

Abstract
Transcription factors (TFs) have to find their binding sites, which are distributed throughout the genome. Facilitated diffusion is currently the most widely accepted model for this search process. Based on this model the TF alternates between one‐dimensional sliding along the DNA, and three‐dimensional bulk diffusion. In this view, the non‐specific associations between the proteins and the DNA play a major role in the search dynamics. However, little is known about how the DNA properties around the motif contribute to the search. Accumulating evidence showing that TF binding sites are embedded within a unique environment, specific to each TF, leads to the hypothesis that the search process is facilitated by favorable DNA features that help to improve the search efficiency. Here, we review the field and present the hypothesis that TF‐DNA recognition is dictated not only by the motif, but is also influenced by the environment in which the motif resides.
Keywords: facilitated diffusion model, motif environment, motif finding, protein‐DNA recognition, transcription factors, transcription factor search
Abbreviations
- TF
transcription factor
- 1D
one dimensional
- 3D
three dimensional
Introduction
Transcriptional regulation is dependent on the binding of transcription factors (TFs) to specific DNA target sites in the genome, and therefore understanding the determinants that control when and where a TF will bind in the genome is of high importance. Over the years, extensive effort has been devoted to characterizing the preferred binding motif of hundreds of TFs 1, 2, 3, 4, 5, 6, however, these motifs are not sufficient to fully explain TF binding across the genome (Fig. 1A). Recent studies have demonstrated that TF binding sites are characterized not only by the presence of a preferred motif, but also by unique environmental features extending beyond the consensus motif 7, 8, 9. Here we hypothesize that these features that characterize the motif environment can affect the dynamics of the TF search process, making it more efficient. While over the years extensive knowledge has been gained on TF‐DNA recognition (as reviewed in 10, 11), the question of how TFs locate their cognate binding sites (typically spanning over 6–12 nucleotides) scattered over millions to billions of base pairs has remained an enigma. More than two decades ago, von Hippel and co‐workers have proposed the facilitated diffusion model 12, 13. In this model, a TF undergoes random three‐dimensional (3D) diffusion until it collides with the DNA at a random site, most likely forming non‐specific interactions. The TF then undergoes one‐dimensional (1D) diffusion, which involves local sliding and hopping along the non‐specific DNA region surrounding the random collision site. The 1D search continues until the TF either locates its target site (forming specific interactions with the DNA), or dissociates from the DNA and resumes 3D diffusion. This model has now been supported by many in vitro 14, 15, 16, 17, 18, 19 and in vivo studies 20, 21. In this paradigm, the non‐specific interactions between the TF and the DNA at the 1D diffusion stage is expected to play an important role in facilitating the TF to its cognate binding site. However, studies examining the effects of the divergence of DNA sequence and shape in this process are lacking. We propose that the motif environments that possess favorable features, specific to each TF, may help to narrow down the TF search space, and help to attract the TF to its functional site, thus providing a more efficient search process. This could be achieved through different mechanisms, for example by allowing for longer 1D diffusion, thereby increasing the time spent by the TF in regions where functional binding sites exist, or by attracting the protein to relatively proximal regions of the binding site, increasing the thermodynamic probability of binding to a functional site (Fig. 1B). In the following sections we review recent studies providing evidence for the contribution of the motif environment to TF binding and present our hypothesis for the contribution of regions far beyond the core motif and its immediate flanks to the TF search dynamics.
Figure 1.
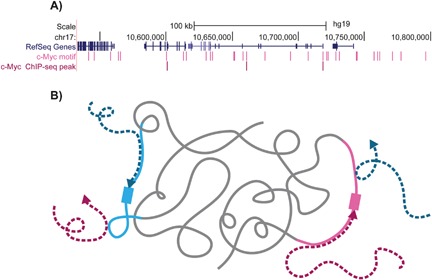
An illustration representing the contribution of the motif environment to TF binding. A: TF motifs appear many times in the genome, however, only a very small fraction of the available motifs are bound by the TF. An example for this is shown for a ∼300 kb region in chromosome 17, where only a small fraction of regions containing the known c‐Myc motif (light pink) are bound by c‐Myc (shown by c‐Myc ChIP‐seq peaks in dark pink). B: How can TFs locate their relatively short binding sites, which constitute only a minuscule fraction of the genome? We hypothesize that the favorable motif environment (light pink and light blue), which is specific to each TF, can help to narrow down the TF search space (dark pink and navy blue dashed arrows), attracting the TFs to their cognate binding sites (light pink and light blue rectangulars).
Evidence for contributions of the motif surroundings to transcription factor binding
Contributions of nucleotides immediately flanking the core motif
While it is well established that TF binding requires the existence of a core consensus binding motif, more recent studies have shown that in addition to the core motif, the nucleotides adjacent to the motif can also have a profound effect on TF binding (Fig. 2, bottom) 3, 22, 23, 24, 25, 26. Quite strikingly, it was shown that the measured binding affinity of a TF to a strong motif with unfavorable flanks can be almost equivalent to the binding affinity of a TF to a weak motif 23. The nucleotides bordering the motif have also been shown to contribute to gene expression 26. For example, by varying the Pho4 binding sites in the yeast promoter of Pho5, and testing Pho5 transcription rate, Rajkumar et al. have shown that changes in the 1–2 base pairs directly flanking the Pho4 enhancer‐box (E‐box) motif can significantly affect the transcription rate. Overall, as previously proposed 23, 26, differences in the nucleotides adjoining the core motif may enable the fine‐tuned regulation of TF binding and gene expression.
Figure 2.
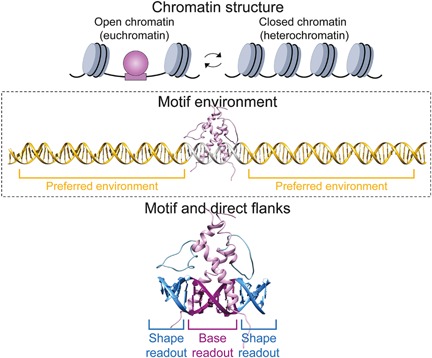
TF‐DNA recognition involves multiple levels of interactions. Top: The first level of recognition involve chromatin accessibility, where nucleosome‐depleted regions (i.e. open chromatin) are associated with TF binding while closed chromatin is often thought as inaccessible to most TFs. Middle: The unique features of the environment in which the motif is embedded can further direct the TF to its cognate binding site. Bottom: Specific TF interactions with the DNA occur through the interplay of base and shape readout at the core motif and its proximal flanks.
A possible explanation for the role of flanking sequences on TF binding involves the recognition of the intrinsic 3D structure of the DNA (i.e. DNA shape) 22. In this study, Gordân et al. examined the binding sites of two paralogous yeast TFs from the basic helix‐loop‐helix (bHLH) family, Cbf1, and Tye7. These two TFs have virtually identical DNA binding site motifs (the E‐box CACGTG), both in vitro and in vivo, yet they bind to different genomic regions within the yeast genome. Using in vitro genomic‐context Protein Binding Microarray (gcPBM) assays, it was shown that the two TFs prefer binding to sequences with significant differences in nucleotides directly flanking the E‐box motif, which explained the differences in binding specificity observed in vivo. The differences in nucleotides neighboring the core motif were shown to influence the structural properties of the DNA, presumably contributing to the recognition by the TF via DNA shape readout 27. Other protein families have also been suggested to recognize nucleotides directly flanking the core motif using DNA shape readout 23.
Contributions of regions far beyond the core motif and immediate flanks
It is well established that there is a strong interplay between the chromatin state and the binding of TFs to DNA 28, 29 (Fig. 2, top). Moreover, in addition to the importance of positions within and directly flanking the core motif (extended motif), recent studies have shown that more distal regions can also contribute to TF binding, even in regions of open chromatin 7, 8, 9 (Fig. 2, middle). However, while the motif and its direct flanks (Fig. 2, bottom) likely represent direct interactions with the TF, either via base or shape readout, preferences for the local environment surrounding the TF binding site likely represent nonspecific interactions of the protein with the DNA. By testing the binding of six TFs (three belonging to the bHLH family, and three to the E2 TF (E2F) family) using in vitro gcPBMs, to sequences containing the exact same extended motif but differing in the nucleotide composition farther away from the binding site, Afek et al. demonstrated a large variation in signal intensities 8. In a recent study, we examined the contribution of the sequence environment to TF binding for hundreds of TFs 7. We analyzed in vitro HT‐SELEX data of 239 TFs from different families, comparing the sequence composition in distal regions surrounding bound and unbound sequences, both containing the same motif. While considering limitations due to read coverage, we found that the majority of TFs show significant preferences for a specific GC composition within the extended regions 7. Differences in GC content between bound and unbound sequences were observed 10 bp up‐ and downstream from the extended motif. Importantly, direct interactions between the TF and the DNA are not plausible at these distances. As both the gcPBM 8 and HT‐SELEX 7 studies were conducted in vitro, these results indicate that TFs may have an intrinsic preference to bind within regions with specific nucleotide content.
By further analyzing ChIP‐seq data for 56 TFs 7, comparing bound and unbound sequences that contain the same motif (both in open chromatin regions), we found that the GC content in the extended regions around TF bound motifs, stretching far beyond the core motif, differ significantly from the GC content in regions flanking unbound motifs. The differences in GC content that were observed far beyond the extended motif showed high agreement between the in vivo and in vitro data. Although there are limitations for both in vivo and in vitro experiments, TFs belonging to similar protein families showed similar GC preferences at distal regions. Beyond the contribution to TF binding, the sequence environment distant from the motif was also shown to influence the expression of the gene regulated by the TF. Using massively parallel enhancer assay in living mouse retinas, White and co‐workers 9 have tested the binding of the TF Cone‐rod homeobox (Crx) to 84 bp sequences containing the known Crx motif. They have found that while sequences bound by Crx can activate transcription, other sequences with an equivalent number of Crx motifs could not. Interestingly, they have found that the GC content surrounding the Crx motifs strongly distinguishes between functional and non‐functional Crx sites, demonstrating the contribution of the motif environment to the regulatory potential of Crx. Moreover, they have found that Crx bound regions lacking a Crx motif have particularly high GC content, suggesting that the sequence features from the motif's distal regions can compensate for the lack of a Crx motif.
Overall, the evidence provided above supports the hypothesis that the motif environment has an important role in TF‐DNA recognition. This assumption was recently reinforced by novel findings, showing that many quantitative trait loci (QTLs) fall within the motif environment of TFs 30. Notably, while the nucleotide positions of the core motif show high information content representing a strong preference of the TF to a specific sequence, the distal regions demonstrate very low information content but have an overall nucleotide content preference over the entire region, as illustrated in a hypothetical example in Fig. 3. This example emphasizes that current approaches aimed at identifying preferences extending beyond the core motif − such as examining the information content at each position surrounding sequences aligned by the consensus motif − are less suitable for identifying environmental preferences of TFs. Instead, examining the environmental features such as GC content, over the entire region can allow detecting regional preferences
Figure 3.
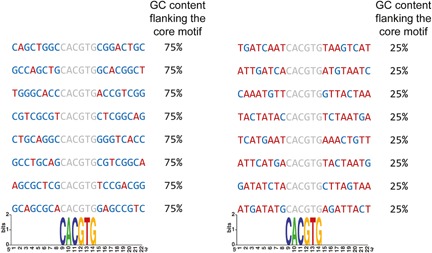
Illustration emphasizing the drawback of position specific information content based approaches to identify preferences for the motif environment. In this example the sequences are aligned by the shared E‐box motif CACGTG (marked in gray), represented by very high information content at each position. However, while the motif is surrounded by high GC content in the left (marked in blue, 75% GC content flanking the core motif in each sequence), and high AT content in the right (marked in red, 75% AT content flanking the core motif in each sequence), both groups have very low (undetectable) information content in these regions.
How can the motif environment contribute to transcription factor search mechanisms?
As discussed above, the local environment surrounding TF binding sites possesses different features, which we hypothesize, may help direct the TFs to their cognate sites. Below, we present current knowledge of the different features of the motif environment, and discuss possible mechanisms for how they might contribute to TF search dynamics.
Homotypic clusters may help in directing transcription factors to their binding sites
The presence of multiple copies of adjacent binding sites of the same TF, often referred to as homotypic cluster, has been widely observed across divergent species ranging from invertebrates 31, 32 to vertebrates 33, 34, 35: Clusters of closely spaced binding sites of the same TF (four on average) were found to be highly enriched in Escherichia coli 36. Lifanov et al. 31 reported that the developmental network in Drosophila is highly enriched in homotypic motifs. In mammals, homotypic clusters have been reported to cover a relatively large part of the genome (approximately 1.6%), with half of the promoters and enhancers containing at least one homotypic cluster 33. Furthermore, comprehensive analysis of the binding sites of diverse TFs from in vitro and in vivo data demonstrated an enrichment in homotypic clusters around the bound sites 7.
The contribution of homotypic clusters to in vitro TF binding has been recently studied by Levo et al. 23. In this study the authors have quantitatively measured the binding of two yeast TFs (Gcn4 and Gal4) to thousands of sequences, testing the effect of the number of potential binding sites on TF binding. By separating the bound sequences on a gel before measuring TF binding, they were able to separate an individual binding event from multiple binding events. Importantly, even in the case of a single binding event, increasing numbers of binding sites resulted in enhanced TF binding. These observations emphasize that the number of potential binding sites can contribute not only to multiple binding events, but can also increase the probability for a single TF to bind one target site.
Beyond the contribution of homotypic clusters to TF binding, early studies have suggested that the presence of multiple binding sites of a TF in gene promoters can affect the expression of a gene 37, 38. Recent massively parallel gene expression assays have expanded these observations 39, 40, 41. Notably, these studies demonstrated a positive correlation between the number of binding sites of a single TF and the gene expression. In agreement with the observation that regulatory regions contain on average between 3 and 5 motifs of a given TF binding site 7, 33, 36, the aforementioned studies 39, 41 found that the contribution of the number of binding sites saturates at about three to four sites. Such organization of multiple, closely spaced, binding sites is not a unique feature of TFs. Splicing factors such as NOVA 42 and PTB 43 have also been shown to preferentially bind to regions enriched in homotypic clusters. This knowledge has significantly contributed to the prediction accuracy of the binding sites of diverse splicing factors 44, 45, 46 and TFs 7.
Overall, several mechanistic explanations have been suggested for the functional role of homotypic clusters in TF binding: (i) a higher number of sites can increase the probability of binding to the target sequence 23; (ii) multiple sites can create barriers or traps that either block or direct the sliding of the TF to its functional binding site 47, 48, 49, 50; (iii) the existence of several binding sites enables cooperative binding of several proteins 47; (iv) multiple sites can facilitate competition with histones, where high concentration of TFs bound to these sites could displace histones or prevent their binding 51.
An environment possessing a high similarity to the motif may facilitate genome scanning
As aforementioned, the contribution of homotypic clusters has been widely studied. However, an intriguing question is whether homotypic clusters represent numerous independent motifs or if these multiple motifs are rather found within regions that are generally characterized by high similarity to the TF binding sites. The two possible scenarios are illustrated in Fig. 4. When examining the regions surrounding bound motifs, we noticed that the majority of analyzed TFs prefer binding to sequences that show an overall high similarity to their binding motif 7. The preference of TFs to bind to regions that possess favorable nucleotide composition was observed for the vast majority of TFs from diverse families, including the C2H2 zinc finger proteins which tend to bind GC‐rich motifs and homeodomain proteins which preferentially bind to AT‐rich motifs. Interestingly, the overall high similarity between the motif environment and the preferred motifs was not simply a consequence of enrichment of homotypic clusters around the motifs, as the same phenomenon was observed when removing all positions with significant similarity to the motif. These results reinforce the hypothesis that homotypic clusters are usually embedded within regions that show overall high similarity to the preferred TF motif, rather than isolated low‐affinity binding sites embedded in a genomic‐context environment (shown in Fig. 4 as black and cyan lines, respectively). Taken together, we propose that the established homotypic cluster model, suggesting that TF binding sites are surrounded by clusters of closely spaced motifs, is a part of a more general phenomenon where the binding site environment is characterized by an overall high similarity to the preferred TF motif.
Figure 4.
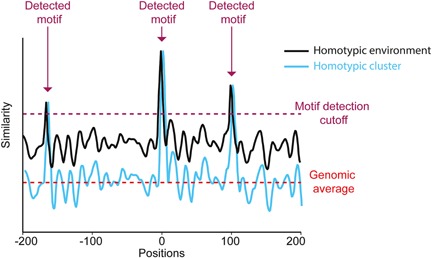
A simplified scheme demonstrating the conceptual differences between a sequence possessing homotypic clusters (cyan) and a sequence characterized by a homotypic environment (black). Plot representing motif similarity (i.e. similarity between the position weight matrix (PWM) of a TF and a PWM‐length window, where high values represent high similarity to the motif) in each position of two hypothetical sequences. In this example, both sequences possess three detectible motifs (i.e. positions with similarity scores higher than the required detection cutoff, marked by purple arrows). However, while the sequence in cyan (representing homotypic clusters) shows an overall low similarity to the motif, equivalent to the average similarity score found in the genome (marked by pink dashed line), the sequence in black (representing a homotypic environment) possesses high similarity scores across the entire sequence (higher than the genomic average).
We hypothesize that the overall similarity of the sequence environment to the motif could be related to the efficiency of the TF search process, where the favorable environment helps in guiding the TFs to their targets in the genome. It is widely believed that the TF search process is a two level process that combines a random 3D diffusion mode and a 1D diffusion mode along the DNA 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 46. In this facilitated diffusion model, the protein undergoes 3D diffusion until it collides with the DNA, most likely at a non‐specific site, and then undergoes 1D diffusion involving sliding and hopping along the DNA. The sliding process is characterized by an association and dissociation rate, where the equilibrium between the two determines the average distance scanned by the protein before dissociating from the DNA and resuming 3D diffusion. It has been suggested that the lower the dissociation rate, the longer the distance the protein can scan along the DNA 52. While scanning does not require sequence specificity, the properties of the environment surrounding a TF binding site may facilitate this process by changing the association and dissociation rate. According to the aforementioned model, the dissociation rate is expected to be lower for sequences that are more similar to the preferred binding site of the protein, leading to a longer residency of the TF in the vicinity of its binding site (Fig. 1B). In this view, while the 3D diffusion process results in random interactions with the DNA, a favorable environment could possibly allow the TF to spend more time around its cognate binding sites. In contrast, an unfavorable environment will result in a shorter 1D diffusion time, possibly reducing the time spent by the TF in regions where no functional binding sites exist. The favorable environment around binding sites could also contribute to binding via a funnel effect, where the core binding motif is flanked by weaker sites that would direct the TF to the binding site, moving from low affinity to higher affinity binding sites 8, 53.
DNA shape beyond the core binding site can influence transcription factor binding site search
It is becoming increasingly evident that the DNA structural properties of the extended motif, including its intrinsic curvature and flexibility, mainly arising from the stacking interactions between base pairs, has a strong contribution to TF binding 22, 27, 54, 55, 56, 57, 58, 59. But can the DNA shape in more distal regions from the core motif also contribute to the binding site search?
By examining the 3D properties of the DNA surrounding binding sites extracted from in vitro and in vivo data, utilizing our high‐throughput DNA shape prediction method 60, we found that diverse TFs have a high preference for specific propeller twist angles surrounding their binding sites 7. Interestingly, TFs from different families demonstrated opposite preferences. For example, TFs belonging to the C2H2 zinc finger family prefer binding to motifs surrounded by a higher, less negative, propeller twist, while TFs from the homeodomain family prefer binding to sequences with an enhanced negative propeller twist. The propeller twist, representing the intra‐base pair rotation with respect to the Watson–Crick base pairing axis, has been suggested to be a good predictor of DNA flexibility and rigidity 61, 62, 63. Specifically, regions with enhanced negative propeller twist are expected to be more rigid compared to regions with less negative propeller twist 64, 65. An intriguing conjecture is that the preference of TFs to bind in an environment that possesses unique propeller twist properties reflects the contribution of the DNA flexibility on TF binding. Previous studies have suggested that structural features of gene promoters, such as rigidity, can influence TF binding preferences 66, 67 and contribute to gene expression 68. By designing 70 different variations of the wild‐type yeast His3 promoter, Raveh‐Sadka et al. 69 have shown that long poly(dA:dT) tracts, which are expected to increase DNA rigidity, increase His3 expression. Furthermore, Tirosh and co‐workers 70 found that regions showing abundant TF binding sites are located in the proximity to a region of rigid DNA. Notably, regulatory regions such as enhancers have been shown to be more conserved at the 3D structural level compared to the sequence level 71.
These independent observations support the assumption that the DNA flexibility beyond the core motif can affect TF binding to regulatory regions of the DNA and, as a consequence, alter the expression pattern of downstream genes. A possible explanation is that DNA flexibility can alter DNA accessibility. DNA flexibility has been known to contribute to nucleosome positioning (as reviewed in 72): whereas rigid DNA can prevent histone binding, making the DNA more accessible for TF binding, flexible DNA can readily facilitate nucleosome formation, thereby making the DNA less accessible to TFs. Indeed, it was previously suggested that some TFs (such as TFs from the homeodomain family) are more frequently bound in regions depleted of nucleosomes, while other TFs (such as from the C2H2 family) prefer binding in regions enriched in nucleosomes 51. Consistently, we have shown that the regions surrounding motifs bound by TFs from the C2H2 zinc finger or homeodomain families are characterized by significantly high and low propeller twist values, respectively 7. While we caution that propeller twist is closely related to GC content due to the number of hydrogen bonds in A/T versus G/C base pairs, this observation suggests that some TFs have intrinsic properties that allow them to better compete with nucleosomes by binding to similar DNA sequences, providing a possible role for the DNA shape preferences of different TFs on the binding dynamics between histones and TFs.
Another possible explanation is that the DNA rigidity can influence the sliding and hopping of TFs across the DNA 73. While extensive studies over the last decades have greatly extended our current understanding of the different molecular determinants that control TF‐target site search, the role of the DNA sequence‐dependent structure on the search efficiency is still poorly understood. It is likely that the DNA structural features can contribute to the rate of 1D diffusion by affecting the rate of sliding on the DNA. Interestingly, it has been observed that binding sites that show lower similarity to the core motif, yet are still bound by the protein, are surrounded by favorable structural features, which presumably enable initial DNA contacts of the protein 74. This observation supports the assumption that the DNA shape is potentially scanned by the protein, before the TF forms stable protein–DNA interactions at the binding site.
Further studies
With recent advances in single‐molecule methods, the basic kinetic principles that govern TF search mechanisms for their functional binding sites are starting to unveil. However, how can the DNA sequence and shape surrounding the cognate binding site alter the search dynamics still remain elusive. TF search efficiency is thought to be dictated by multiple parameters, such as 3D diffusion time, number of binding trials to the DNA before detecting the functional site, residence times on specific and non‐specific sites, length scanned by 1D diffusion, etc. These features coordinate the efficiency of the search process. We propose that changes in the DNA sequence of the motif environment will have a substantial effect on these parameters. A favorable environment might, for example, allow for a longer residence times at non‐specific sites, perhaps increasing the length scanned by 1D diffusion, or reducing the number of trials needed before finding the functional site, while having no effect on the time spent in 3D diffusion between collisions. Different approaches for single‐molecule dynamics, such as force‐based detection and manipulation, and fluorescence imaging and spectroscopy allow to track individual protein molecules and monitor their diffusion along the DNA 19, 20, 75, 76, 77, 78. These methods could be used to dissect different aspects of the TF binding dynamics. For example, in Chen et al. 75 the authors have used single‐molecule imaging to measure Sox2 residence times on different probe lengths and found that the Sox2 residence time on the DNA was elevated as the probe length increased. An interesting approach would be to use such methods to study how differences in GC content and other DNA properties surrounding the core motif of a fixed length probe will affect the TF's residence time (Fig. 5). In such experiment, in vitro single‐molecule imaging will be used to measure the fluorescently tagged Sox2 protein dwelling time on sequences containing the Sox2 binding motif surrounded by an AT‐rich environment, compared to sequences containing the same motif but surrounded by GC‐rich environment. Sox2‐specific and ‐nonspecific residence time on DNA would be quantified and compared between the two sequences. We propose that since Sox2 binds to AT‐rich motifs 79, dwelling time on the AT‐rich sequences will increase. These in vitro experiments will help to decipher the contribution of the motif environment on different parameters that dictate the efficiency of the search process.
Figure 5.
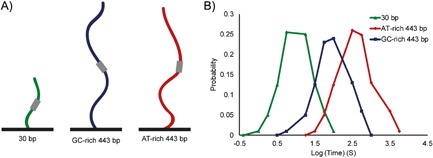
Schematic of a suggested experiment for testing the contribution of the motif environment to the dynamics of Sox2 binding. A: Different sequences will be tested: 30 bp sequence containing Sox2 known motif, GT‐rich 443 bp sequences containing the known motif, and AT‐rich 443 bp sequences containing the motif. B: We propose that Sox2 dwell times will increase for the preferred AT‐rich, long sequences.
Conclusions
Given the enormous number of potential TF binding sites in the genome, the TF search for its cognate binding site can be viewed as finding a needle in a haystack. This problem has been the subject of immense interest for decades. Evidence, accumulated over the years, supports the notion that TF binding sites are embedded within unique environments that are characterized by different features, such as high or low GC content, homotypic clusters, preferred DNA shape, etc. We have recently shown that these features allow the differentiation between TF bound motifs and similar motifs that are not bound by the protein. Moreover, we found that TFs belonging to the same protein family tend to bind to motifs surrounded by environments that share similar features, while TFs from different families can have diverged in their preferences for features of the motif environment. The unique features surrounding TF binding sites, reaching substantially beyond the motif, may add another important layer of information contributing to TF‐DNA recognition. This level of information can be considered as intermediate layer between the higher‐order recognition involving the chromatin architecture (identifying regions of open vs. closed chromatin) to the specific recognition of the binding site, involving sequence and shape readout of the motif and its proximal flanks. An intriguing possibility is that the unique and favorable environment helps to attract the TFs to their cognate binding sites, therefore narrowing down the TF's search space. In this view, the facilitated diffusion model, which has been considered a wasteful process involving mostly random interactions between the TF and the DNA, presents a much more efficient way for specific TFs to locate their binding sites in the genome. We propose that future models characterizing the search process of TFs should take into account the motif environment.
Acknowledgments
This work was supported by the Israeli Science Foundation [grant 1623/12 to Y.M.G.], the National Institutes of Health [grants R01GM106056 and U01GM103804 to R.R.], and the USC‐Technion Visiting Fellows Program [to I.D. and R.R.].
The authors have declared no conflict of interest.
References
- 1. Badis G, Berger MF, Philippakis AA, Talukder S, et al. 2009. Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berger MF, Badis G, Gehrke AR, Talukder S, et al. 2008. Variation in homeodomain DNA‐binding revealed by high‐resolution analysis of sequence preferences. Cell 133: 1266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jolma A, Yan J, Whitington T, Toivonen J, et al. 2013. DNA‐binding specificities of human transcription factors. Cell 152: 327–39. [DOI] [PubMed] [Google Scholar]
- 4. Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, et al. 2008. Analysis of homeodomain specificities allows the family‐wide prediction of preferred recognition sites. Cell 133: 1277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan J, Enge M, Whitington T, Dave K, et al. 2013. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 154: 801–13. [DOI] [PubMed] [Google Scholar]
- 6. Zhu C, Byers KJRP, McCord RP, Shi Z, et al. 2009. High‐resolution DNA‐binding specificity analysis of yeast transcription factors. Genome Res 19: 556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dror I, Golan T, Levy C, Rohs R, et al. 2015. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res 25: 1268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Afek A, Schipper JL, Horton J, Gordân R, et al. 2014. Protein − DNA binding in the absence of specific base‐pair recognition. Proc Natl Acad Sci USA 111: 17140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White MA, Myers CA, Corbo JC, Cohen BA. 2013. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis‐regulatory function of ChIP‐seq peaks. Proc Natl Acad Sci 110: 11952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levo M, Segal E. 2014. In pursuit of design principles of regulatory sequences. Nat Rev Genet 15: 453–68. [DOI] [PubMed] [Google Scholar]
- 11. Slattery M, Zhou T, Yang L, Dantas Machado AC, et al. 2014. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci 39: 381–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berg OG, Winter RB, von Hippel PH. 1981. Diffusion‐driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry (Mosc) 20: 6929–48. [DOI] [PubMed] [Google Scholar]
- 13. von Hippel PH, Berg OG. 1989. Facilitated target location in biological systems. J Biol Chem 264: 675–8. [PubMed] [Google Scholar]
- 14. Blainey PC, van Oijen AM, Banerjee A, Verdine GL, et al. 2006. A base‐excision DNA‐repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci 103: 5752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonnet I, Biebricher A, Porté P‐L, Loverdo C, et al. 2008. Sliding and jumping of single EcoRV restriction enzymes on non‐cognate DNA. Nucleic Acids Res 36: 4118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Broek B, Lomholt MA, Kalisch S‐MJ, Metzler R, et al. 2008. How DNA coiling enhances target localization by proteins. Proc Natl Acad Sci 105: 15738–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gowers DM, Wilson GG, Halford SE. 2005. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc Natl Acad Sci USA 102: 15883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leith JS, Tafvizi A, Huang F, Uspal WE, et al. 2012. Sequence‐dependent sliding kinetics of p53. Proc Natl Acad Sci 109: 16552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang YM, Austin RH, Cox EC. 2006. Single molecule measurements of repressor protein 1D diffusion on DNA. Phys Rev Lett 97: 048302. [DOI] [PubMed] [Google Scholar]
- 20. Elf J, Li G‐W, Xie XS. 2007. Probing transcription factor dynamics at the single‐molecule level in a living cell. Science 316: 1191–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammar P, Leroy P, Mahmutovic A, Marklund EG, et al. 2012. The lac repressor displays facilitated diffusion in living cells. Science 336: 1595–8. [DOI] [PubMed] [Google Scholar]
- 22. Gordân R, Shen N, Dror I, Zhou T, et al. 2013. Genomic regions flanking E‐box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep 3: 1093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levo M, Zalckvar E, Sharon E, Dantas Machado AC, et al. 2015. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res 25: 1018–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maerkl SJ, Quake SR. 2007. A systems approach to measuring the binding energy landscapes of transcription factors. Science 315: 233–7. [DOI] [PubMed] [Google Scholar]
- 25. Nutiu R, Friedman RC, Luo S, Khrebtukova I, et al. 2011. Direct measurement of DNA affinity landscapes on a high‐throughput sequencing instrument. Nat Biotechnol 29: 659–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajkumar AS, Dénervaud N, Maerkl SJ. 2013. Mapping the fine structure of a eukaryotic promoter input‐output function. Nat Genet 45: 1207–15. [DOI] [PubMed] [Google Scholar]
- 27. Rohs R, West SM, Sosinsky A, Liu P, et al. 2009. The role of DNA shape in protein‐DNA recognition. Nature 461: 1248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thurman RE, Rynes E, Humbert R, Vierstra J, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barozzi I, Simonatto M, Bonifacio S, Yang L, et al. 2014. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell 54: 844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tehranchi AK, Myrthil M, Martin T, Hie BL, et al. 2016. Pooled ChIP‐seq links variation in transcription factor binding to complex disease risk. Cell 165: 730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lifanov AP, Makeev VJ, Nazina AG, Papatsenko DA. 2003. Homotypic regulatory clusters in Drosophila. Genome Res 13: 579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Markstein M, Markstein P, Markstein V, Levine MS. 2002. Genome‐wide analysis of clustered dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc Natl Acad Sci 99: 763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gotea V, Visel A, Westlund JM, Nobrega MA. et al. 2010. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res 20: 565–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sinha S, Adler AS, Field Y, Chang HY, et al. 2008. Systematic functional characterization of cis‐regulatory motifs in human core promoters. Genome Res 18: 477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang C, Xuan Z, Otto S, Hover JR, et al. 2006. A clustering property of highly‐degenerate transcription factor binding sites in the mammalian genome. Nucleic Acids Res 34: 2238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ezer D, Zabet NR, Adryan B. 2014. Physical constraints determine the logic of bacterial promoter architectures. Nucleic Acids Res 42: 4196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Driever W, Thoma G, Nüsslein‐Volhard C. 1989. Determination of spatial domains of zygotic gene expression in the Drosophila embryo by the affinity of binding sites for the bicoid morphogen. Nature 340: 363–7. [DOI] [PubMed] [Google Scholar]
- 38. Struhl G, Struhl K, Macdonald PM. 1989. The gradient morphogen bicoid is a concentration‐dependent transcriptional activator. Cell 57: 1259–73. [DOI] [PubMed] [Google Scholar]
- 39. Sharon E, Kalma Y, Sharp A, Raveh‐Sadka T, et al. 2012. Inferring gene regulatory logic from high‐throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30: 521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharon E, van Dijk D, Kalma Y, Keren L, et al. 2014. Probing the effect of promoters on noise in gene expression using thousands of designed sequences. Genome Res 24: 1698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith RP, Taher L, Patwardhan RP, Kim MJ, et al. 2013. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet 45: 1021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ule J, Stefani G, Mele A, Ruggiu M, et al. 2006. An RNA map predicting Nova‐dependent splicing regulation. Nature 444: 580–6. [DOI] [PubMed] [Google Scholar]
- 43. Xue Y, Zhou Y, Wu T, Zhu T, et al. 2009. Genome‐wide analysis of PTB‐RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 36: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paz I, Akerman M, Dror I, Kosti I, et al. 2010. SFmap: a web server for motif analysis and prediction of splicing factor binding sites. Nucleic Acids Res 38: W281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paz I, Kosti I, Ares M, Cline M, et al. 2014. RBPmap: a web server for mapping binding sites of RNA‐binding proteins. Nucleic Acids Res 42: W361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang C, Lee K‐Y, Swanson MS, Darnell RB. 2013. Prediction of clustered RNA‐binding protein motif sites in the mammalian genome. Nucleic Acids Res 41: 6793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ezer D, Zabet NR, Adryan B. 2014. Homotypic clusters of transcription factor binding sites: a model system for understanding the physical mechanics of gene expression. Comput Struct Biotechnol J 10: 63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Afek A, Cohen H, Barber‐Zucker S, Gordân R, et al. 2015. Nonconsensus protein binding to repetitive DNA sequence elements significantly affects eukaryotic genomes. PLOS Comput Biol 11: e1004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lange M, Kochugaeva M, Kolomeisky AB. 2015. Dynamics of the protein search for targets on DNA in the presence of traps. J Phys Chem B 119: 12410–6. [DOI] [PubMed] [Google Scholar]
- 50. Shvets AA, Kolomeisky AB. 2015. Sequence heterogeneity accelerates protein search for targets on DNA. J Chem Phys 143: 245101. [DOI] [PubMed] [Google Scholar]
- 51. Charoensawan V, Janga SC, Bulyk ML, Babu MM, et al. 2012. Dna sequence preferences of transcriptional activators correlate more strongly than repressors with nucleosomes. Mol Cell 47: 183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Halford SE, Marko JF. 2004. How do site‐specific DNA‐binding proteins find their targets? Nucleic Acids Res 32: 3040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brackley CA, Cates ME, Marenduzzo D. 2012. Facilitated diffusion on mobile DNA: configurational traps and sequence heterogeneity. Phys Rev Lett 109: 168103. [DOI] [PubMed] [Google Scholar]
- 54. Abe N, Dror I, Yang L, Slattery M, et al. 2015. Deconvolving the recognition of DNA shape from sequence. Cell 161: 307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dror I, Zhou T, Mandel‐Gutfreund Y, Rohs R. 2014. Covariation between homeodomain transcription factors and the shape of their DNA binding sites. Nucleic Acids Res 42: 430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Joshi R, Passner JM, Rohs R, Jain R, et al. 2007. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell 131: 530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Slattery M, Riley T, Liu P, Abe N, et al. 2011. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147: 1270–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang L, Zhou T, Dror I, Mathelier A, et al. 2014. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res 42: D148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou T, Shen N, Yang L, Abe N, et al. 2015. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc Natl Acad Sci 112: 4654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou T, Yang L, Lu Y, Dror I, et al. 2013. DNAshape: a method for the high‐throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res 41: W56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Comoglio F, Schlumpf T, Schmid V, Rohs R, et al. 2015. High‐resolution profiling of Drosophila replication start sites reveals a dna shape and chromatin signature of metazoan origins. Cell Rep 11: 821–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hancock SP, Ghane T, Cascio D, R Rohs, et al. 2013. Control of DNA minor groove width and Fis protein binding by the purine 2‐amino group. Nucleic Acids Res 41: 6750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. el Hassan MA, Calladine CR. 1996. Propeller‐twisting of base‐pairs and the conformational mobility of dinucleotide steps in DNA. J Mol Biol 259: 95–103. [DOI] [PubMed] [Google Scholar]
- 64. Crothers DM, Shakked Z. 1999. DNA bending by adenine‐thymine tracts Oxford Handook of Nucleic Acid Structures. Oxford UK: Oxford University Press; pp. 455–70. [Google Scholar]
- 65. Nelson HCM, Finch JT, Luisi BF, Klug A. 1987. The structure of an oligo(dA) · oligo(dT) tract and its biological implications. Nature 330: 221–6. [DOI] [PubMed] [Google Scholar]
- 66. Grove A, Galeone A, Mayol L, Geiduschek EP. 1996. Localized DNA flexibility contributes to target site selection by DNA‐bending proteins. J Mol Biol 260: 120–5. [DOI] [PubMed] [Google Scholar]
- 67. Starr DB, Hoopes BC, Hawley DK. 1995. DNA bending is an important component of site‐specific recognition by the TATA binding protein. J Mol Biol 250: 434–46. [DOI] [PubMed] [Google Scholar]
- 68. Bansal M, Kumar A, Yella VR. 2014. Role of DNA sequence based structural features of promoters in transcription initiation and gene expression. Curr Opin Struct Biol 25: 77–85. [DOI] [PubMed] [Google Scholar]
- 69. Raveh‐Sadka T, Levo M, Shabi U, Shany B, et al. 2012. Manipulating nucleosome disfavoring sequences allows fine‐tune regulation of gene expression in yeast. Nat Genet 44: 743–50. [DOI] [PubMed] [Google Scholar]
- 70. Tirosh I, Berman J, Barkai N. 2007. The pattern and evolution of yeast promoter bendability. Trends Genet 23: 318–21. [DOI] [PubMed] [Google Scholar]
- 71. Parker SCJ, Hansen L, Abaan HO, Tullius TD, et al. 2009. Local DNA topography correlates with functional noncoding regions of the human genome. Science 324: 389–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Struhl K, Segal E. 2013. Determinants of nucleosome positioning. Nat Struct Mol Biol 20: 267–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mondal A, Bhattacherjee A. 2015. Searching target sites on DNA by proteins: role of DNA dynamics under confinement. Nucleic Acids Res 43: 9176–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zentner GE, Kasinathan S, Xin B, Rohs R, et al. 2015. ChEC‐seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun 6: 8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen J, Zhang Z, Li L, B‐C Chen, et al. 2014. Single‐molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156: 1274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Granéli A, Yeykal CC, Robertson RB, Greene EC. 2006. Long‐distance lateral diffusion of human Rad51 on double‐stranded DNA. Proc Natl Acad Sci 103: 1221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tafvizi A, Huang F, Leith JS, Fersht AR, et al. 2008. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys J 95: L01–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tafvizi A, Huang F, Fersht AR, Mirny LA, et al. 2011. A single‐molecule characterization of p53 search on DNA. Proc Natl Acad Sci 108: 563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML. 2015. UniPROBE, update 2015: new tools and content for the online database of protein‐binding microarray data on protein‐DNA interactions. Nucleic Acids Res 43: D117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]