

Abstract
Objective:
We sought to estimate the causal effect of low serum 25(OH)D on multiple sclerosis (MS) susceptibility that is not confounded by environmental or lifestyle factors or subject to reverse causality.
Methods:
We conducted mendelian randomization (MR) analyses using an instrumental variable (IV) comprising 3 single nucleotide polymorphisms found to be associated with serum 25(OH)D levels at genome-wide significance. We analyzed the effect of the IV on MS risk and both age at onset and disease severity in 2 separate populations using logistic regression models that controlled for sex, year of birth, smoking, education, genetic ancestry, body mass index at age 18–20 years or in 20s, a weighted genetic risk score for 110 known MS-associated variants, and the presence of one or more HLA-DRB1*15:01 alleles.
Results:
Findings from MR analyses using the IV showed increasing levels of 25(OH)D are associated with a decreased risk of MS in both populations. In white, non-Hispanic members of Kaiser Permanente Northern California (1,056 MS cases and 9,015 controls), the odds ratio (OR) was 0.79 (p = 0.04, 95% confidence interval (CI): 0.64–0.99). In members of a Swedish population from the Epidemiological Investigation of Multiple Sclerosis and Genes and Environment in Multiple Sclerosis MS case-control studies (6,335 cases and 5,762 controls), the OR was 0.86 (p = 0.03, 95% CI: 0.76–0.98). A meta-analysis of the 2 populations gave a combined OR of 0.85 (p = 0.003, 95% CI: 0.76–0.94). No association was observed for age at onset or disease severity.
Conclusions:
These results provide strong evidence that low serum 25(OH)D concentration is a cause of MS, independent of established risk factors.
Multiple sclerosis (MS) is an immune-mediated, demyelinating disease that leads to a wide variety of symptoms and disability. Both genetic and environmental factors have been implicated in its etiology, including vitamin D deficiency. Observational studies have consistently shown an association of low serum 25(OH)D and increased risk of MS, but it has not been shown that low 25(OH)D is actually a cause of MS.1 The apparent beneficial effects of 25(OH)D on MS might alternately be explained by reverse causation (i.e., MS could be leading to low 25(OH)D) or by confounding by sun exposure, obesity, or some other unknown factors.
Mendelian randomization (MR), equivalently, instrumental variable (IV) analysis using a genetic instrument, is a technique that can overcome the problems of both reverse causation and confounding when assessing the causal relationship between an exposure and an outcome.2 Single nucleotide polymorphisms (SNPs) known to be associated with 25(OH)D levels, rather than measured 25(OH)D, can be used as an IV to estimate the effect of low 25(OH)D on MS. Because SNP genotypes are determined at birth and are not likely to be influenced by potential confounding variables, the effect estimate from MR analysis should not be confounded, and reverse causation is unlikely because MS does not determine which 25(OH)D-associated SNPs are inherited (figure). We used MR analysis to estimate the causal relationship between serum 25(OH)D levels and MS susceptibility in 2 large case-control studies. We also investigated 2 clinical phenotypes for MS: age at onset and disease severity.
Figure. Relationship of exposure, outcome, confounding variables, and genetic instrumental variable used in mendelian randomization.

IV = instrumental variable; SNP = single nucleotide polymorphism.
METHODS
KPNC participants.
Data were collected from members of Kaiser Permanente Medical Care Plan, Northern California Region (KPNC). KPNC is an integrated health service delivery system with a membership of 3.2 million that comprises about 25–30% of the population of a 22-county service area and is the largest health care provider in northern California. Membership is largely representative of the general population in the service area; however, persons in impoverished neighborhoods are underrepresented.3
Eligible KPNC cases were defined as individuals with a diagnosis of MS by a neurologist (ICD-9 code 340.xx), age 18–69 years, and membership in KPNC at initial contact. The study was restricted to self-identified white (non-Hispanic) race/ethnicity, the population with the highest prevalence of MS. The treating neurologist was contacted for approval to contact each case as a potential MS study participant. A total of 3,293 potential MS cases were reviewed by KPNC neurologists, who approved contact with 2,823 (86%) at the time of the data freeze (August 2014). Diagnoses were validated using electronic health record (EHR) review and according to published diagnostic criteria.4 Multiple Sclerosis Severity Scores (MSSS) were calculated for each case at the time of study entry (mean disease duration = 17.7 years), as described,5 and participants were asked to recall the age of first MS symptom onset which was validated using EHR data when possible.
Controls were white (non-Hispanic) current KPNC members without a diagnosis of MS or related condition (optic neuritis, transverse myelitis, or demyelinating disease; ICD-9 codes: 340, 341.0, 341.1, 341.2, 341.20, 341.21, 341.22, 341.8, 341.9, 377.3, 377.30, 377.39, and 328.82) confirmed through EHR data. Potential study participants were contacted by email with a follow-up phone call. The participation rate was 80% for cases and 66% for controls. Genetic data were available for approximately 80% of study participants.
Additional controls were individuals of the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort participating in the KPNC Research Program on Genes, Environment, and Health, which is described elsewhere (dbGaP phs000674.v2.p2).6,7 Respondents completed a written consent form and provided a saliva sample for DNA extraction. A total of 110,266 participant samples were initially collected. Approximately 103,000 samples were successfully genotyped, and 77% of participants subsequently returned new consent forms for placement in dbGaP (NIH), resulting in a final sample size of 78,486 participants. From these participants, we selected a subset of 12,605 self-reported non-Hispanic white individuals matched to MS cases for sex and age (±2 years) at a 10:1 ratio.
EIMS/GEMS participants.
Data were collected from 2 population-based case-control studies on incident MS patients (the Epidemiological Investigation of Multiple Sclerosis [EIMS] study) and prevalent MS patients (the Genes and Environment in Multiple Sclerosis [GEMS] study). In the EIMS study, the inclusion criteria were age 16–70 years, recently (within 2 years) diagnosed MS according to the McDonald criteria,4 and ability to understand Swedish. For the GEMS study, the participants were identified from the Swedish National MS registry and recruited during 2009–2011. In both studies, controls (without MS) were randomly chosen from the population register and matched to the patients with MS by sex, age at inclusion in the study, and region of residence (2 controls per case in EIMS and 1 control per case in GEMS). Information on age at onset was self-reported. Data on MSSS5 were retrieved from the Swedish National MS registry for all participants, in both EIMS and GEMS studies as per September 2014 (mean disease duration = 20.1 years). All participants in the EIMS study were distinct from those in the GEMS study. Details of the study design have been described elsewhere.8,9 The participation rate was 92% for cases and 67% for controls in EIMS, and 82% for cases and 66% for controls in GEMS. Genotyping data were available for 75% of EIMS and 91% of GEMS participants.
Standard protocol approvals, registrations, and patient consents.
Study protocols for KPNC participants were approved by the Institutional Review Boards of KPNC and the University of California, Berkeley. Ethical approval for both EIMS and GEMS was obtained from the Regional Ethical Review Board in Stockholm at Karolinska Institutet, and participants provided written informed consent.
Exposure assessment in KPNC.
KPNC participants completed a computer-assisted telephone interview comprised questions related to various events and exposures, as described elsewhere.10,11 GERA controls were mailed a survey consisting of questions related to health behaviors, sociodemographic information, and diagnoses (dbGaP phs000674.v2.p2).
Whole blood was collected and processed using the Gentra Puregene protocol. Saliva was collected using Oragene kits. Medium resolution HLA-DRB1 and genome-wide SNP genotyping were performed as previously described12,13 using Illumina Infinium 660K and Human Omni Express BeadChip arrays for KPNC cases and controls, and Axiom (Affymetrix) custom chips for the additional GERA controls. SNPs with a minor allele frequency (MAF) <1%, success rate <90%, or not in Hardy-Weinberg equilibrium (HWE) among controls (p < 0.000001) were removed from analysis. Samples with >10% failed genotype calls and related individuals were also removed. Imputation against reference haplotypes from Phase I of the 1000 Genomes Project was conducted using SHAPEIT and IMPUTE2. To ensure high-quality imputation results, only SNPs with info score >0.8 on all 3 genotyping platforms and with MAF in controls with SD <0.03 across all 3 platforms were retained for analysis. Cross-platform association tests were conducted to remove SNPs associated with the genotype array (FDR q < 0.05). Population outliers were identified using multidimensional scaling (MDS) and removed from analysis. Two MDS components were used to adjust for population stratification due to genetic ancestry differences between cases and controls in the association analysis.
Exposure assessment in EIMS and GEMS.
The exposure assessment was done through an extensive questionnaire that covered demographic and environmental factors; details are described elsewhere.8,9 Participants were asked to provide blood samples, which were genotyped on an Illumina custom array (MS replication chip). HLA-DRB1 information was imputed with a modified version of HLA*IMP:0214 with 400 Swedish controls added to the reference panel for MHC class II genes. SNPs with <2% MAF, success rate <98%, or not in HWE among controls (p < 0.0001) were removed from analysis. Individuals with >2% failed genotype calls, with increased heterozygosity (>mean + 2SD), related individuals, or whose recorded sex differed from genotype were removed from analysis. We removed population outliers identified using the smartpca program15 with standard settings using 3,736 ancestry informative markers. Two principal component analysis vectors, those with p <0.05, were used to adjust for population stratification in the association analysis, similar to KPNC comparisons described above.
Statistical analyses.
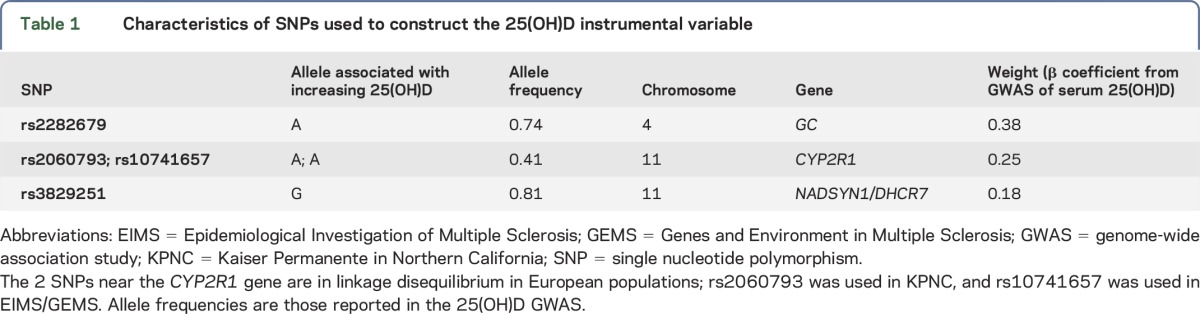
A 25(OH)D IV was constructed for each individual using the β coefficients for 3 published genetic variants associated at genome-wide significance with higher serum 25(OH)D: rs2282679-A, in an intron of GC; rs2060793-A, upstream of CYP2R1; and rs3829251-G, in an intron of NADSYN1 and upstream of DHCR7.16 The IV was calculated by multiplying the number of alleles that correspond to an increase in 25(OH)D for each locus by the β coefficient from the genome-wide association study (GWAS) for that variant and then taking the sum across the 3 loci (table 1). For KPNC, either the genotyped alleles or imputed allele probabilities were used to calculate the IV. For EIMS/GEMS, genotyped alleles were used to calculate the IV, and missing genotypes (2 for rs2282679 and 243 for rs3822951) were replaced with 2 times the MAF. In addition, rs10741657, which is in perfect linkage disequilibrium (LD) with rs2060793 (r2 = 1 in 1,000 Genomes Phase I European population), was used in the IV calculation for EIMS/GEMS, as the latter SNP was not genotyped. A weighted genetic risk score (wGRS) for 110 non-HLA MS susceptibility loci identified through the most recent MS GWAS was calculated for each individual.17 The wGRS was calculated by multiplying the number of risk alleles for each locus by the logarithm of the odds ratio (OR) for that variant, then taking the sum across the 110 loci. One SNP was missing for KPNC (rs201202118), and 2 SNPs were missing for EIMS/GEMS (rs2028597 and rs6874308).
Table 1.
Characteristics of SNPs used to construct the 25(OH)D instrumental variable
After quality control and removal of population outliers, 1,655 individuals from KPNC (1,056 cases and 599 controls) with genetic data were available as well as an additional 8,416 controls from the GERA study, for a total of 1,056 cases and 9,015 controls. Data for 6,335 cases and 5,762 controls were available from the Swedish studies (EIMS and GEMS).
Demographic differences between cases and controls were compared using χ2 and independent sample t tests where appropriate. Linear regression was used to test the assumption that the IV is not associated with confounding factors. MR analysis, in this case a separate-sample IV analysis, was performed by regressing MS case status on the 25(OH)D IV in logistic models, and regressing MSSS and age at onset on the 25(OH)D IV in linear regression models. All analyses were controlled for sex, year of birth, ever smoking, college education, HLA-DRB1*15:01 carrier status, wGRS of non-HLA MS risk variants, and genetic ancestry (as derived from the first 2 components from MDS for KPNC or smartpca for Sweden). Swedish analyses were additionally controlled for region of residency and study type (EIMS vs GEMS). Matching variables were included as covariates because the final study populations included all participants with genotyping data available, and a matched analysis was not performed. ORs with 95% confidence intervals (CIs) were estimated. Meta-analysis assuming random effects was performed.
All analyses were conducted using PLINK,18 STATA (StataCorp, College Station, TX), SAS (SAS Institute, Cary, NC), or R.19
RESULTS
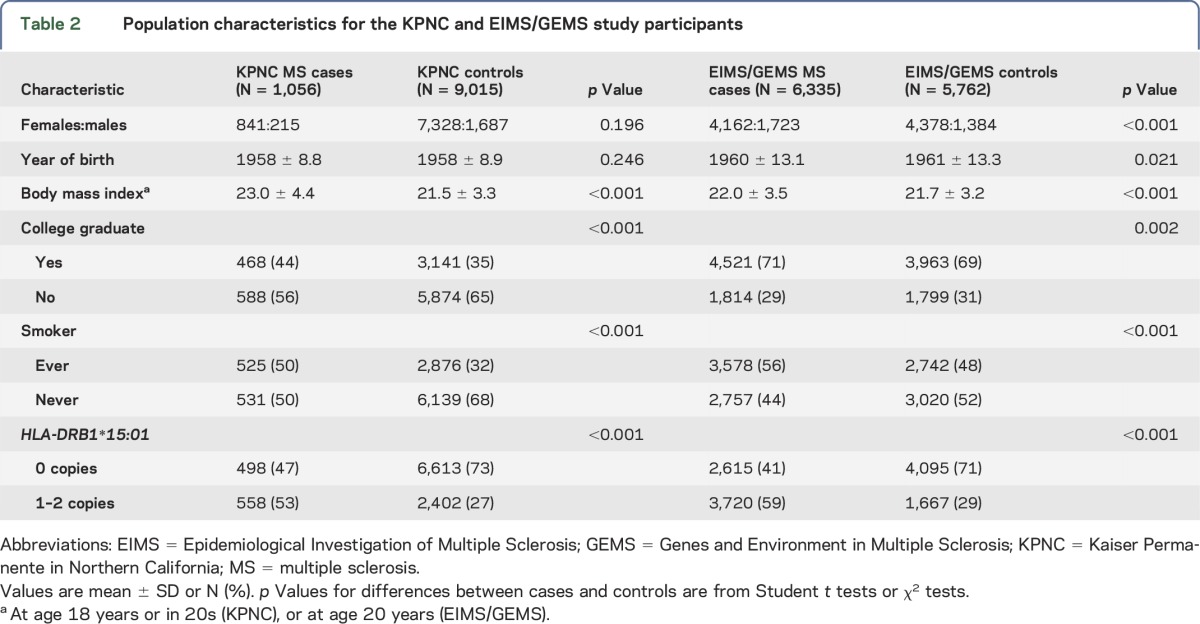
Demographic and disease characteristics of MS cases and controls are found in table 2 for both KPNC and Sweden studies. There were significant differences between cases and controls with respect to smoking, college graduation, obesity at age 18–20 years or in 20s, and HLA-DRB1*15:01 status for both KPNC and EIMS/GEMS, as expected, and for age and sex in EIMS/GEMS. The 25(OH)D IV was lower among cases than that in controls in KPNC (1.026 vs 1.045, respectively; p = 0.049) and in EIMS/GEMS (1.044 vs 1.056, respectively; p = 0.043). In both KPNC and EIMS/GEMS, the IV was not associated with year of birth, sex, smoking, college education, obesity at age 18–20 years or in 20s, HLA-DRB1*15:01 status, or the wGRS of 110 non-HLA MS risk variants, or with study type in the Swedish population. In EIMS/GEMS, it was associated with 2 of the 6 geographic regions.
Table 2.
Population characteristics for the KPNC and EIMS/GEMS study participants
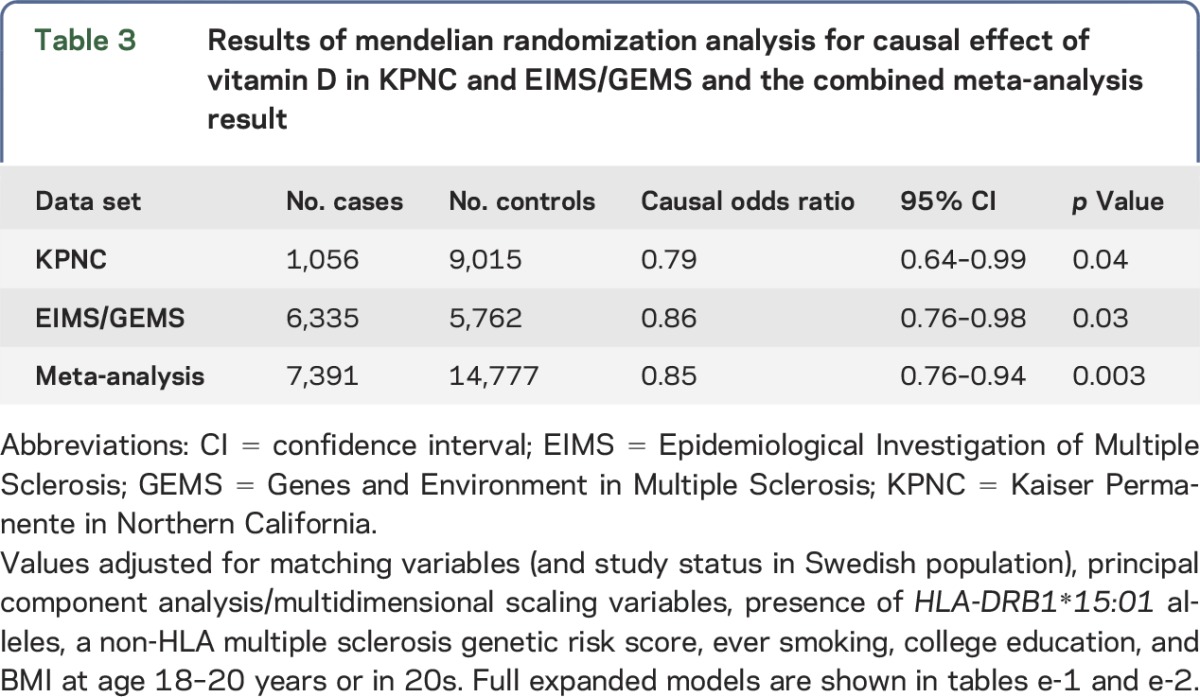
The 25(OH)D IV was associated with MS susceptibility after controlling for sex, year of birth, ancestry, smoking, wGRS of 110 non-HLA MS risk variants, and number of HLA-DRB1*15:01 alleles, as well as study type and region of residency in the Swedish study. An increasing IV score corresponding to increasing 25(OH)D levels was protective against MS (OR 0.79, 95% CI: 0.64–0.99; p = 0.04) in the KPNC study. An association was also found for the Swedish study (OR 0.86, 95% CI: 0.76–0.98; p = 0.03). Adjustment did not alter the findings substantially; however, for both KPNC and EIMS/GEMS data sets, p values and ORs were both lower in the full models compared with those in the unadjusted models (data not shown). Expanded models are shown for KPNC (table e-1 at Neurology.org/ng) and EIMS/GEMS (table e-2). Both studies were combined into a meta-analysis: the association remained (OR 0.85, 95% CI: 0.76–0.94; p = 0.003). No evidence of heterogeneity between populations was observed (I2 = 0.0%, heterogeneity χ2 p = 0.51). Results are summarized in table 3. The 25(OH)D IV was not associated with MSSS or age at onset in either study (data not shown).
Table 3.
Results of mendelian randomization analysis for causal effect of vitamin D in KPNC and EIMS/GEMS and the combined meta-analysis result
Validating additional assumptions.
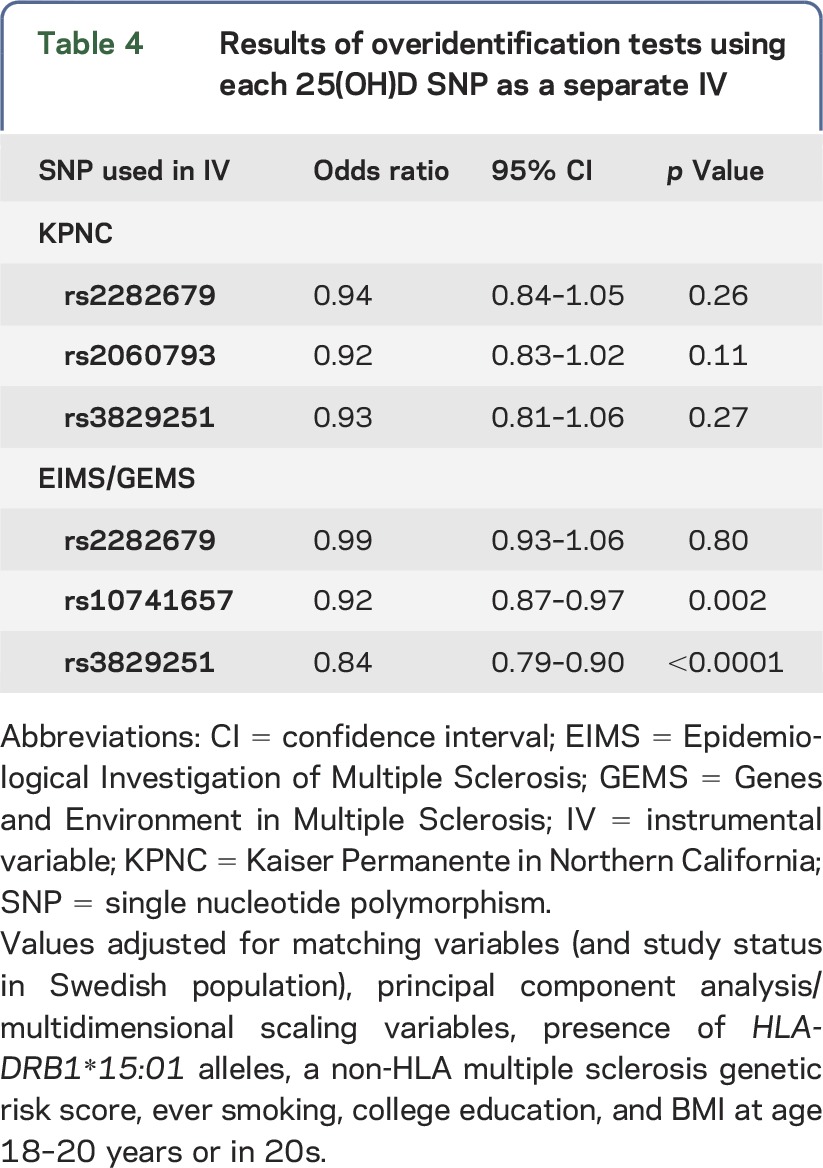
To test the MR model assumptions for the 25(OH)D IV, we conducted overidentification tests to evaluate the null hypothesis that effect estimates from multiple IVs are identical.20 Each variant was split into a separate instrument. Estimates suggested the same direction of causal effect, with ORs ranging from 0.92 to 0.94 in KPNC and 0.84 to 0.99 in EIMS/GEMS (table 4).
Table 4.
Results of overidentification tests using each 25(OH)D SNP as a separate IV
DISCUSSION
This study, using data from 2 different populations, provides strong evidence that low 25(OH)D is causally associated with MS susceptibility. Numerous observational studies have shown an association between low serum 25(OH)D level and increased risk of MS, and some have provided evidence supporting a causal association. In a study that examined patients with clinically isolated syndrome, a first event suggestive of MS, low 25(OH)D levels early in the disease course predicted higher MS activity.21 Two prospective, nested case-control studies, which collected serum prior to MS onset, showed a significantly reduced risk of MS in those with high 25(OH)D.22,23 Results from the current study rule out the most plausible alternative (noncausal) interpretations of these observational studies, thus strengthening evidence of a causal link between 25(OH)D and MS, even after accounting for other risk factors and potential confounders.
Molecular studies also suggest that low serum 25(OH)D may be a causal risk factor for MS. Studies examining the location of vitamin D receptor–binding sites (also known as vitamin D response elements or VDREs) in the genome have shown that MS-associated loci are substantially enriched for VDREs, including in the promoter region of HLA-DRB1,24,25 and that VDREs are more often exposed in open chromatin regions in immune cells compared with nonimmune cells.26 These findings reinforce the plausibility that vitamin D regulates genes that play important roles in the development or progression of MS. Furthermore, increased exposure to vitamin D leads to changes in immune cells that lead to decreased production of inflammatory cytokines, a decrease in Th1 and Th17 cell differentiation, and an increase in T regulatory cells, suggesting that low vitamin D is acting on MS by shifting the balance of the immune system toward a more proinflammatory state.27–29
The results of this study are in agreement with another recent MR study that used summary-level data only from the International Multiple Sclerosis Genetics Consortium and showed a causal effect for low 25(OH)D on MS risk.30 There is some data overlap between previous and current studies; specifically, 2,812 EIMS/GEMS participants included here were also in the summary data analysis.30 However, because individual-level data were not available for the prior study, the authors were unable to control for established genetic and environmental risk factors associated with MS. Importantly, the current study shows that measured variables did not contribute substantially to confounding of the causal relationship between low 25(OH)D and MS risk. Furthermore, the previous analysis relied on stronger assumptions30 compared with a traditional MR analysis as presented in the current study. Finally, a CYP24A1 SNP described in the previous report30 was not included in our analysis, as it did not demonstrate association with 25(OH)D at genome-wide significance,16 which was among the criteria for SNPs to be included in our IV for MR analysis. It is possible that the association between a new IV that included the CYP24A1 SNP and risk of MS might show a stronger magnitude of effect. The number of SNPs that capture variation in serum 25(OH)D levels is likely to grow as GWAS become larger.
An MR study relies on the following assumptions regarding the validity of the IV: (1) the genotype (IV) is associated with the phenotype (25(OH)D levels), (2) it is independent of measured or unmeasured confounders, and (3) it can only influence the outcome via the causal effect of the exposure.20 We were able to meet most model assumptions by using a 25(OH)D IV established to be strongly associated with serum 25(OH)D in an independent population through a large GWAS and testing whether the 25(OH)D IV is independent of measured confounders. However, associations with unmeasured or unknown confounders cannot be ruled out, leaving one assumption not fully testable.
Common reasons for violations of MR assumptions are population stratification, LD (between the loci under study and other polymorphisms associated with the outcome), pleiotropy, and developmental canalization.2 The potential for population stratification has been handled rigorously in the data analysis stage, and it is unlikely that it has caused confounding in our study. We have evaluated possible loci that might be associated with the 25(OH)D SNPs and with MS and have not found any basis for LD being the explanation for our findings. Pleiotropy concerns the possibility of polymorphisms having different biological effects that may influence the outcome through other pathways than the one studied. To our knowledge, the SNPs do not have other biological effects than in vitamin D metabolism, with the exception of rs3892951, upstream of DHCR7, which plays a role in cholesterol synthesis. Cholesterol metabolism has been associated with MS, although the exact mechanisms are not yet understood; a role for pleiotropy cannot be excluded.31 On the other hand, the SNPs are near genes that have a clear, biologically plausible role in determining 25(OH)D levels. GC codes for the vitamin D–binding protein, which transports vitamin D to target tissues; CYP2R1 converts vitamin D to its main circulating form, 25(OH)D; and DHCR7 converts 7-dehydrocholesterol, the compound that is converted to vitamin D3 in the skin in the presence of UV radiation, to cholesterol. In addition, a 2012 study evaluated SNPs in these 3 genes to look for evidence of pleiotropic effects and found that there were no associations with several biomarkers.32 A study of 1,500 Danish MS patients found that SNPs in GC and CYP2R1 have significant effects on 25(OH)D levels, further indicating that variants in those 2 genes are affecting MS via serum vitamin D.33 Canalization refers to the buffering of the effects of genetic variants against changing 25(OH)D levels; if this effect is present, the association between the IV and MS risk would be biased toward the null.
Strengths and limitations.
Our study included non-Hispanic whites, which limits the generalizability of our findings. This limitation is particularly relevant in light of recent findings indicating that 25(OH)D levels may not be associated with MS in Hispanic populations.34 We assume that the 3 alleles associated with 25(OH)D levels in the previous GWAS on Europeans are also associated with 25(OH)D levels in our populations. However, we were able to test this assumption in a subset of 2,077 EIMS participants for whom serum samples were collected at the time of inclusion in the study: a linear regression model adjusted for the same confounders as the main analyses showed that the IV was associated with 25(OH)D levels (p < 0.0001) among MS cases and healthy controls. Other limitations include the assumption of linearity and the assumption that the 3 alleles used to construct the IV have additive effects on 25(OH)D levels. Furthermore, our analysis of severity was restricted to a cross-sectional measure, the MSSS, and models could not account for the use of disease-modifying therapies. Additional studies should aim to replicate findings, specifically in populations of other races/ethnicities, and better examine how specific 25(OH)D-related variants might influence MS susceptibility and progressive disease. The strengths of this MR study are that results are very unlikely to be due to reverse causation or confounded by unmeasured variables, that we were able to adjust for genetic ancestry, which could feasibly still confound the relationship between 25(OH)D SNPs and MS, and that similar results were found in 2 independent populations from 2 areas with different sunlight exposure levels.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Allan Bernstein for his contributions to the Kaiser Permanente MS Research Program. They also thank the International MS Genetics Consortium for providing the genotype data for the vitamin D–related SNPs and SNPs in the MHC region used for HLA imputation for the Swedish population.
GLOSSARY
- CI
confidence interval
- EHR
electronic health record
- EIMS
Epidemiological Investigation of Multiple Sclerosis
- GERA
Genetic Epidemiology Research on Adult Health and Aging
- GEMS
Genes and Environment in Multiple Sclerosis
- GWAS
genome-wide association study
- HWE
Hardy-Weinberg equilibrium
- ICD-9
International Classification of Diseases, 9th Revision
- IV
instrumental variable
- KPNC
Kaiser Permanente in Northern California
- LD
linkage disequilibrium
- MAF
minor allele frequency
- MDS
multidimensional scaling
- MR
mendelian randomization
- MS
multiple sclerosis
- MSSS
Multiple Sclerosis Severity Scores
- SNP
single nucleotide polymorphism
- VDRE
vitamin D response element
- wGRS
weighted genetic risk score
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Brooke Rhead and Maria Bäärnhielm: analysis and interpretation and manuscript drafting. Milena Gianfrancesco, Amanda Mok, and Ingrid Kockum: interpretation of data and revision of manuscript. Xiaorong Shao: analysis and interpretation of data. Hong Quach: acquisition of data. Ling Shen, Catherine Schaefer, Jenny Link, Alexandra Gyllenberg, Tomas Olsson, and Jan Hillert: acquisition of data and revision of manuscript. Anna Karin Hedström and M. Maria Glymour: revision of manuscript. Lars Alfredsson and Lisa F. Barcellos: acquisition of data, revision of manuscript, and study supervision.
STUDY FUNDING
This study was funded by NIH grants R01AI076544, R01NS049510, and R01ES017080. Development of the Kaiser Permanente Research Program on Genes, Environment, and Health was supported by grants from the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, the Robert Wood Johnson Foundation, and the Kaiser Permanente Community Benefit Program. Genotyping of the GERA cohort was supported by a grant (RC2 AG036607; C.S. and Neil Risch, PIs).
DISCLOSURE
B. Rhead is funded by NIH Training Grant 5T32HG000047 and received support from the Rheumatology Research Foundation in 2014. M. Bäärnhielm reports no disclosures. M. Gianfrancesco has received research support from NIH/National Institute of Neurological Disorders and Stroke. A. Mok received support from the Rheumatology Research Foundation in 2015. X. Shao, H. Quach, and L. Shen report no disclosures. C. Schaefer has received research support from GlaxoSmithKline, NIEHS, NIMH, NIAID, NIA, and the National Multiple Sclerosis Society. J. Link has received research support from the Karolinska Institutet and Neuro Sweden. A. Gyllenberg has received funding from the Swedish NEURO Foundation 2014–2015. A.K. Hedström reports no disclosures. T. Olsson has received lecture and/or advisory board honoraria and unrestricted MS research grant support from Biogen, Novartis, Genzyme, SanofiAventis, Medimmune, and Merck-Serono; has received academic grant support from the Swedish Research Council, the AFA Foundation, the Knut and Alice Wallenberg Foundation, and the Swedish Brain Foundation; and has received research support from Merck, Biogen, SanofiAventis, Bayer, Novartis, AstraZeneca, Euratrans Neurinox, combiMS, the Swedish Brain Foundation, and the AFA Foundation. J. Hillert has received honoraria for serving on advisory boards for BiogenIdec, Novartis, and Sanofi-Genzyme; has received travel funding/speaker honoraria from BiogenIdec, Merck-Serono, Bayer-Schering, TEVA, Novartis, Sanofi-Genzyme, and Sanofi-Aventis; has served on the editorial board of the Journal of Neuroimmunology; has served as principle investigator for projects or received unrestricted research support from BiogenIdec, Merck-Serono, TEVA, Sanofi-Aventis, Novartis, and Bayer-Schering; and has received research support from the Swedish Research Council, Biogen, Novartis, Sanofi-Genzyme, Merck-Serono, Bayer-Schering, the Karolinska Institutet, and the Swedish Brain Foundation. I. Kockum has received speaker honoraria from Merck-Serono; has held stock in several Swedish companies, including Novo Nordisk (none related to this study); and has received research support from the “100års fonden,” the NHR Foundation, Karolinska University Hospital, and the Swedish Childhood Diabetes Foundation. M.M. Glymour has served on the scientific advisory boards of the MRC Centre for Causal Analyses in Translational Epidemiology and the Society for Epidemiologic Research; has received travel funding from Institut de Santé Publique, d'Epidémiologie et de Developpement, Université Victor Segalen Bordeaux; has served on the editorial boards of Epidemiology, PLoS One, the Journal of Causal Inference, and the American Journal of Epidemiology; has received publishing royalties from Oxford University Press; is an employee of the University of California, San Francisco; and has received research support from NIH/NIA, NIH/NIMH, NIA/Harvard Program on the Global Demography of Aging Pilot, NIH/NICHD, the Office for Minority Health and Health Disparities (NIH), the Milton Fund for Harvard University Junior Faculty, the American Heart Association, and the Robert Wood Johnson Foundation. L. Alfredsson receives research support from the Swedish Medical Research Council (521-2012-2917), the Swedish Research Council for Health, Working Life and Welfare (2012-0325 and 2015-00195), AFA Insurance, and the Swedish Brain Foundation; and has received speaker honoraria from TEVA and Biogen Idec. L.F. Barcellos has received research support from NIH/NIEHS, NIH/National Institute of Neurological Disorders and Stroke, USEPA, NIH/NCI, NIH/NIAMS, NIH/NIAID, the National Multiple Sclerosis Society, the Peder Sather Grant for Collaborative Research, and the Robert Wood Johnson Health and Society Program Pilot Grant. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Sundström P, Salzer J. Vitamin D and multiple sclerosis-from epidemiology to prevention. Acta Neurol Scand 2015;132:56–61. [DOI] [PubMed] [Google Scholar]
- 2.Smith GD, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32:1–22. [DOI] [PubMed] [Google Scholar]
- 3.Krieger N. Overcoming the absence of socioeconomic data in medical records: validation and application of a census-based methodology. Am J Public Health 1992;82:703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polman CH, Reingold SC, Banwell B, et al. . Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roxburgh RHSR, Seaman SR, Masterman T, et al. . Multiple Sclerosis Severity Score: using disability and disease duration to rate disease severity. Neurology 2005;64:1144–1151. [DOI] [PubMed] [Google Scholar]
- 6.Kvale MN, Hesselson S, Hoffmann TJ, et al. . Genotyping informatics and quality control for 100,000 subjects in the genetic epidemiology research on Adult health and aging (GERA) cohort. Genetics 2015;200:1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banda Y, Kvale MN, Hoffmann TJ, et al. . Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics 2015;200:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedström AK, Hillert J, Olsson T, Alfredsson L. Alcohol as a modifiable lifestyle factor affecting multiple sclerosis risk. JAMA Neurol 2014;71:300–305. [DOI] [PubMed] [Google Scholar]
- 9.Hedström AK, Åkerstedt T, Olsson T, Alfredsson L. Shift work influences multiple sclerosis risk. Mult Scler 2015;21:1195–1199. [DOI] [PubMed] [Google Scholar]
- 10.Gianfrancesco MA, Acuna B, Shen L, et al. . Obesity during childhood and adolescence increases susceptibility to multiple sclerosis after accounting for established genetic and environmental risk factors. Obes Res Clin Pract 2014;8:e435–e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs FBS, Acuña BS, Shen L, et al. . Adverse socioeconomic position during the life course is associated with multiple sclerosis. J Epidemiol Community Health 2014;68:622–629. [DOI] [PubMed] [Google Scholar]
- 12.Barcellos LF, Sawcer S, Ramsay PP, et al. . Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet 2006;15:2813–2824. [DOI] [PubMed] [Google Scholar]
- 13.Sawcer S, Hellenthal G, Pirinen M, et al. . Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dilthey A, Leslie S, Moutsianas L, et al. . Multi-population classical HLA type imputation. PLoS Comput Biol 2013;9:e1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 16.Ahn J, Yu K, Stolzenberg-solomon R, et al. . Genome-wide association study of circulating vitamin D levels. Hum Mol Genet 2010;19:2739–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.International Multiple Sclerosis Genetics Consortium (IMSGC), Beecham AH, Patsopoulos NA, et al. . Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013;45:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, et al. . PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 20.Glymour MM, Tchetgen EJT, Robins JM. Credible mendelian randomization studies: approaches for evaluating the instrumental variable assumptions. Am J Epidemiol 2012;175:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ascherio A, Munger KL, White R, et al. . Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol 2014;71:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006;296:2832–2838. [DOI] [PubMed] [Google Scholar]
- 23.Salzer J, Hallmans G, Nyström M, Stenlund H, Wadell G, Sundström P. Vitamin D as a protective factor in multiple sclerosis. Neurology 2012;79:2140–2145. [DOI] [PubMed] [Google Scholar]
- 24.Disanto G, Sandve GK, Berlanga-Taylor AJ, et al. . Vitamin d receptor binding, chromatin states and association with multiple sclerosis. Hum Mol Genet 2012;21:3575–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramagopalan SV, Maugeri NJ, Handunnetthi L, et al. . Expression of the multiple sclerosis-associated MHC class II Allele HLA-DRB1*1501 is regulated by vitamin D. PLoS Genet 2009;5:e1000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramagopalan SV, Heger A, Berlanga AJ, et al. . A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res 2010;20:1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart PH, Gorman S, Finlay-Jones JJ. Modulation of the immune system by UV radiation: more than just the effects of vitamin D? Nat Rev Immunol 2011;11:584–596. [DOI] [PubMed] [Google Scholar]
- 28.Aranow C. Vitamin D and the immune system. J Rheumatol 2010;37:491–495. [DOI] [PubMed] [Google Scholar]
- 29.Prietl B, Treiber G, Pieber TR, Amrein K. Vitamin D and immune function. Nutrients 2013;5:2502–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mokry LE, Ross S, Ahmad OS, et al. . Vitamin D and risk of multiple sclerosis: A Mendelian Randomization Study. PLoS Med 2015;12:e1001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tettey P, Simpson S, Taylor BV, van der Mei IAF. Vascular comorbidities in the onset and progression of multiple sclerosis. J Neurol Sci 2014;347:23–33. [DOI] [PubMed] [Google Scholar]
- 32.Berry DJ, Vimaleswaran KS, Whittaker JC, Hingorani AD, Hyppönen E. Evaluation of genetic markers as instruments for Mendelian randomization studies on vitamin D. PLoS One 2012;7:e37465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laursen JH, Sondergaard HB, Albrechtsen A, et al. . Genetic and environmental determinants of 25-hydroxyvitamin D levels in multiple sclerosis. Mult Scler J 2014;1414–1422. [DOI] [PubMed] [Google Scholar]
- 34.Rito Y, Flores J, Fernández Aguilar Á, et al. . Vitamin D in multiple sclerosis patients: not the same risk for everybody. Mult Scler 2016;22:126–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.