we evaluated whether this lowered the expression of the mRNA-encoded protein as described by several other groups.18,19,22,23 Using IFNAR−/− mice and i.v. delivery of Fluc mRNA, we showed that the type I IFN response indeed negatively affected Fluc expression. It was previously shown that the bioavailability of the antigen is a key factor that determines the induction of antigen-specific CTLs after mRNA vaccination.9,50 Therefore, we were surprised that despite higher protein expression, the lysis of target cells in LMP-vaccinated IFNAR−/− mice was significantly lower when compared with the target cell lysis in LMP-vaccinated wild-type mice. Interestingly, DCs generated from bone marrow from IFNAR−/− mice are less mature.20 This might in part explain our results since immature DCs have a higher phagocytic capacity and as such ability to engulf (at least) naked mRNA.50 However, immature DCs possess a lower capacity to activate CTLs.51 Alternatively, the lower vaccine efficacy despite higher protein expression in IFNAR−/− mice could be explained in light of several studies, which show that presentation of low amounts of antigens results in the stimulation of T cells with a high avidity T-cell receptor. These T cells have a better functionality.25,52,53 One could argue that CD8+ T cells, activated in wild-type mice might have a higher avidity T-cell receptor, an issue that should be addressed experimentally.
Clearly, more research needs to be performed to study the role of type I IFNs in the induction of immunity by particulate mRNA-based cancer vaccines, as subtle differences in DC activation and T-cell receptor avidity are unlikely to fully explain the difference in vaccination outcome in wild type versus IFNAR−/− mice. In this regard, a lot can be learned from studies on infection with viruses carrying a single-stranded RNA genome. Both mRNA in particulate form54,55,56 and RNA viruses57,58,59 trigger pattern-recognition receptors, like TLR3 and TLR7. From studies with RNA viruses, we learned that type I IFNs have direct and indirect effects on multiple immune cells. The direct effects involve their ability to activate APCs and to promote among others the production of IFN-γ by and survival of CD8+ T cells.49,60,61,62 Its indirect effects are often mediated through the induction of chemokines and cytokines to recruit and regulate the activity of various immune cells.63,64 Moreover, we learned that type I IFN production can be attributed to various cell types like stromal cells, macrophages, and DCs, showing that there are disparate type I IFN sources.59 The impact of different type I IFN sources, as well as the impact of its timing and amount, was studied using lymphocytic choriomeningitis virus, a noncytopathic RNA virus as a model.65 In this study, it was suggested that prolonged production of type I IFNs upon infection of macrophages is required alongside its transient production by DCs to induce strong anti-lymphocytic choriomeningitis virus CTLs. However, we showed that even the transient presence of type I IFN has a significant impact on the activation of CTLs. The latter might be explained by the fact that besides the timing, also the amount can determine the outcome of type I IFN stimulation. In this regard, it was shown that low concentrations of type I IFNs induce production of stimulatory cytokines, whereas higher levels of type I IFNs can dampen their production.66,67 Consequently, in vaccine-mediated immunity, the amount of type I IFN and its presence at the appropriate time and anatomic location might be critical.
In conclusion, we showed that Lipofectamine RNAiMAX is able to package and protect mRNA from degradation. We showed that LMPs can be delivered intravenously to various APCs and that simultaneous or separate delivery of even small amounts of antigen mRNA results in strong antigen-specific CTL responses. These are supported by type I IFNs that are transiently produced upon LMP delivery. Therefore, this study highlights the need to study methods for mRNA encapsulation in detail, in order to provide future groundwork in developing mRNA-based therapeutics that promote antitumor immunity.
Materials and methods
Mice and cell cultures. Female, 6 to 12 weeks old, C57BL/6 mice were purchased from Charles River (Lyon, France). CD11c-diphteria toxin receptor mice in which CD11c+ cells are depleted upon treatment with 16 ng diphteria toxin/g mouse body weight (Sigma-Aldrich, Ghent, Belgium) were kindly provided by B. Lambrecht (University of Ghent, Ghent, Belgium). Mice lacking the type I IFN receptor (IFNAR−/−) were obtained from C. Libert (University of Ghent, Belgium). All animals were treated according to the European guidelines for animal experimentation. Experiments were approved by the Ethical Committee for the use of laboratory animals of the Vrije Universiteit Brussel (Jette, Belgium). HEK 293T cells and RAW246.7 cells were cultured as recommended by the American Type Culture Collection. Single-cell suspensions were prepared from spleens and lymph nodes as described.9 Primary mouse bone marrow-derived DCs were generated from C57BL/6 mice as described.9
Packaging of mRNA. The vectors pGEM-Ii80tOVA, -truncated nerve growth factor receptor, and -deltaUTsig-tyrosinase-related protein-2-DCL were described.9 A codon-optimized version of the Mus musculus Thy1.1, the Fluc, the HPV E7, and the eGFP gene were purchased from GeneArt (Life Technologies, Ghent, Belgium) and cloned as a NcoI-XhoI fragment in the vector pEtheRNA. All enzymes were purchased from Fermentas (Thermo Scientific, Brussels, Belgium). Prior to in vitro transcription, pGEM and pEtheRNA vectors were linearized with SpeI and BfuAI, respectively. In vitro transcription was performed as described.9 One microgram of mRNA was packaged in 2 µl of Lipofectamine RNAiMAX (Life Technologies). Packaging was performed according to manufacturer's instructions.
Electrophoretic mobility shift assay, size, and zeta potential measurement. One microgram Fluc mRNA was mixed with 2 µl Lipofectamine RNAiMAX (Life Technologies) in a volume of 10 µl OptiMem (Life Technologies). After 20 minutes of incubation, 50% fetal clone I serum (HyClone, Erembodegem, Belgium) was added. mRNA formulated in 0.8 lactated Ringers' solution served as a control. Complexation of the mRNA in RNAiMAX and its degradation upon serum exposure were analyzed by gel electrophoresis on a 2.4% agarose gel that was run for 1 hour at 7 V/cm in the presence of ethidium bromide. The size and charge of 3 µg of eGFP mRNA packaged in Lipofectamine RNAiMAX at different mRNA:lipid ratios in a total volume of 100 µl OptiMem and diluted to 1 ml with HEPES buffer were assessed using the Malvern Zetasizer 3000HS (Worcestershire, UK).
In vitro transfection with packaged mRNA. Five hundred thousand HEK293T cells or RAW246.7 cells were transfected with 2.5 µg eGFP mRNA complexed with 5 µl of Lipofectamine RNAiMAX diluted in OptiMem to obtain a total volume of 100 µl. Cells were plated 24 hours before transfection in a six-well plate. Before transfection, cells were washed with OptiMem. The transfection mix was added to the cells and cells were incubated for 1 hour at 37°C, 5% carbon dioxide. Thereafter, cells were diluted in 2 ml of the appropriate medium. RAW246.7 cells were also transfected in the presence of serum. Therefore, LMPs were added directly to cell culture medium containing 5% fetal clone I serum. Transfection of bone marrow-derived DCs was performed at day 6 of culture in a 24-well plate. The cell culture medium was removed and the LMPs containing 1 µg eGFP mRNA were added to 5 × 105 cells. After 2 hours incubation, the cells were recultured for 24 hours in the appropriate cell culture medium. To perform transfection in the presence of serum, LMPs were added directly into the cell culture medium containing 5% fetal clone I serum. Transfection efficiency was evaluated as eGFP expression 24 hours after transfection using flow cytometry.
Intravenous, s.c., and i.m. administration of mRNA C57BL/6 mice, CD11c-diphtheria toxin receptor mice or IFNAR−/− mice were injected intravenously (250 μl), subcutaneously (250 μl), or intramuscularly (25 μl) with the indicated amount of LMPs.
In vivo bioluminescence imaging. In vivo bioluminescence imaging was performed as previously described.68
Flow cytometry. To characterize cells that were transfected in situ with Thy1.1 mRNA after i.v. injection of Thy1.1 LMPs, spleen was isolated, single-cell suspension prepared, and cells stained with a phycoerythrin-conjugated anti-Thy1.1 antibody (clone OX7) combined with fluorescein isothiocynate-conjugated antibodies against CD11b (clone M1/70) and AlexaFluor 647 (AF647)-conjugated antibodies against CD11c (clone N418) (Becton Dickinson, Erembodegem Belgium). To evaluate the efficiency of transfection of HEK293T cells with eGFP LMPs, the percentage of eGFP-positive cells was evaluated. To evaluate the efficiency of transfection of DCs and RAW246.7 cells with eGFP LMPs, DCs and RAW246.7 cells were stained with an allophycocyanin-conjugated anti-CD11c and a Peridinin-chlorophyll proteins-Cyanine 5.5 (PerCP-Cy5.5) anti-CD11b antibody respectively. The expression of CD11c or CD11b and eGFP were measured using flow cytometry. Untreated cells served as a negative control. MHC Dextramers were used for the detection of H-2Kb/SIINFEKL-specific T cells. The staining was performed according to the manufacturer's instructions (Immudex, Copenhagen, Denmark). Data were collected using a FACSCanto flow cytometer (Becton Dickinson) and analyzed using FACSDiva (Becton Dickinson) or FlowJo (Treestar, Ashland, Oregon) software.
In vivo CTL assay. Splenocytes from naive syngeneic mice were pulsed or not with 5 μM of the appropriate peptide for 1 hour. Peptide-pulsed cells were labeled with 2.5 μM carboxyfluorescein succinimidyl ester after which they were mixed at a 1:1 ratio with 0.25 μM carboxyfluorescein succinimidyl ester-labeled nonpulsed cells. At least 107 cells were injected intravenously. Spleen and lymph nodes were analyzed 18 hours later by flow cytometry. The percentage-specific lyses were calculated as described.9 The following peptides were purchased from Eurogentec (Liège, Belgium) and used: SIINFEKL (OVA), SVYDFFVWL (tyrosinase-related protein-2), and H2NRAHYNIVTFCOOH (HPV E7).
Quantitative real-time polymerase chain reaction. Isolation of total RNA from the spleen, its reverse transcription, and the quantitative real-time polymerase chain reaction analysis performed to evaluate the upregulation of the type I IFN-responsive gene Mx1 were performed as previously described.49
Enzyme-linked immunosorbent assay. The enzyme-linked immunosorbent assay to measure IFN-α was performed according to the manufacturer's protocol (PBL Assay Science Orléans, France).
Statistical analysis. The unpaired Student's t-test was performed to compare the data sets. Sample sizes are indicated in the figure legends. Number of asterisks in the figures indicates the level of statistical significance: *P < 0.05, **P < 0.01, and ***P < 0.001. The results are shown in column graph as mean ± SEM.
Acknowledgments
This work was supported by grants from the Interuniversity Attraction Poles Program-Belgian State-Belgian Science Policy, the National Cancer Plan of the Federal Ministry of Health, the Belgian Federation against Cancer, the “Vlaamse Liga tegen Kanker (VLK),” an IWT-TBM program, the Research Foundation Flanders (FWO-V), an EU FP7-funded Network of Excellence and the Scientific Fund Willy Gepts of the University Hospital Brussels. The production of mRNA was supported by eTheRNA immunotherapies, a spin-off company of the Vrije Universiteit Brussel, founded by Kris Thielemans. K.B. and K.V.J. are funded by the IWT. H.D. is funded by the FWO-V. The authors thank Petra Roman and Elsy Vaeremans for the production of the mRNA and Angelo Willems for the assistance with the FACS analyses. We furthermore thank Claude Libert for providing the IFNAR−/− mice and Bart Lambrecht for providing the CD11c-diphtheria toxin receptor mice.
The work was performed in Brussels and Ghent, which are both cities in Belgium.
References
- Benteyn, D, Heirman, C, Bonehill, A, Thielemans, K and Breckpot, K (2015). mRNA-based dendritic cell vaccines. Expert Rev Vaccines 14: 161–176. [DOI] [PubMed] [Google Scholar]
- Van Lint, S, Renmans, D, Broos, K, Dewitte, H, Lentacker, I, Heirman, C et al. (2015). The ReNAissanCe of mRNA-based cancer therapy. Expert Rev Vaccines 14: 235–251. [DOI] [PubMed] [Google Scholar]
- Van Lint, S, Heirman, C, Thielemans, K and Breckpot, K (2013). mRNA: from a chemical blueprint for protein production to an off-the-shelf therapeutic. Hum Vaccin Immunother 9: 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weide, B, Pascolo, S, Scheel, B, Derhovanessian, E, Pflugfelder, A, Eigentler, TK et al. (2009). Direct injection of protamine-protected mRNA: results of a phase ½ vaccination trial in metastatic melanoma patients. J Immunother 32: 498–507. [DOI] [PubMed] [Google Scholar]
- Goyvaerts, C, De Groeve, K, Dingemans, J, Van Lint, S, Robays, L, Heirman, C et al. (2012). Development of the nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther 19: 1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyvaerts, C, Dingemans, J, De Groeve, K, Heirman, C, Van Gulck, E, Vanham, G et al. (2013). Targeting of human antigen-presenting cell subsets. J Virol 87: 11304–11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyvaerts, C, de Kurt, G, Van Lint, S, Heirman, C, Van Ginderachter, JA, De Baetselier, P et al. (2014). Immunogenicity of targeted lentivectors. Oncotarget 5: 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciré, S, Da Rocha, S, Yao, R, Fisson, S, Buchholz, CJ, Collins, MK et al. (2014). Immunization of mice with lentiviral vectors targeted to MHC class II+ cells is due to preferential transduction of dendritic cells in vivo. PLoS One 9: e101644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint, S, Goyvaerts, C, Maenhout, S, Goethals, L, Disy, A, Benteyn, D et al. (2012). Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res 72: 1661–1671. [DOI] [PubMed] [Google Scholar]
- Kreiter, S, Selmi, A, Diken, M, Koslowski, M, Britten, CM, Huber, C et al. (2010). Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res 70: 9031–9040. [DOI] [PubMed] [Google Scholar]
- Perche, F, Benvegnu, T, Berchel, M, Lebegue, L, Pichon, C, Jaffrès, PA et al. (2011). Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 7: 445–453. [DOI] [PubMed] [Google Scholar]
- Zhou, WZ, Hoon, DS, Huang, SK, Fujii, S, Hashimoto, K, Morishita, R et al. (1999). RNA melanoma vaccine: induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum Gene Ther 10: 2719–2724. [DOI] [PubMed] [Google Scholar]
- Martinon, F, Krishnan, S, Lenzen, G, Magné, R, Gomard, E, Guillet, JG et al. (1993). Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol 23: 1719–1722. [DOI] [PubMed] [Google Scholar]
- Pascolo S. Vaccination with messenger RNA (mRNA). Handb Exp Pharmacol. 2008: 221–235. [DOI] [PubMed]
- Geall, AJ, Mandl, CW and Ulmer, JB (2013). RNA: the new revolution in nucleic acid vaccines. Semin Immunol 25: 152–159. [DOI] [PubMed] [Google Scholar]
- Van der Jeught, K, Bialkowski, L, Daszkiewicz, L, Broos, K, Goyvaerts, C, Renmans, D et al. (2015). Targeting the tumor microenvironment to enhance antitumor immune responses. Oncotarget 6: 1359–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phua, KK, Staats, HF, Leong, KW and Nair, SK (2014). Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci Rep 4: 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikó, K, Buckstein, M, Ni, H and Weissman, D (2005). Suppression of RNA recognition by toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23: 165–175. [DOI] [PubMed] [Google Scholar]
- Kormann, MS, Hasenpusch, G, Aneja, MK, Nica, G, Flemmer, AW, Herber-Jonat, S et al. (2011). Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol 29: 154–157. [DOI] [PubMed] [Google Scholar]
- Pollard, C, Rejman, J, De Haes, W, Verrier, B, Van Gulck, E, Naessens, T et al. (2013). Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol Ther 21: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikó, K, Ni, H, Capodici, J, Lamphier, M and Weissman, D (2004). mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem 279: 12542–12550. [DOI] [PubMed] [Google Scholar]
- Devoldere, J, Dewitte, H, De Smedt, SC and Remaut, K (2016). Evading innate immunity in nonviral mRNA delivery: don't shoot the messenger. Drug Discov Today 21: 11–25. [DOI] [PubMed] [Google Scholar]
- Karikó, K, Muramatsu, H, Welsh, FA, Ludwig, J, Kato, H, Akira, S et al. (2008). Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16: 1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung, V, Ellegast, J, Kim, S, Brzózka, K, Jung, A, Kato, H et al. (2006). 5'-Triphosphate RNA is the ligand for RIG-I. Science 314: 994–997. [DOI] [PubMed] [Google Scholar]
- Zeh, HJ 3rd, Perry-Lalley, D, Dudley, ME, Rosenberg, SA and Yang, JC (1999). High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol 162: 989–994. [PubMed] [Google Scholar]
- Palmowski, MJ, Choi, EM, Hermans, IF, Gilbert, SC, Chen, JL, Gileadi, U et al. (2002). Competition between CTL narrows the immune response induced by prime-boost vaccination protocols. J Immunol 168: 4391–4398. [DOI] [PubMed] [Google Scholar]
- Ma, B, Zhang, S, Jiang, H, Zhao, B and Lv, H (2007). Lipoplex morphologies and their influences on transfection efficiency in gene delivery. J Control Release 123: 184–194. [DOI] [PubMed] [Google Scholar]
- Sheng, Y, Yuan, Y, Liu, C, Tao, X, Shan, X and Xu, F (2009). In vitro macrophage uptake and in vivo biodistribution of PLA-PEG nanoparticles loaded with hemoglobin as blood substitutes: effect of PEG content. J Mater Sci Mater Med 20: 1881–1891. [DOI] [PubMed] [Google Scholar]
- Conner, SD and Schmid, SL (2003). Regulated portals of entry into the cell. Nature 422: 37–44. [DOI] [PubMed] [Google Scholar]
- Hansen, CG and Nichols, BJ (2009). Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci 122(Pt 11): 1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay, G, Alakhova, DY and Kabanov, AV (2010). Endocytosis of nanomedicines. J Control Release 145: 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, W, Kim, BY, Rutka, JT and Chan, WC (2008). Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol 3: 145–150. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia, MA, Aggarwal, P, Hall, JB and McNeil, SE (2008). Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm 5: 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, JP and Huang, L (1997). Overcoming the inhibitory effect of serum on lipofection by increasing the charge ratio of cationic liposome to DNA. Gene Ther 4: 950–960. [DOI] [PubMed] [Google Scholar]
- Zelphati, O, Uyechi, LS, Barron, LG and Szoka, FC Jr (1998). Effect of serum components on the physico-chemical properties of cationic lipid/oligonucleotide complexes and on their interactions with cells. Biochim Biophys Acta 1390: 119–133. [DOI] [PubMed] [Google Scholar]
- Simberg, D, Weisman, S, Talmon, Y, Faerman, A, Shoshani, T and Barenholz, Y (2003). The role of organ vascularization and lipoplex-serum initial contact in intravenous murine lipofection. J Biol Chem 278: 39858–39865. [DOI] [PubMed] [Google Scholar]
- Wasungu, L, Scarzello, M, van Dam, G, Molema, G, Wagenaar, A, Engberts, JB et al. (2006). Transfection mediated by pH-sensitive sugar-based gemini surfactants; potential for in vivo gene therapy applications. J Mol Med (Berl) 84: 774–784. [DOI] [PubMed] [Google Scholar]
- Dewitte, H, Verbeke, R, Breckpot, K, De Smedt, S, Lentacker, I. (2014). Nanoparticle design to induce tumor immunity and challenge the suppressive tumor microenvironment. Nano Today 9: 743–758. [Google Scholar]
- Marchini, C, Montani, M, Amici, A, Amenitsch, H, Marianecci, C, Pozzi, D et al. (2009). Structural stability and increase in size rationalize the efficiency of lipoplexes in serum. Langmuir 25: 3013–3021. [DOI] [PubMed] [Google Scholar]
- Hoekstra, D, Rejman, J, Wasungu, L, Shi, F and Zuhorn, I (2007). Gene delivery by cationic lipids: in and out of an endosome. Biochem Soc Trans 35(Pt 1): 68–71. [DOI] [PubMed] [Google Scholar]
- Thiele, L, Diederichs, JE, Reszka, R, Merkle, HP and Walter, E (2003). Competitive adsorption of serum proteins at microparticles affects phagocytosis by dendritic cells. Biomaterials 24: 1409–1418. [DOI] [PubMed] [Google Scholar]
- Dewitte, H, Verbeke, R, Breckpot, K, Vandenbroucke, RE, Libert, C, De Smedt, SC et al. (2014). Choose your models wisely: how different murine bone marrow-derived dendritic cell protocols influence the success of nanoparticulate vaccines in vitro. J Control Release 195: 138–146. [DOI] [PubMed] [Google Scholar]
- Phua, KK, Nair, SK and Leong, KW (2014). Messenger RNA (mRNA) nanoparticle tumour vaccination. Nanoscale 6: 7715–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavarra, RP, Taylor, L, Greene, AR, Yousefieh, N, Horeth, D, van Rooijen, N et al. (2005). Impact of macrophage and dendritic cell subset elimination on antiviral immunity, viral clearance and production of type 1 interferon. Virology 342: 177–189. [DOI] [PubMed] [Google Scholar]
- Schaft, N, Wellner, V, Wohn, C, Schuler, G and Dörrie, J (2013). CD8(+) T-cell priming and boosting: more antigen-presenting DC, or more antigen per DC? Cancer Immunol Immunother 62: 1769–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmüller, K, Wille-Reece, U, Lindsay, RW, Trager, LR, Darrah, PA, Flynn, BJ et al. (2011). Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest 121: 1782–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes, MB, Kacha, AK, Kline, J, Woo, SR, Kranz, DM, Murphy, KM et al. (2011). Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 208: 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, JZ, Kurche, JS, Burchill, MA and Kedl, RM (2011). TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 118: 3028–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Jeught, K, Joe, PT, Bialkowski, L, Heirman, C, Daszkiewicz, L, Liechtenstein, T et al. (2014). Intratumoral administration of mRNA encoding a fusokine consisting of IFN-β and the ectodomain of the TGF-β receptor II potentiates antitumor immunity. Oncotarget 5: 10100–10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diken, M, Kreiter, S, Selmi, A, Britten, CM, Huber, C, Türeci, Ö et al. (2011). Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther 18: 702–708. [DOI] [PubMed] [Google Scholar]
- Breckpot, K and Escors, D (2009). Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets 9: 328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S, Linette, GP, Longerich, S and Haluska, FG (2002). Antimelanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209-2M is correlated to TCR avidity. J Immunol 169: 531–539. [DOI] [PubMed] [Google Scholar]
- Viganò, S, Utzschneider, DT, Perreau, M, Pantaleo, G, Zehn, D and Harari, A (2012). Functional avidity: a measure to predict the efficacy of effector T cells? Clin Dev Immunol 2012: 153863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel, B, Teufel, R, Probst, J, Carralot, JP, Geginat, J, Radsak, M et al. (2005). Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur J Immunol 35: 1557–1566. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek, M, Duchardt, KM, Lorenz, C, Pfeiffer, R, Ojkić-Zrna, S, Probst, J et al. (2011). Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother 34: 1–15. [DOI] [PubMed] [Google Scholar]
- Van Lint, S, Thielemans, K and Breckpot, K (2011). mRNA: delivering an antitumor message? Immunotherapy 3: 605–607. [DOI] [PubMed] [Google Scholar]
- Tan, PH, Beutelspacher, SC, Xue, SA, Wang, YH, Mitchell, P, McAlister, JC et al. (2005). Modulation of human dendritic-cell function following transduction with viral vectors: implications for gene therapy. Blood 105: 3824–3832. [DOI] [PubMed] [Google Scholar]
- Breckpot, K, Escors, D, Arce, F, Lopes, L, Karwacz, K, Van Lint, S et al. (2010). HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol 84: 5627–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero, I, Quetglas, JI, Reboredo, M, Dubrot, J, Rodriguez-Madoz, JR, Mancheño, U et al. (2015). Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res 75: 497–507. [DOI] [PubMed] [Google Scholar]
- Ceppi, M, Ruggli, N, Tache, V, Gerber, H, McCullough, KC and Summerfield, A (2005). Double-stranded secondary structures on mRNA induce type I interferon (IFN alpha/beta) production and maturation of mRNA-transfected monocyte-derived dendritic cells. J Gene Med 7: 452–465. [DOI] [PubMed] [Google Scholar]
- Hervas-Stubbs, S, Perez-Gracia, JL, Rouzaut, A, Sanmamed, MF, Le Bon, A and Melero, I (2011). Direct effects of type I interferons on cells of the immune system. Clin Cancer Res 17: 2619–2627. [DOI] [PubMed] [Google Scholar]
- Nguyen, KB, Cousens, LP, Doughty, LA, Pien, GC, Durbin, JE and Biron, CA (2000). Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat Immunol 1: 70–76. [DOI] [PubMed] [Google Scholar]
- Zhang, X, Sun, S, Hwang, I, Tough, DF and Sprent, J (1998). Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8: 591–599. [DOI] [PubMed] [Google Scholar]
- Nguyen, KB, Salazar-Mather, TP, Dalod, MY, Van Deusen, JB, Wei, XQ, Liew, FY et al. (2002). Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol 169: 4279–4287. [DOI] [PubMed] [Google Scholar]
- Wang, Y, Swiecki, M, Cella, M, Alber, G, Schreiber, RD, Gilfillan, S et al. (2012). Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11: 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier, G, Humbert, M, Deauvieau, F, Scuiller, M, Hiscott, J, Bates, EE et al. (2005). A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med 201: 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes, AA, Ma, X, Cuomo, P, Park, K, Wahl, L, Wolf, SF et al. (2001). Type I interferons and IL-12: convergence and cross-regulation among mediators of cellular immunity. Eur J Immunol 31: 2026–2034. [DOI] [PubMed] [Google Scholar]
- Keyaerts, M, Verschueren, J, Bos, TJ, Tchouate-Gainkam, LO, Peleman, C, Breckpot, K et al. (2008). Dynamic bioluminescence imaging for quantitative tumour burden assessment using IV or IP administration of D: -luciferin: effect on intensity, time kinetics and repeatability of photon emission. Eur J Nucl Med Mol Imaging 35: 999–1007. [DOI] [PubMed] [Google Scholar]