

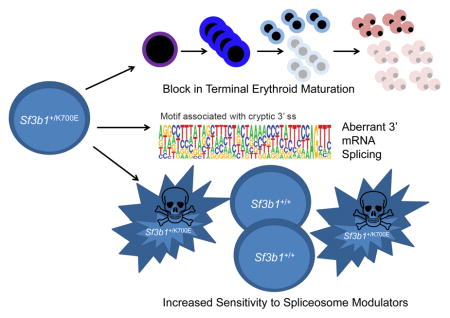
Summary
Over 80% of patients with the refractory anemia with ring sideroblasts subtype of myelodysplastic syndrome (MDS) have mutations in Splicing Factor 3B, Subunit 1 (SF3B1). We generated a conditional knock-in mouse model of the most common SF3B1 mutation, Sf3b1K700E. Sf3b1K700E mice develop macrocytic anemia due to a terminal erythroid maturation defect, erythroid dysplasia, and long-term hematopoietic stem cell (LT-HSC) expansion. Sf3b1K700E myeloid progenitors and SF3B1-mutant MDS patient samples demonstrate aberrant 3’ splice-site selection associated with increased nonsense-mediated decay. Tet2 loss cooperates with Sf3b1K700E to cause a more severe erythroid and LT-HSC phenotype. Furthermore, the spliceosome modulator, E7017, selectively kills SF3B1K700E-expressing cells. Thus, SF3B1K700E expression reflects the phenotype of the mutation in MDS and may be a therapeutic target in MDS.
In Brief/eTOC Blurb
Obeng et al. generate knockin mice with SF3B1K700E, a prevalent mutation in myelodysplastic syndrome (MDS). Sf3b1+/K700E mice display characteristics of MDS. Mouse and human MDS cells expressing SF3B1K700E exhibit aberrant 3’ splice site selection, and SF3B1K700E sensitizes cells to a spliceosome modulator.
Introduction
Recurrent point mutations in components of the 3’ mRNA splicing machinery are the most common mutations in patients with myelodysplastic syndromes (MDS) (Papaemmanuil et al., 2011; Visconte et al., 2012a; Welch et al., 2012; Yoshida et al., 2011). MDS is characterized by clonal hematopoiesis, peripheral blood cytopenias, and a variable risk of transformation to acute myeloid leukemia (AML). Splicing factor gene mutations in MDS are typically heterozygous, missense, and mutually exclusive of one another. The three most commonly mutated splicing factors in MDS, SF3B1, U2AF1, and SRSF2, are all involved in the initial steps of spliceosome assembly (Kramer, 1996; Will and Luhrmann, 2011; Yoshida et al., 2011).
The spliceosome is a large macromolecular complex composed of five small nuclear ribonucleoproteins (snRNPs) and over 200 additional proteins (Chen and Manley, 2009). Over 90% of human genes undergo alternative splicing of precursor messenger RNA (pre-mRNA), a tightly regulated process that dramatically increases the complexity of the protein repertoire encoded by the genome (Becerra et al., 2015). The 5’ splice site of an intron is typically defined by a GU dinucleotide. Three sequence elements define the 3’ splice site: an AG dinucleotide at the 3' end of the intron, a variable length of pyrimidine nucleotides within the intron called the polypyrimidine tract, and a branchpoint sequence upstream of the polypyrimidine tract that generally contains a conserved adenosine. Recognition of these sequences by different components of the spliceosome machinery is required for the initial step of pre-mRNA splicing (Will and Luhrmann, 2011).
SF3B1 encodes subunit 1 of the splicing factor 3b (SF3b) protein complex. Together with the SF3a protein complex and a 12S rRNA, SF3b forms the U2 snRNP (Kramer, 1996). Base pairing between the U2 snRNP and the branch point sequence is essential for pre-mRNA splicing (Gozani et al., 1998). The SF3b/SF3a complex anchors the U2 snRNP to the pre-mRNA (Gozani et al., 1996), and SF3B1 is a critical component of the activated spliceosome that helps position the branch point adenosine for nucleophilic attack from the 5’ splice site (Gozani et al., 1998).
SF3B1 point mutations observed in MDS are confined to exons 14 through 16. The most common SF3B1 mutation is an A to G transition that results in a lysine to glutamic acid substitution at amino acid position 700 (K700E) (Papaemmanuil et al., 2011; Yoshida et al., 2011). The absence of nonsense or frameshift mutations and the presence of specific amino acid substitutions in each of the three most commonly mutated spliceosome genes suggest specific alterations of function are required for their pathogenesis in MDS.
SF3B1 mutations are present in ~25% of all MDS cases (Garcia-Manero, 2012; Malcovati et al., 2011) and in over 85% of cases of Refractory Anemia with Ring Sideroblasts (RARS), a form of MDS characterized by an isolated anemia, erythroid dysplasia, and the presence of at least 15% ring sideroblasts in the bone marrow (Malcovati et al., 2011; Mufti et al., 2008; Papaemmanuil et al., 2011; Vardiman et al., 2002; Yoshida et al., 2011). Ring sideroblasts are erythroid precursors with pathologically iron-laden mitochondria encircling the nucleus (Cartwright and Deiss, 1975; Mufti et al., 2008).
Several studies have attempted to decipher the role of MDS-associated splicing factor mutations in malignant hematopoiesis in vivo. Mouse models of MDS-associated splicing factor mutations in Srsf2 (Kim et al., 2015) and U2AF1 (Shirai et al., 2015) exhibit features of MDS, including cytopenias and an increase in hematopoietic stem and progenitor cells (HSPCs). Transcriptome analysis has revealed distinct splicing abnormalities between the two genotypes related to their function within the spliceosome. In this paper, we describe a hematopoietic-specific conditional knock-in mouse model of Sf3b1K700E and characterize its effects on malignant hematopoiesis, pre-mRNA splicing, and the pathogenesis of MDS.
Results
Physiologic, hematopoietic-specific SF3B1K700E expression causes a progressive macrocytic anemia
In order to study the role of SF3B1 mutations in the pathogenesis of MDS, we generated a Sf3b1K700E conditional knock-in mouse (Figure 1A). Mice heterozygous for the allele (denoted Sf3b1+/K700E) were crossed with Mx1-Cre transgenic animals, and expression of Cre recombinase was induced with pIpC injection (Jaisser, 2000; Kuhn et al., 1995). We confirmed recombination by PCR performed on genomic DNA isolated from peripheral blood 3 weeks post-pIpC (Figure 1B). Expression of the A2213G mutation was confirmed by Sanger sequencing of cDNA isolated from peripheral blood (Figure 1C) and by RNA sequencing (Figure S1A). We confirmed that total Sf3b1 expression was similar between wild-type Mx1-cre positive littermates (denoted Sf3b1+/+), Sf3b1+/K700E mice, and Mx1-cre negative mice that carried the targeting construct (denoted Flox Cre Negative) by qRT-PCR (Figure S1B).
Figure 1. Heterozygous conditional knock-in of Sf3b1K700E results in a progressive macrocytic anemia.

(A) Targeting allele for conditional expression of Sf3b1K700E. Upon induction of the Cre recombinase, the inverted exons 15 and 16 containing a guanine mutation at position 2213 are flipped into the proper orientation. (B) PCR analysis of genomic DNA isolated from the peripheral blood of 2 wild-type (Sf3b1+/+) and 2 heterozygous mutant (Sf3b1+/K700E) mice 3 weeks post- pIpC. (C) Expression of the A2213G mutation was analyzed by Sanger sequencing of cDNA isolated from the peripheral blood of Sf3b1+/+ and Sf3b1+/K700E mice 3 weeks post-pIpC. Data are representative of at least 9 mice per group. (D-E) Analysis of peripheral blood hemoglobin (Hb, D) and mean corpuscular volume (MCV, E) over the course of 64 weeks post-pIpC (n = 9 Sf3b1+/+ and 11 Sf3b1+/K700E mice). (F) Plasma erythropoietin (Epo) levels measured 12 weeks post-pIpC (n = 9 Sf3b1+/+ and 11 Sf3b1+/K700E mice). (G–H) Peripheral blood Hb (G) and MCV (H) 4 and 12 weeks after noncompetitive transplantation of 1.0 × 106 unfractionated bone marrow cells from Sf3b1+/K700E and Sf3b1+/+ donors into B6.SJL congenic recipient mice (n = 5 mice per group; horizontal lines indicate the mean). Data presented as mean ± SEM. * p < 0.05; ** p < 0.001. See also Figure S1.
We analyzed 11 Sf3b1+/K700E mice and 9 littermate controls for a period of 64 weeks following pIpC treatment. Compared to Sf3b1+/+ littermate controls, Sf3b1+/K700E mice developed a progressive anemia. Anemia was present as early as 4 to 8 weeks post-pIpC and was more severe by 20 weeks post-pIpC (Figure 1D). Macrocytosis, an abnormal increase in the erythrocyte mean corpuscular volume (MCV), was apparent at 20 weeks post-pIpC (Figure 1E). The total red blood cell number was also consistently lower in Sf3b1+/K700E mice compared to Sf3b1+/+ littermates beginning at 20 weeks post-pIpC (Figure S1C). In response to the anemia, plasma erythropoietin levels in Sf3b1+/K700E mice increased as early as 12 weeks post-pIpC (Figure 1F) and persisted for over 60 weeks post-pIpC (Figure S1D). However, the reticulocyte counts in Sf3b1+/K700E mice remained inappropriately normal over the course of the 64 weeks (Figure S1E). These findings suggest a red blood cell production defect is associated with SF3B1K700E expression. No significant differences were observed in the total white blood cell count, mature white blood cell lineages, or platelet counts between the two groups (Figures S1F–S1H). All of the Sf3b1+/+ mice were alive at 64 weeks post-pIpC, whereas two of the Sf3b1+/K700E mice died (18%, Figure S1I). No animals developed acute leukemia.
To determine whether the erythroid-specific effects we observed with SF3B1K700E expression are cell intrinsic, we performed noncompetitive bone marrow transplantation experiments. Four weeks after pIpC treatment, unfractionated bone marrow from Sf3b1+/K700E and Sf3b1+/+ CD45.2+ littermate donors was transplanted into lethally irradiated congenic CD45.1+ B6.SJL wild-type recipients. Donor chimerism was > 95% in both recipient groups over the course of the experiment (Figure S1J). Similar to the phenotype of Sf3b1+/K700E primary mice, Sf3b1+/K700E recipient mice developed a macrocytic anemia within 12 weeks post-transplantation (Figures 1G and 1H). White blood cell and platelet counts remained normal (Figures S1K and S1L). Taken together, these findings demonstrate that heterozygous expression of SF3B1K700E causes a progressive, macrocytic anemia associated with a compensatory increase in plasma erythropoietin levels and relative preservation of the white blood cell and platelet counts.
SF3B1K700E expression is associated with a block in terminal erythroid maturation and erythroid dysplasia
To characterize the pathogenesis of the anemia seen in Sf3b1+/K700E mice, we examined committed erythroid precursor maturation in the spleen and bone marrow of Sf3b1+/+ and Sf3b1+/K700E mice based on CD71 and Ter119 expression, which subdivides cells into stages R1-R4 of erythroblast maturation (Figure S2A) (Socolovsky et al., 2001; Zhang et al., 2003). We observed a block in erythroblast development, characterized by an accumulation of cells in stage R2 of erythroblast maturation and a concomitant decrease in the stage R4 population in both the spleen (Figure 2A) and bone marrow (Figure S2B) of Sf3b1+/K700E animals, but not in littermate controls, 64 weeks post-pIpC. These changes are most prominent in the spleen, the primary site of stress erythropoiesis in mice. We did not observe this block in erythroid maturation in the spleen or bone marrow of young mice (12 weeks post-pIpC, Figures S2C and S2D), consistent with the onset of primary MDS in older patients (Garcia-Manero, 2012). A similar block in erythroid differentiation was also apparent using a more recently reported methodology for characterizing terminal erythropoiesis (Liu et al., 2013) using forward scatter, Ter119 and CD44 expression (Figures S2E–S2G).
Figure 2. Heterozygous conditional knock-in of Sf3b1K700E causes a cell intrinsic block in terminal erythroid maturation and erythroid dysplasia.

(A) Analysis of terminal erythroid maturation, defined as progression from immature R1 to more mature R4 erythroblasts, in the spleens of Sf3b1+/+ and Sf3b1+/K700E mice, 64 weeks post-pIpC (n = 9 Sf3b1+/+ and 11 Sf3b1+/K700E mice). (B) Hb measured 4 days before and 3, 6, and 9 days after the first injection of Sf3b1+/+ and Sf3b1+/K700E animals with phenylhydrazine (PHZ, n = 11 Sf3b1+/+ and 10 Sf3b1+/K700E mice). (C) Analysis of terminal erythroid maturation in the spleens of 11-week old Sf3b1+/+ and Sf3b1+/K700E mice, 9 days after PHZ treatment (n = 6 Sf3b1+/+ and 5 Sf3b1+/K700E mice). (D–F) 2.0 × 105 bone marrow c-Kit+ progenitor cells from 3 Sf3b1+/+ and 3 Sf3b1+/K700E mice were isolated 24 weeks post-pIpC and plated in erythroid differentiation medium. The number of Sf3b1+/+ or Sf3b1+/K700E cells in culture 48 and 72 hr after plating was quantified in triplicate samples from each mouse (D). The number of cells in each stage of terminal erythroid maturation 48 hr after plating was quantified in triplicate samples from each mouse (E). Cell cycle analysis by combined proliferation (Ki67) and cell cycle (Hoechst33342) staining in permeabilized Sf3b1+/+ or Sf3b1+/K700E cells, 48 hr after cells were plated (F, G0: Ki67−/Hoechst−; G1: Ki67+/Hoechst−; S/G2/M: Ki67+/Hoechst+; duplicate samples were analyzed from 3 separate mice per group). (G) Spleens from Sf3b1+/+ and Sf3b1+/K700E mice, 64 weeks post-pIpC, stained with an anti-CD71 antibody to mark erythroid progenitor cells. Scale bar indicates 125 μM. (H) Hematoxylin and eosin stains of spleens from Sf3b1+/+ and Sf3b1+/K700E mice, 47 weeks post-pIpC. Arrows indicate dysplastic erythroid precursors. Scale bar indicates 10 μM. Data presented as mean ± SEM. * p < 0.05; ** p < 0.01. See also Figure S2
We next evaluated the effects of the SF3B1K700E mutation on stress erythropoiesis by treating mice with phenylhydrazine (PHZ), a drug that induces rapid intravascular hemolysis (Socolovsky et al., 2001). We injected primary Sf3b1+/+ and Sf3b1+/K700E mice subcutaneously with 30 mg/kg PHZ on days 0 and 1 and then monitored the hemoglobin (Hb) and absolute reticulocyte counts. Experiments were performed on 11 week-old mice, 3 weeks post-pIpC, in order to assess stress erythropoiesis at a time when the mice did not have a significant baseline anemia. Following PHZ treatment, the Hb nadir of Sf3b1+/K700E mice occurred earlier and was significantly lower than the Hb nadir of Sf3b1+/+ controls (Figure 2B). Although not statistically significant, the rise in the reticulocyte count in response to PHZ was lower in Sf3b1+/K700E mice compared to Sf3b1+/+ mice on days 3 and 6 (Figure S2H). We also assessed erythroblast maturation with CD71 and Ter119 staining, nine days after PHZ treatment and noted impaired erythroblast maturation in the spleens of PHZ-treated Sf3b1+/K700E mice (Figure 2C). This is in contrast to the spleens of similarly aged, young Sf3b1+/K700E mice that were not treated with PHZ (Figure S2C).
Although several studies have reported that Sf3b1 heterozygous (Sf3b1+/−) mice do not develop progressive macrocytic anemia (Matsunawa et al., 2014; Visconte et al., 2012b; Wang et al., 2014), erythroid maturation was only specifically measured in one study where no differences were seen (Wang et al., 2011). Visconte et al. subsequently observed an intermittent increase in MCV and a statistically significant decrease in Hb at 11 months in Sf3b1+/− mice (Visconte et al., 2014). We therefore sought to compare Sf3b1+/+ and Sf3b1+/K700E erythroblast maturation using an in vitro culture system that closely mimics the in vivo terminal proliferation and maturation of erythroid cells. Equal numbers of c-Kit+ HSPCs were isolated from the bone marrow of Sf3b1+/+ and Sf3b1+/K700E mice, and the cells were cultured in the presence of cytokines that induce erythroid differentiation (Ishikawa et al., 2014). Although both the Sf3b1+/+ and Sf3b1+/K700E HSPCs differentiated in culture, the total number of cells (Figure 2D) and the absolute number of cells at each stage of erythroid maturation (Figure 2E) were significantly lower in cells derived from the Sf3b1+/K700E animals compared to Sf3b1+/+ animals. Cell cycle analysis revealed a significantly lower percentage of Sf3b1+/K700E cells in G1 and a higher percentage of Sf3b1+/K700E cells in G0 compared to Sf3b1+/+ erythroblasts (Figure 2F). There was no difference in apoptosis based on Annexin V staining (Figure S2I). In contrast to the Sf3b1+/K700E HSPCs, in vitro erythroid differentiation of Sf3b1+/− HSPCs was not impaired (Figures S2J and S2K).
There are also conflicting reports in the literature regarding whether Sf3b1 haploinsufficiency causes dyserythropoiesis and ring sideroblast formation. Visconte et al. noted rare ring sideroblasts and some dyserythropoietic features in the bone marrow of Sf3b1+/− mice (Visconte et al., 2012b; Visconte et al., 2014). In contrast, two independent groups quantified the number of ring sideroblasts in the bone marrow of Sf3b1+/+ and Sf3b1+/− mice and found no differences between these animals (Matsunawa et al., 2014; Wang et al., 2014). We performed an extensive review of the peripheral blood and bone marrow of Sf3b1+/+ and Sf3b1+/K700E mice and did not find a statistically significant increase in ring sideroblasts or circulating siderocytes, enucleated red blood cells with one or more Perls’ stain positive iron granules, in the Sf3b1+/K700E animals (data not shown). This is consistent with the absence of ring sideroblasts in mouse models of congenital sideroblastic anemias, including a conditional knockout model of Abcb7 (Friedman et al., 2004; Keyhani et al., 1974a; Pondarre et al., 2007). However, we did observe an increase in erythroid precursors and erythroid dysplasia in the spleens of Sf3b1+/K700E mice (Figures 2G and 2H). No significant differences in bone marrow cellularity were observed between Sf3b1+/+ and Sf3b1+/K700E mice (data not shown). In aggregate, these data demonstrate that hematopoietic specific expression of the Sf3b1K700E allele in mice leads to impaired terminal erythroid maturation, which is exacerbated by age and stress erythropoiesis.
SF3B1K700E expression results in expansion of the long-term hematopoietic stem cell compartment
We next determined whether expression of SF3B1K700E during adult hematopoiesis affects different HSPC compartments by performing multi-parameter flow cytometry on the bone marrow of Sf3b1+/+ and Sf3b1+/K700E mice, 64 weeks after pIpC treatment. Although we found no significant difference in the frequency of Lin− Sca-1+ c-Kit+ (LSK) cells (data not shown), we observed a significant increase in the frequency of CD150+ CD48− Lin− Sca-1+ c-Kit+ long term repopulating hematopoietic stem cells (LT-HSCs) and a statistically significant decrease in the percentage of granulocyte/monocyte progenitors (GMPs) in aged Sf3b1+/K700E mice (Figures 3A and 3B). We observed no significant differences in the frequency of short-term hematopoietic stem cells (ST-HSCs), multipotent progenitors (MPPs), common myeloid progenitors (CMPs), megakaryocyte/erythroid progenitors (MEPs), or colony forming units in methylcellulose assays (Figures 3A–3C).
Figure 3. Heterozygous expression of Sf3b1K700E selectively enriches for long-term hematopoietic stem cells but is associated with decreased repopulating ability.

(A–B) Number of LT-HSCs, ST-HSCs, and MPPs (A) and CMPs, GMPs, and MEPs (B) in the bone marrow of Sf3b1+/+ and Sf3b1+/700E mice, 64 weeks post-pIpC (n = 9 Sf3b1+/+ and 11 Sf3b1+/K700E mice). (C) Absolute number of colonies (BFU-E, CFU-G, CFU-M, CFU-GM, CFU-GEMM) 7 days after 2.0 × 105 unfractionated bone marrow cells isolated from Sf3b1+/+ or Sf3b1+/K700E mice 24 weeks post-pIpC were plated in M3434 methylcellulose. Triplicate samples were plated from 3 mice per group. (D) Percentage of CD45.2 donor chimerism in the peripheral blood 4 to 20 weeks after competitive repopulation assay. 1 × 106 unfractionated bone marrow cells from CD45.2+ Sf3b1+/+ or Sf3b1+/700E mice were mixed 1:1 with CD45.1+ B6.SJL unfractionated bone marrow cells and transplanted into lethally irradiated (10.5 Gy) B6.SJL recipients (n = 5 mice per group). CD45.2 chimerism of the input bone marrow was measured at time 0 (n = 1 sample per group). (E) Percentage of CD45.2+ donor LT-HSC, ST-HSC, MPP, and LK chimerism in the bone marrow of Sf3b1+/+ or Sf3b1+/K700E recipient mice 20 weeks after competitive repopulation assay (n = 4–5 mice per group). Data presented as mean ± SEM. * p < 0.05. See also Figure S3.
To investigate whether this increase in LT-HSCs was due to increased self-renewal in vivo, we performed competitive repopulation experiments. Four weeks after pIpC treatment, one million unfractionated bone marrow cells from CD45.2+ Sf3b1+/K700E or Sf3b1+/+ littermates were mixed in a 1:1 ratio with unfractionated bone marrow cells from congenic CD45.1+ B6.SJL mice and injected into lethally irradiated CD45.1+ B6.SJL recipients. At all time points analyzed, we observed significantly lower peripheral blood chimerism in the Sf3b1+/K700E recipient mice compared to the Sf3b1+/+ recipient mice (Figure 3D). CD45.2 chimerism was significantly lower in all stem and progenitor cells (Figure 3E), and we did not observe any skewing of mature white blood cells derived from Sf3b1+/K700E HSPCs (Figures S3A–S3C). Given the low level of peripheral blood chimerism noted as early as 4 weeks post-transplant, we cannot completely rule out an engraftment defect in the Sf3b1+/K700E cells. However, we have observed similar engraftment of Sf3b1+/K700E and Sf3b1+/+ bone marrow cells in noncompetitive transplant experiments (Figure S1J). Taken together, these data suggest that hematopoietic-specific expression of SF3B1K700E is associated with an increase in the frequency of immunophenotypic LT-HSCs in the bone marrow, but a significantly impaired capacity to reconstitute hematopoiesis in a competitive transplantation setting, relative to Sf3b1+/+ cells.
SF3B1K700E expression leads to increased alternative 3’ splice site usage
To determine whether SF3B1K700E expression is associated with mutation-specific alterations in pre-mRNA splicing, we performed RNA sequencing (RNAseq) on myeloid progenitor (Lin− c-Kit+, LK) cells sorted from three Sf3b1+/K700E and three Sf3b1+/+ mice, 4 weeks after pIpC administration. Sf3b1K700E mutant allele expression was 27–32% (Figure S1A). We identified 72 aberrant splicing events that were specific to Sf3b1+/K700E myeloid progenitors [False Discovery Rate (FDR) <0.1], with 48/72 (66%) of these events representing alternative 3’ splicing events (Figure 4A).
Figure 4. Aberrant 3’ splicing due to recognition of an alternative branch point adenosine motif is conserved between murine and human cells expressing mutant SF3B1.

(A) Number and type of alternative splicing events in Sf3b1+/K700E LK myeloid progenitors compared to littermate controls (3 mice per group; n = 72 events, FDR < 0.1). (B) Number and type of alternative splicing events in unfractionated bone marrow mononuclear cells isolated from SF3B1-mutant (n = 6) compared to wild-type SF3B1 (n = 4) MDS patient samples (n = 134 events, FDR < 0.05). (C) Density plot of the relative positions of cryptic 3’ splice sites (ss) compared to their canonical 3’ ss in SF3B1-mutant MDS patient samples and Sf3b1+/K700E myeloid progenitors. The distance to the first AG from all Reference Sequence (RefSeq) canonical 3’ ss is included for comparison (n = number of alternative 3’ splicing events analyzed). (D–E) Consensus 3’ ss motif near the canonical (bottom) 3’ AG dinucleotide and cryptic (top) 3’ AG dinucleotide for 21 alternative 3’ splicing events identified in Sf3b1+/K700E compared to Sf3b1+/+ myeloid progenitor cells (D) and 67 alternative 3’ splicing events identified in SF3B1-mutant compared to wild-type SF3B1 bone marrow mononuclear cells (E). Motif components are underlined and defined in the key. (F) The number of adenosines found 8 to 18 nucleotides upstream of the cryptic AG compared to the canonical AG in six SF3B1-mutant MDS patient samples. Box plots and whiskers are represented as per the Tukey method. See also Figure S4 and Tables S1–S3. .
To extend these findings to human samples, we performed RNAseq on unfractionated bone marrow mononuclear cells isolated from six SF3B1-mutant and four SF3B1-wild-type MDS patient samples matched for MDS-subtype, prognostic score, karyotype, and the presence or absence of additional MDS-associated mutations (Table 1). In total, 134 aberrant splicing events were identified in patient samples with SF3B1 mutations (FDR < 0.05, Figure 4B). Although comparison of the differentially-spliced genes identified in human SF3B1-mutant MDS and Sf3b1+/K700E myeloid progenitor cells revealed minimal overlap (Tables S1 and S2), the types of splicing events were remarkably similar. As observed in murine Sf3b1-mutant cells, the most common aberrant splicing event in SF3B1-mutated MDS samples was alternative 3’ splice site (ss) selection (91/134, 67.9%; Figure 4B). The locations of the cryptic 3’ ss in mouse and human cells were both between -15 and -24 nucleotides upstream of the canonical 3′ ss (Figure 4C), similar to recently published reports studying 3’ ss selection in other SF3B1-mutant cancers (Alsafadi et al., 2016; Darman et al., 2015; DeBoever et al., 2015). Finally, the sequence contexts associated with the cryptic 3’ ss in both the Sf3b1+/K700E myeloid progenitors (Figure 4D) and SF3B1-mutant MDS patient samples (Figure 4E) are both characterized by upstream adenosine enrichment and a shorter/weaker polypyrimidine tract (Figure 4F and data not shown), motifs which are in accord with mutant SF3B1-specific cryptic ss previously reported in chronic lymphocytic leukemia (CLL) and several solid tumor patient samples and cell lines (Alsafadi et al., 2016; Darman et al., 2015).
Table 1.
MDS Patient Sample Characteristics
| Sample ID | SF3B1 | Mutant Allele Frequency | Sex | FAB1 | IPSS2 | Karyotype | Additional Mutations |
|---|---|---|---|---|---|---|---|
| MDS_149 | - | - | M | RARS | Int1 | Normal |
TET2 ASXL1 |
| MDS_314 | p.K700E | 42.69 | M | RA | Int1 | Normal | |
| MDS_228 | - | - | M | RA | Low | Normal | ASXL1 |
| MDS_260 | p.K666T | 44.60 | M | RA | Low | Normal | |
| MDS_328 | - | - | M | RARS | Int1 | Normal | TET2 |
| MDS_361 | p.K700E | 38.32 | M | RARS | Int1 | Normal | |
| MDS_292 | - | - | M | RA | Low | Normal | None |
| MDS_253 | p.K666R | 42.62 | M | RA | Low | Normal | |
| MDS_334 | p.K700E | 36.81 | M | RA | Low | Normal | |
| MDS_341 | p.K700E | 32.82 | M | RARS | Low | Normal |
French-American-British Classification
MDS International Prognistic Scoring System Score
Additional computational analysis of the sequences between each cryptic and canonical 3’ ss revealed eight genes which were differentially spliced in the presence of mutant SF3B1 in both human and murine cells (FDR < 0.05, Table 2). Three of the cryptic 3’ ss were in junctions that were ≥ 95% conserved between mice and humans. Two of these genes are predicted to be involved in RNA processing: SKIV2L encodes a putative RNA helicase predicted to block translation of poly(A)-deficient mRNA; SERBP1 is predicted to encode an RNA binding protein with a role in the regulation of mRNA stability.
Table 2.
Genes with Conserved Aberrant 3’ Splice Sites Between Sf3b1+/K700E Myeloid Progenitors and SF3B1-Mutant MDS Patient Samples
| Mouse Aberrant Junction1 | Mouse Gene Symbol | Human Gene Symbol | Human Aberrant Junction2 | Mouse NMD3 | Human NMD |
|---|---|---|---|---|---|
| chr1:156041134- 156051278:+:start |
Tor1aip2 | TOR1AIP2 | chr1:179835004- 179846373:-:end |
No | No |
| chr1:156041002- 156051278:+:start |
Tor1aip2 | TOR1AIP2 | chr1:179835004- 179846373:-:end |
No | No |
| chr1:92918126- 92918965:+:start |
Rnpepl1 | RNPEPL1 | chr2:241515027- 241516055:+:start |
No | Yes |
| chr19:41773142- 41774065:-:end |
Arhgap19 | ARHGAP19 | chr10:99023246- 99024582:-:end |
No | Yes |
| chr17:34846173- 34846521:-:end |
Skiv2l | SKIV2L | chr6:31936315- 31936399:+:start |
No | Yes |
| chr6:67272228- 67272863:+:start |
Serbp1 | SERBP1 | chr1:67890660- 67890765:-:end |
No | No |
| chr2:59933730- 59935214:-:end |
Baz2b | BAZ2B | chr2:160253915- 160255339:-:end |
No | No |
| chr12:84295335- 84297989:+:start |
Ptgr2 | PTGR2 | chr14:74358911- 74360478:+:start |
Ambiguous | No |
| chr6:67272027- 67272138:+:start |
Serbp1 | SERBP1 | chr1:67890660- 67890765:-:end |
No | No |
| chr18:36613976- 36614714:+:start |
Ankhd1 |
ANKHD1- EIF4EBP3 |
chr5:139815842- 139818078:+:start |
No | No |
Sequences were aligned to hg19
Sequences were aligned to mm10
NMD, aberrant transcript predicted to undergo nonsense-mediated decay Bold indicates junctions that are ≥ 95% conserved between mouse and human
The lack of overlapping genes between human and murine samples is likely due to the poor conservation of intronic DNA sequences between species (Roy and Gilbert, 2006). This is illustrated by ABCB7, a heme transporter known to be mutated in X-linked sideroblastic anemia with ataxia (Allikmets et al., 1999) and reported to be downregulated in RARS (Boultwood et al., 2008; Pellagatti et al., 2006). In our human MDS data, we also observed the aberrant splicing of ABCB7 described by Darman et al. in an isogenic B cell line expressing the SF3B1K700E mutation (Figure S4A) (Darman et al., 2015). However, the sequence for the ABCB7 cryptic ss is not conserved in mice, and we did not observe aberrant splicing of Abcb7 in the Sf3b1-mutant murine cells.
Gene set enrichment analysis (GSEA) of the differentially expressed genes revealed significant changes in genes involved in RNA processing and metabolism, cell cycle, heme metabolism, and nonsense-mediated decay (NMD, Table S3). NMD was predicted to occur in 36.8% of the altered murine transcripts with an FDR < 0.1, and 32.7% of the altered human transcripts with an FDR < 0.05 (Tables S1 and S2). Gene expression analysis of the murine (Figure S4B) and human (Figure S4C) genes with altered transcripts predicted to undergo NMD revealed that their expression was significantly less than the expression of genes with altered transcripts that were not predicted to undergo NMD (p = 0.0003). These data demonstrate that Sf3b1+/K700E myeloid progenitors and SF3B1-mutant MDS samples faithfully recapitulate the alterations in pre-mRNA splicing observed in other cancer cells with SF3B1 mutations.
Additive effect of Sf3b1K700E mutation and Tet2 deletion on MDS pathogenesis
Loss of function mutations in TET2 occur in over 20% of MDS patient samples (Bejar et al., 2011; Langemeijer et al., 2009), and SF3B1 and TET2 mutations commonly co-occur in patients with MDS (Bejar et al., 2012; Haferlach et al., 2014). To study the effect of this combination of mutations on MDS pathogenesis, we crossed Sf3b1+/K700E mice with a previously characterized conditional Tet2 knockout mouse (Moran-Crusio et al., 2011; Quivoron et al., 2011).
Both Sf3b1+/K700E and Sf3b1+/K700E Tet2−/− mice developed a progressive macrocytic anemia (Figures 5A and 5B), with Sf3b1+/K700E Tet2−/− mice exhibiting a more severe anemia and macrocytosis. Moreover, the double mutant mice display an accelerated terminal erythroid maturation block compared to mice harboring either mutation alone, as early as 12 weeks post-pIpC (Figure 5C). By 45 weeks post-pIpC, this block in erythroid maturation is apparent in all of the mutant animals (Figure 5D).
Figure 5. The combination of the Sf3b1K700E mutation and Tet2 deletion causes an earlier onset of MDS characteristics and rescues the competitive disadvantage of Sf3b1+/K700E stem cells.

(A–B) Peripheral blood Hb (A) and MCV (B) from Sf3b1+/+, Sf3b1+/K700E, Tet2−/−, and Sf3b1+/K700E Tet2−/− mice 45 weeks post-pIpC (n = 7–10 mice per group). (C–D) Analysis of terminal erythroid maturation in the spleens of Sf3b1+/+, Sf3b1+/K700E, Tet2−/−, and Sf3b1+/K700E Tet2−/− mice 12 (C) and 45 (D) weeks post-pIpC (n = 7–10 mice per group). (E–F) Number of LT-HSCs, ST-HSCs, and MPPs in the bone marrow of Sf3b1+/+, Sf3b1+/K700E, Tet2−/− and Sf3b1+/K700E Tet2−/− mice 12 (E) and 45 (F) weeks post-pIpC (n = 7–10 mice per group). (G) Periodic acid-Schiff stained spleens from Sf3b1+/+, Sf3b1+/K700E, Tet2−/−, and Sf3b1+/K700E Tet2−/− mice, 45 weeks post-pIpC. Arrows indicate dysplastic erythroid precursors. Scale bar indicates 5 μM. (H) Percentage of CD45.2 donor chimerism in the peripheral blood 4 to 24 weeks after competitive repopulation assay with 1:1 unfractionated bone marrow from Sf3b1+/+, Sf3b1+/K700E, Tet2−/−, or Sf3b1+/K700E Tet2−/− CD45.2+ mice and CD45.1+ B6.SJL mice transplanted into lethally irradiated B6.SJL recipients (n = 5 mice per group). CD45.2 chimerism of the input bone marrow was measured at time 0 (n = 1 sample per group). Data presented as mean ± SEM. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. See also Figure S5.
Analysis of mature white blood cells at 45 weeks post-pIpC was notable for an increased percentage of granulocytes associated with homozygous Tet2 loss. A statistically significant increase in the percentage of granulocytes was noted in the peripheral blood of Tet2−/− mice (Figure S5A). Compared to Sf3b1+/+ and Sf3b1+/K700E mice, both Tet2 −/− and Sf3b1+/K700E Tet2−/− mice had a statistically significant increase in the percentage of granulocytes and a statistically significant decrease in the percentage of B cells in both the spleen (Figure S5B) and the bone marrow (Figure S5C).
At 12 weeks, we found a small but reproducible increase in the frequency of LT-HSCs in Sf3b1+/K700E Tet2−/− mice compared to Sf3b1+/+ mice (Figure 5E). By 45 weeks this expansion of LT-HSCs in Sf3b1+/K700E Tet2−/− mice was more marked when compared to the increase observed in Sf3b1+/K700E (p = 0.028) or Tet2−/− (p = 0.00768) mice (Figure 5F). The spleens of Sf3b1+/K700E Tet2−/− mice were enlarged, similar to Tet2−/− mice (Figure S5D). Consistent with an additive effect of these mutations on MDS pathogenesis, histopathological analysis of the spleens of the double mutant animals was notable for dysplastic erythroid progenitors and megakaryocytes (Figure 5G). We did not observe a statistically significant decrease in survival in the double mutant animals (Figure S5E) and none of the animals developed leukemia during the course of these experiments.
We next compared the function of double mutant to single mutant stem cells in competitive repopulation experiments. Four weeks post-pIpC, a one-to-one ratio of Sf3b1+/+, Sf3b1+/K700E, Tet2−/− or Sf3b1+/K700E Tet2−/− to congenic B6.SJL unfractionated bone marrow was transplanted into lethally irradiated B6.SJL recipients. In contrast to the Sf3b1+/K700E bone marrow recipients, the initial chimerism of Sf3b1+/K700E Tet2−/− recipients was similar to Sf3b1+/+ recipients (Figure 5H). Additionally, the peripheral blood chimerism in Sf3b1+/K700E Tet2−/− recipients increased over time with a delayed onset, but similar slope, compared to Tet2−/− recipients. Analysis of mature lineages was notable for a multilineage competitive advantage in Tet2−/− recipients, as reported previously (Quivoron et al., 2011); however, Sf3b1+/K700E Tet2−/− recipients showed myeloid skewing that was most notable in the peripheral blood and spleen (Figure S5F–S5H). Flow cytometric analysis of the stem and progenitor cells 24 weeks post-transplantation demonstrated increasing donor chimerism in Sf3b1+/K700E Tet2−/− recipient mice as their bone marrow cells matured from LT-HSCs (46.5% CD45.2+ cells) to more committed myeloid progenitors (80 – 90% CD45.2+ cells; Figures S5I and S5J). This is in contrast to Tet2−/− recipients that had nearly 100% donor chimerism in all stem and progenitor cell compartments by 24 weeks and Sf3b1+/K700E bone marrow recipients that had low levels of donor chimerism in all stem and progenitor cell compartments.
Taken together, these data demonstrate that Tet2 loss exacerbates the macrocytic anemia and impaired terminal erythroid maturation caused by SF3B1K700E expression and causes an earlier expansion of LT-HSCs in the double mutant bone marrow. Furthermore, loss of Tet2 rescues the impaired competitive repopulating activity conferred by Sf3b1+/K700E. These phenotypes of the Sf3b1+/K700E Tet2−/− mice faithfully recapitulate cardinal features of MDS (Beachy and Aplan, 2010).
Cells expressing SF3B1K700E have an increased sensitivity to pharmacologic spliceosome modulation
Screens to identify naturally occurring products with anti-tumor activity led to the identification of microbial products that specifically inhibit SF3b complex function (Kaida et al., 2007; Kotake et al., 2007; Mizui et al., 2004; Nakajima et al., 1996a; Nakajima et al., 1996b). E7107 is a derivative of naturally occurring pladenolides that has been shown to inhibit spliceosome assembly (Eskens et al., 2013; Folco et al., 2011; Hong et al., 2014). As splicing factor mutations are uniformly heterozygous and mutually exclusive (Yoshida et al., 2011), we hypothesized that survival of Sf3b1-mutant cells may be dependent on the activity of the residual wild-type Sf3b1 allele and that cells heterozygous for Sf3b1 mutations may therefore have increased sensitivity to spliceosome modulators.
We tested this hypothesis in vitro using c-Kit+ HSPCs isolated from the bone marrow of Sf3b1+/+ or Sf3b1+/K700E mice. We treated the cells with E7107 for 72 hr and observed that although Sf3b1+/+ murine HSPCs are sensitive to nanomolar concentrations of E7107 (IC50 = 1.249 nM), Sf3b1+/K700E HSPCs are sensitive to even lower concentrations of the drug (IC50 = 0.619 nM, Figure 6A).
Figure 6. SF3B1K700E expressing cells are sensitive to the spliceosome modulator, E7107, in vitro and in vivo.

(A) 2.0 × 104 c-Kit+ bone marrow HSPCs were isolated from Sf3b1+/+ and Sf3b1+/K700E mice and cultured in DMSO or increasing concentrations of E7107. Cell viability was assessed using CellTiter-Glo® (n = 2 mice per group; triplicate samples per dose). Data are representative of at least 5 independent experiments with cells harvested 4 to 24 weeks post-pIpC. (B) Schematic of in vivo E7107 treatment. (C) Change in the percentage of CD45.2 donor chimerism in the peripheral blood of B6.SJL recipient mice transplanted with a 20:1 ratio of Sf3b1+/+ or Sf3b1+/K700E to B6.SJL unfractionated bone marrow 4 weeks after competitive transplantation (day 0) and 12 days after treatment with E7107 or vehicle (n = 5 mice per group). (D–F) Unfractionated bone marrow (D), LK myeloid progenitor (E), and LSK (F) CD45.2 donor chimerism in Sf3b1+/+ or Sf3b1+/K700E recipients treated with E7107 or vehicle as indicated (n = 5 mice per group). Data presented as mean ± SEM. * p < 0.05. See also Figure S6.
To evaluate the activity of E7107 against Sf3b1+/K700E HSPCs in vivo, we treated competitive transplant recipients with E7107 for a total of 10 days (Figures 6B). Lethally irradiated B6.SJL mice were transplanted with a 1:20 ratio of B6.SJL to either Sf3b1+/+ or Sf3b1+/K700E unfractionated bone marrow. We assessed initial engraftment 4 weeks post-transplant and found the peripheral blood CD45.2 chimerism was above 50% in both recipient groups, although lower in the Sf3b1+/K700E recipients (Figure 6C). We observed a statistically significant decrease in CD45.2 chimerism in the peripheral blood, bone marrow, and spleens of Sf3b1+/K700E recipients treated with E7107 compared to those treated with vehicle alone (Figures 6C, 6D, S6A). A similar decrease in chimerism was not seen in Sf3b1+/+ recipients treated with E7107. Treatment of Sf3b1+/K700E recipients with E7107 was also associated with a statistically significant decrease in lymphoid and myeloid CD45.2 chimerism, LK chimerism, and a lower, although not statistically significant decrease in LSK chimerism (Figures S6B, 6E, 6F). Taken together, these data show that HSPCs expressing mutant SF3B1 have an increased sensitivity to pharmacologic spliceosome modulation compared to wild-type HSPCs.
Discussion
We demonstrate that heterozygous, hematopoietic-restricted expression of SF3B1K700E is sufficient to cause characteristic features of MDS, including a macrocytic anemia due to a block in terminal erythropoiesis, erythroid dysplasia and expansion of LT-HSCs in the bone marrow. The finding that SF3B1 mutations in patients with MDS are uniformly heterozygous missense mutations at highly restricted amino acid residues, rather than a range of inactivating mutations, suggests that these mutations confer an alteration of function instead of a loss of protein function. RNA sequencing of Sf3b1+/K700E myeloid progenitor cells and SF3B1-mutant bone marrow mononuclear cells from MDS patients showed that mutant SF3B1 expression is most commonly associated with aberrant 3’ mRNA splicing. The motif associated with the cryptic 3’ ss is conserved between SF3B1-mutant human and murine samples and is characterized by an enrichment of adenosines and a short polypyrimidine tract upstream of the cryptic 3’ ss. This splicing abnormality has also been reported in SF3B1-mutant CLL samples and solid tumors including breast carcinoma and melanoma (Alsafadi et al., 2016; Darman et al., 2015).
The hematopoietic phenotype of the Sf3b1+/K700E conditional knock-in mouse is distinct from that reported in studies of heterozygous deletion of Sf3b1 (Matsunawa et al., 2014; Visconte et al., 2012b; Visconte et al., 2014; Wang et al., 2014). Haploinsufficiency of Sf3b1 is not associated with anemia, a block in terminal erythroid maturation or statistically significant differences in HSPC frequencies in mice followed for up to 70 weeks (Matsunawa et al., 2014; Wang et al., 2014). We did not observe substantial numbers of ring sideroblasts in the bone marrow of Sf3b1+/K700E animals, as seen in MDS patients with SF3B1 mutations. Murine models of other genetic mutations that lead to sideroblastic anemia in humans also do not produce ring sideroblasts in mice, including loss of Abcb7 (Friedman et al., 2004; Keyhani et al., 1974b; Pondarre et al., 2007).
We found that over 30% of both the murine and human genes with aberrant splicing associated with SF3B1 mutations were predicted to undergo NMD. Genes predicted to undergo NMD had decreased expression compared to genes that were not predicted to undergo NMD. Of note, the splicing abnormality in SF3B1-mutant cancers is distinct from the aberrant splicing associated with mutations in Srsf2 (Kim et al., 2015) and U2AF1 (Shirai et al., 2015). This may contribute to the mutual exclusivity as well as the MDS subtype specificity of splicing factor mutations in MDS.
As SF3B1 and TET2 mutations co-occur in patients with MDS (Bejar et al., 2012; Haferlach et al., 2014), we generated double mutant mice to evaluate the effect of the combination of these mutations. We found that Sf3b1+/K700E Tet2−/− mice develop an earlier and more pronounced anemia, a more profound and accelerated expansion of the LT-HSC compartment, and erythroid and megakaryocyte dysplasia. We also found that the combination of Tet2 loss and SF3B1K700E expression is sufficient to rescue the competitive repopulation disadvantage induced by expression of SF3B1K700E alone. The additive effects of Tet2 loss and SF3B1K700E expression on the HSPCs from the double mutant animals may explain why mutations in both of these genes are well tolerated in MDS patient samples.
Finally, we demonstrated that HSPCs expressing SF3B1K700E have an increased sensitivity to the spliceosome modulator, E7107, both in vitro and in vivo. Sf3b1 mutation therefore sensitizes cells to pharmacologic targeting of wild-type SF3B1, consistent with the observation that the growth of SF3B1-mutant endometrial cancer and uveal melanoma cell lines is impaired by deletion of wild-type, but not mutant, SF3B1 (Zhou et al., 2015). These findings suggest that there may be a therapeutic window for the use of spliceosome modulators in the treatment of hematologic malignancies with SF3B1 mutations.
Experimental Procedures
Generation of a Sf3b1K700E Conditional Knock-In Mouse
Generation of the Sf3b1K700E conditional knock-in mouse is described in the Supplementary Experimental Procedures. All experiments and procedures were conducted in the Children’s Hospital Boston animal facility and were approved by the Children’s Hospital Institutional Animal Care and Use Committee.
Patient Samples and Sequencing
Patients included in the RNA sequencing were diagnosed between 1994 and 2008. Patients were diagnosed with MDS according to the French-American-British and 2008 World Health Organization classifications. Samples were de-identified at the time of inclusion. This study was approved by the Columbia University Institutional Review Board and performed in accordance with the Declaration of Helsinki. All patients gave their informed written consent. RNA sequencing was performed using paired-end reads generated from cDNA libraries prepared from MDS samples (bone marrow mononuclear cells).
RNA Sequencing and Analysis
For sorted mouse cell populations and MDS samples, RNA was extracted using PrepEase RNA Spin Kit (Ammymetrix, Santa Clara, CA). For mouse RNAs, nondirectional libraries were prepared using the NEBNext® Ultra™ RNA (poly A) Library Preparation Kit for Illumina® (New England Biolabs, Ipswitch, MA). 300-bp DNA fragments were isolated and sequenced on the NextSeq PE150 chip. For MDS samples, cDNA library preparation, sequencing, and raw read filtering methods were executed at BGI as described previously (Ren et al., 2012). Quantification of RNAseq data, identification of differentially spliced junctions, and motif analysis were conducted as described previously (Darman et al., 2015).
E7107 Drug Treatment
Increasing concentrations of E7107 (H3 Biomedicine, Cambridge, Massachusetts) were added to 20,000 c-Kit+ cells per well cultured in 96-well plates. Cells were cultured in StemSpan™ Serum-Free Expansion Medium (Stem Cell Technology, Vancouver, Canada) supplemented with 50 ng/mL m-tpo and 50 ng/mL m-scf). In vivo drug treatment was performed via daily intravenous tail vein injection of 4 mg/kg/dose E7107 or vehicle (10% ethanol, 5% Tween-80, and saline) for 10 days (5 days on, 2 days off, then repeat). Treated mice were lethally irradiated (10.5 Gy) and transplanted with 20:1 ratio of Sf3b1+/+ or Sf3b1+/K700E BL6 unfractionated bone marrow to B6.SJL unfractionated bone marrow. Four weeks post-transplant, peripheral blood CD45.2 chimerism was assessed. Four hr after the last drug treatment, peripheral blood cells, splenocytes, and bone marrow cells were isolated and the donor cell chimerism was again assessed.
Statistical Analyses
All data are reported as mean ± SEM. We performed analysis for statistically significant differences with the 2-tailed Student t-test and One Way ANOVA. Box plots and whiskers are presented as per the Tukey method where the center is the median, and the first and third quartiles define the top and bottom parts of the box. Interquartile range (IQR) is third quartile - first quartile (the box). Outliers are defined as 1.5xIQR outside the first and third quartiles. All flow cytometry data were analyzed with FlowJo Software.
Supplementary Material
Significance.
Somatic point mutations in components of the pre-mRNA spliceosome are the most common mutations in myelodysplastic syndromes (MDS). Here we demonstrate that mutation of the most commonly mutated splicing factor in MDS, Sf3b1, causes impaired red blood cell differentiation leading to anemia, an increase in long-term hematopoietic stem cells in the bone marrow, and myelodysplasia. We also show that mutant Sf3b1 causes increased aberrant 3’ splice site selection leading to increased nonsense-mediated decay. Sf3b1 mutations cooperate with Tet2 loss to cause an accelerated MDS phenotype. Cells expressing mutant SF3B1 have increased sensitivity to the spliceosome modulator, E7107. These data show how mutant SF3B1 affects hematopoiesis and mRNA splicing and identify spliceosome inhibitors as a potential new therapy for MDS.
Highlights.
Sf3b1+/K700E mice develop a progressive macrocytic anemia and myelodysplasia.
Mutant SF3B1 causes aberrant 3’ splice site selection.
Sf3b1+/K700E and Tet2 loss cause an earlier onset of MDS characteristics.
Spliceosome modulators selectively kill cells expressing SF3B1K700E.
Acknowledgments
We gratefully acknowledge Ronald Mathieu and Mahnaz Paktinat from the Harvard Stem Cell Institute/Boston Children’s Hospital Flow Cytometry Core Facility, and Renee Rubio and Yaoyu Wang from the Center for Cancer Computational Biology at the Dana-Farber Cancer Institute. We thank Edwin Chen, Julie Losman, Takahiro Maeda, Mohandas Narla, Rafael Bejar, Stanley Lee, Andrew Guirguis, and Damien Wilpitz for their scientific insights and critical review of the manuscript. This work was supported by the NIH (R01 HL082945), the Leukemia and Lymphoma Society, and a STARR Cancer Consortium award to B.L.E. and the NIH R24 DK099808 award to M.D.F. and B.L.E. E.A.O. was supported by an American Society of Hematology/Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Award.
Footnotes
Accession Numbers
The RNA sequencing datasets have been deposited to the NCBI Gene Expression Omnibus (GEO) and are available under accession number GEO:GSE72790.
Author Contributions
E.A.O, A.M., S.B., P.J.S., M.D.F., and B.L.E. designed the study. A.M. and C.J.W. helped generate mice. E.A.O., R.C., D.R.C., A.M.L, A.M., and M.E.C. performed experiments and generated mice. E.A.O. and B.L.E. analyzed and interpreted the data. L.Y. and M.S. performed the RNA sequencing analysis. A.M.A. and A. R. provided primary patient MDS samples. S.B. and P.G.S. provided reagents and expertise for the spliceosome modulator experiments. C.J.W., L.W. and R.G.G. provided reagents and expertise. E.A.O., M.D.F and B.L.E. prepared the manuscript. All authors provided critical review of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A) Human molecular genetics. 1999;8:743–749. doi: 10.1093/hmg/8.5.743. [DOI] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nature communications. 2016;7:10615. doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachy SH, Aplan PD. Mouse models of myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010;24:361–375. doi: 10.1016/j.hoc.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra S, Andres-Leon E, Prieto-Sanchez S, Hernandez-Munain C, Sune C. Prp40 and early events in splice site definition. Wiley interdisciplinary reviews RNA. 2015 doi: 10.1002/wrna.1312. [DOI] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. The New England journal of medicine. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, Raza A, Kantarjian H, Levine RL, Neuberg D, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:3376–3382. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boultwood J, Pellagatti A, Nikpour M, Pushkaran B, Fidler C, Cattan H, Littlewood TJ, Malcovati L, Della Porta MG, Jadersten M, et al. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PloS one. 2008;3:e1970. doi: 10.1371/journal.pone.0001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright GE, Deiss A. Sideroblasts, siderocytes, and sideroblastic anemia. The New England journal of medicine. 1975;292:185–193. doi: 10.1056/NEJM197501232920405. [DOI] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3? Splice Site Selection through Use of a Different Branch Point. Cell reports. 2015;13:1033–1045. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers. PLoS computational biology. 2015;11:e1004105. doi: 10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskens FA, Ramos FJ, Burger H, O'Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, Tabernero J. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- Folco EG, Coil KE, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes & development. 2011;25:440–444. doi: 10.1101/gad.2009411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JS, Lopez MF, Fleming MD, Rivera A, Martin FM, Welsh ML, Boyd A, Doctrow SR, Burakoff SJ. SOD2-deficiency anemia: protein oxidation and altered protein expression reveal targets of damage, stress response, and antioxidant responsiveness. Blood. 2004;104:2565–2573. doi: 10.1182/blood-2003-11-3858. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G. Myelodysplastic syndromes: 2012 update on diagnosis, risk-stratification, and management. American journal of hematology. 2012;87:692–701. doi: 10.1002/ajh.23264. [DOI] [PubMed] [Google Scholar]
- Gozani O, Feld R, Reed R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes & development. 1996;10:233–243. doi: 10.1101/gad.10.2.233. [DOI] [PubMed] [Google Scholar]
- Gozani O, Potashkin J, Reed R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Molecular and cellular biology. 1998;18:4752–4760. doi: 10.1128/mcb.18.8.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O'Brien J, LoRusso P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investigational new drugs. 2014;32:436–444. doi: 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Maeda M, Pasham M, Aguet F, Tacheva-Grigorova SK, Masuda T, Yi H, Lee SU, Xu J, Teruya-Feldstein J, et al. Role of the clathrin adaptor PICALM in normal hematopoiesis and polycythemia vera pathophysiology. Haematologica. 2014 doi: 10.3324/haematol.2014.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaisser F. Inducible gene expression and gene modification in transgenic mice. J Am Soc Nephrol. 2000;11(Suppl 16):S95–S100. [PubMed] [Google Scholar]
- Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nature chemical biology. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- Keyhani M, Giuliani D, Giuliani ER, Morse BS. Erythropoiesis in pyridoxine deficient mice. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine. 1974a;146:114–119. doi: 10.3181/00379727-146-38054. [DOI] [PubMed] [Google Scholar]
- Keyhani M, Giuliani D, Giuliani ER, Morse BS. Erythropoiesis in pyridoxine deficient mice. Proc Soc Exp Biol Med. 1974b;146:114–119. doi: 10.3181/00379727-146-38054. [DOI] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nature chemical biology. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annual review of biochemistry. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nature genetics. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang J, Ginzburg Y, Li H, Xue F, De Franceschi L, Chasis JA, Mohandas N, An X. Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood. 2013;121:e43–49. doi: 10.1182/blood-2012-09-456079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118:6239–6246. doi: 10.1182/blood-2011-09-377275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunawa M, Yamamoto R, Sanada M, Sato-Otsubo A, Shiozawa Y, Yoshida K, Otsu M, Shiraishi Y, Miyano S, Isono K, et al. Haploinsufficiency of Sf3b1 leads to compromised stem cell function but not to myelodysplasia. Leukemia. 2014;28:1844–1850. doi: 10.1038/leu.2014.73. [DOI] [PubMed] [Google Scholar]
- Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. The Journal of antibiotics. 2004;57:188–196. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufti GJ, Bennett JM, Goasguen J, Bain BJ, Baumann I, Brunning R, Cazzola M, Fenaux P, Germing U, Hellstrom-Lindberg E, et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica. 2008;93:1712–1717. doi: 10.3324/haematol.13405. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. The Journal of antibiotics. 1996a;49:1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. The Journal of antibiotics. 1996b;49:1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. The New England journal of medicine. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellagatti A, Cazzola M, Giagounidis AA, Malcovati L, Porta MG, Killick S, Campbell LJ, Wang L, Langford CF, Fidler C, et al. Gene expression profiles of CD34+ cells in myelodysplastic syndromes: involvement of interferon-stimulated genes and correlation to FAB subtype and karyotype. Blood. 2006;108:337–345. doi: 10.1182/blood-2005-12-4769. [DOI] [PubMed] [Google Scholar]
- Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood. 2007;109:3567–3569. doi: 10.1182/blood-2006-04-015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X, Cui Z, Zhang J, Yi K, Xu W, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell research. 2012;22:806–821. doi: 10.1038/cr.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SW, Gilbert W. The evolution of spliceosomal introns: patterns, puzzles and progress. Nature reviews Genetics. 2006;7:211–221. doi: 10.1038/nrg1807. [DOI] [PubMed] [Google Scholar]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer cell. 2015;27:631–643. doi: 10.1016/j.ccell.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, O'Keefe C, Rogers HJ, Sekeres MA, Maciejewski JP, Tiu RV. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012a;26:542–545. doi: 10.1038/leu.2011.232. [DOI] [PubMed] [Google Scholar]
- Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska A, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012b;120:3173–3186. doi: 10.1182/blood-2012-05-430876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Tabarroki A, Zhang L, Parker Y, Hasrouni E, Mahfouz R, Isono K, Koseki H, Sekeres MA, Saunthararajah Y, et al. Splicing factor 3b subunit 1 (Sf3b1) haploinsufficient mice display features of low risk Myelodysplastic syndromes with ring sideroblasts. Journal of hematology & oncology. 2014;7:89. doi: 10.1186/s13045-014-0089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Sashida G, Saraya A, Ishiga R, Koide S, Oshima M, Isono K, Koseki H, Iwama A. Depletion of Sf3b1 impairs proliferative capacity of hematopoietic stem cells but is not sufficient to induce myelodysplasia. Blood. 2014;123:3336–3343. doi: 10.1182/blood-2013-12-544544. [DOI] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. The New England journal of medicine. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Zhang J, Socolovsky M, Gross AW, Lodish HF. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102:3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Derti A, Ruddy D, Rakiec D, Kao I, Lira M, Gibaja V, Chan H, Yang Y, Min J, et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer research. 2015;75:1949–1958. doi: 10.1158/0008-5472.CAN-14-2930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.