

SUMMARY
Effective cancer immunotherapy requires overcoming immunosuppressive tumor microenvironments. We found that local nitric oxide (NO) production by tumor-infiltrating myeloid cells is important for adoptively transferred CD8+ cytotoxic T cells to destroy tumors. These myeloid cells are phenotypically similar to inducible nitric oxide synthase (NOS2)- and tumor necrosis factor (TNF)-producing dendritic cells (DC), or Tip-DCs. Depletion of immunosuppressive, colony stimulating factor 1 receptor (CSF-1R)-dependent arginase 1+ myeloid cells enhanced NO-dependent tumor killing. Tumor elimination via NOS2 required the CD40-CD40L pathway. We also uncovered a strong correlation between survival of colorectal cancer patients and NOS2, CD40 and TNF expression in their tumors. Our results identify a network of pro-tumor factors that can be targeted to boost cancer immunotherapies.
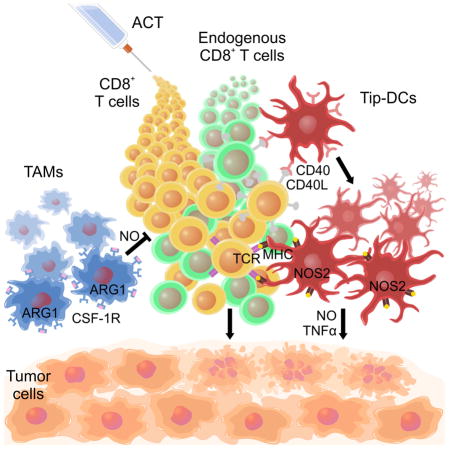
Graphical abstract
Marigo et al. show that nitric oxide produced by Tip-DCs, a subset of tumor-infiltrating myeloid cells, is important for tumor control by adoptive cell therapy (ACT). Tip-DCs require the CD40-CD40L pathway but not CSF-1R, CSF-1R blockade reduces immunosuppressive macrophages and improves tumor control by ACT.
INTRODUCTION
Immunotherapy with antibodies selected to block immune checkpoint signaling molecules such as CTLA-4, PD-1, or PD-L1 or their combinations activates tumor-specific CD8+ T lymphocytes within the tumor stroma, and in some cases drives cancer eradication (Page et al., 2014; Topalian et al., 2015). Adoptive cell therapy (ACT) with CD4+ or CD8+ T lymphocytes specific for tumor antigens is also an emerging approach to treat cancer patients (Rosenberg and Restifo, 2015). The progress to achieve the optimal efficacy following ACT is impeded by incomplete understanding of the cellular and molecular interactions in the tumor microenvironment. For example, although the avidity and affinity of T cell receptor (TCR) toward the target antigen contribute to ACT efficacy, It is much more important the affinity of the target peptide for the presenting MHC class I (MHCI) molecule (Arina and Bronte, 2015). Relatively high binding affinities for the MHCI-peptide complex (less than 10 nM) are necessary for tumor eradication following ACT (Engels et al., 2013; Robbins et al., 2013). Targeting tumor-specific neoantigens with the highest MHCI affinity might be a basic needed for optimal ACT.
Effective immunotherapy is limited in most patients by the immunosuppressive tumor environment. Local immunosuppression of T cells with anti-tumor potential is orchestrated by cells from the mononuclear phagocyte system, such as myeloid–derived suppressor cells (MDSCs) and tumor associated macrophages (TAMs) (Gabrilovich et al., 2012; Ugel et al., 2015). Understanding of how the immunosuppressive milieu develops and persists is central to harnessing the power of immunotherapeutic strategies.
Tumor-associated myeloid cells inhibit anti-tumor T cell responses by overlapping and redundant pathways. A key inhibitory pathway in the tumor microenvironment involves the metabolism of arginine through regulated expression of two enzymes: arginase 1 (ARG1, encoded by ARG1) hydrolyzes arginine while nitric oxide synthase 2 (NOS2, also known as inducible NOS or iNOS, encoded by NOS2) generates nitric oxide (NO) from arginine and oxygen (Colegio et al., 2014; Gabrilovich et al., 2012; Ginhoux et al., 2015; Marigo et al., 2010; Ugel et al., 2015). In cancer the precise roles of ARG1, NOS2 and their reaction products in promoting or controlling cancer remain controversial. For example, in human breast cancers, increased NOS2 is associated with poor clinical outcome (Glynn et al., 2010). By contrast, local low-dose gamma irradiation ‘reprograms’ macrophages to release NO, supporting Th1 responses and CD8+ T lymphocytes (Klug et al., 2013). Irradiation of human carcinomas also causes local NOS2+ macrophage accumulation and increased infiltration of CD4+ and CD8+ T cells (Klug et al., 2013). The net contribution of NOS2 to promote or contain tumors likely depends on the cell types up-regulating NOS2 and the NO concentration reached in tissue microenvironment. ARG1 is immunosuppressive in part through local depletion of arginine, restricting its availability to growing T cells in Th2 and Th1 dominated infections (Duque-Correa et al., 2014; Pesce et al., 2009). ARG1 can also compete with NOS2 for L-arginine and thus limit the overall ability of activated myeloid cells to generate NO (Bronte and Zanovello, 2005; El Kasmi et al., 2008). Collectively, the precise roles of ARG1, NOS2, and NO remain unknown.
Despite the immunosuppressive environment created by MDSCs and TAMs, some protocols of ACT do induce tumor eradication. In tumors responding to ACT, three possible outcomes have been described for tumor-infiltrating myeloid cells: elimination; change towards a more pro-inflammatory and less suppressive phenotype; or no elimination or detectable phenotypic changes (Arina and Bronte, 2015). A key step for the generation of an immune response against cancer is the tumor antigen uptake and presentation in draining lymph nodes to T cells, resulting in the priming and activation of effector CD8+ and CD4+ T cells (Chen and Mellman, 2013). Here, we investigate how the adoptive transfer of CD8+ T cells targeting a tumor-specific antigen with high MHCI affinity fuels this process within the immunosuppressive tumor microenvironment.
RESULTS
Effective adoptive CD8+ anti-tumor T cell therapy requires NOS2
To define when and where ARG1 and NOS2 are expressed in cancer we isolated cells expressing the pan-myeloid marker CD11b from spleens or tumors of EG7 lymphoma tumor-bearing wild-type (WT) mice and analyzed them for the expression of both proteins. ARG1+ and ARG1+NOS2+ cells were found mainly among CD11b+ cells isolated from the tumor compared to the spleen. Moreover, NOS2 expression was significantly higher among CD11b+ cells from the tumor (Figure 1A–C and Figure S1A). This is consistent with data from other solid tumor models (Colegio et al., 2014; Corzo et al., 2010; Haverkamp et al., 2011; Marigo et al., 2010). The lack of ARG1, NOS2, or both did not affect the accumulation of CD11b+ cells in tumors (Figure S1B).
Figure 1. NOS2 is essential for effective ACT.

(A) Representative images of CD11b+ cells sorted from spleens or tumors of EG7 tumor-bearing WT mice stained for NOS2, ARG1 and DAPI. Scale bar, 25 μm.
(B, C) CD11b+ cells sorted from spleens or tumors of EG7 tumor-bearing WT mice were quantified for the percentage of ARG1+, NOS2+, and ARG1+NOS2+ cells (B) or for fluorescence intensity of NOS2 expression (C, Median [50th percentile] represented as a line inside the box. Lines at the bottom and top of the box represent, respectively, the 25th and the 75th quartile, the whiskers represent 10th and 90th percentile. Outliers beyond the whiskers are individually plotted as black dots; *** p ≤ 0.001, unpaired Student t-test analysis). n=357 cells from spleen or from tumor were evaluated.
(D) CD11b+ cells from tumors of WT, ARG KO, NOS KO or ARG NOS KO mice were sorted and co-cultured with OVA-specific, CFSE-labeled, CD8+CD45.1+ T lymphocytes in the presence of OVA peptide. Percent of proliferating T cells within a given number of cell divisions (from 0 to 7 as indicated) is shown in bar graph format relative to positive control (mixed lymphocyte peptide culture [MLPC], lymphocytes stimulated with OVA peptide) or negative controls without OVA (NS, not stimulated).
(E, F) WT and EG7 tumor-bearing WT and NOS KO mice were adoptively transferred with OVA-specific CD8+CD45.1+ T lymphocytes. Cells derived from spleens (E) or tumors (F) were stimulated with OVA peptide and percent of CD45.1+CD8+ cells and IFN-γ+ cells shown in CD45.1+CD8+ gate. Mean ± s.d.; n=4, representative of 3 independent experiments. ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05, by using One Way ANOVA.
(G) Survival percentages of EG7 tumor bearing WT and KO mice either untreated (n=5, for each group) or treated with ACT (WT n=20; ARG KO n= 15; NOS KO n=18; ARG NOS KO n=17). *p ≤ 0.05, logrank test.
See also figure S1.
Despite intense speculation about the roles of ARG1 and NOS2 in cancer, it seems that no study that has used a genetic approach to differentiate the relative roles of them in the same setting. To measure the potential pro- and anti-tumor effects of NOS2 and ARG1, we focused on single and double gene knock out (KO) mice where whole animal Nos2 mutation was used alone or in combination with a macrophage-specific Arg1 deficiency (El Kasmi et al., 2008). We used a system where the tumor expressed a defined antigen (ovalbumin, OVA hereafter) in host backgrounds genetically manipulated for Nos2, Arg1, or both. When we transferred OVA-specific T cells into EG7 tumor-bearing mice we found that the immunosuppressive capacity of tumor-infiltrating CD11b+ cells on antigen-stimulated CD8+ T cells increased during cancer progression and this effect was almost completely ablated by the absence of NOS2 (Figure 1D). ARG1 partially cooperated with NOS2 to suppress T cell proliferation and cytotoxicity but only at later time points, when tumor sizes were larger (Figure 1D and Figure S1C).
To test the influence of host myeloid ARG1 and NOS2 on the outcomes of cellular immunotherapy, we adoptively transferred in vitro activated, congenically-marked OVA-specific CD8+ T cells into EG7 tumor-bearing mice lacking NOS2, ARG1 or both (Figure S1D). OVA is recognized with high affinity and can be used as a model that recapitulates the therapeutic anti-tumor immune response against non-self, mutated epitopes (Gubin et al., 2014; Kreiter et al., 2015). Higher percentages of OVA-specific, CD8+CD45.1+ T cells were recovered in Nos2−/− mice compared to WT mice (Figure 1E, F) consistent with the results in Figure 1D, where NOS2 is required to inhibit T cell proliferation. Transferred T cells also produced interferon (IFN)-γ in Nos2−/− mice compared to WT controls (Figure 1E, F). Despite the expansion of antigen-specific CD8+ T cells in the Nos2−/− mice compared to controls, host NOS2 ablation reduced the therapeutic effect of transferred T cells with or without concomitant ARG1 ablation (Figure 1G). Thus, myeloid expression of NOS2 inhibits T cell proliferation and IFN-γ production but is nevertheless important for the anti-tumor efficacy of these T cells.
In the same setting as above (Figure S1D), elimination of ARG1 improved the efficacy of ACT and increased survival. Given that ARG1 competes with NOS2 for arginine and loss of ARG1 causes myeloid cells to produce more NO (El Kasmi et al., 2008), the most straightforward explanation for these data is that NOS2 and NO are required for the efficacy of therapeutic CD8+ T cells while ARG1 tempers the NO-dependent response. In agreement with this interpretation, lack of ARG1 favored the expansion of intra-tumoral NOS2+ cells following transfer of tumor-specific CD8+ T cells in WT mice (Figure S1E). The 27% increase in NO-producing cells caused by ARG1 absence might have relevant consequences in the dynamic interplay for tumor cell killing, as shown later.
ACT induces the expansion of NOS2+ myeloid cells within tumors
Since the data in Figure 1 implicated local tumor NOS2+ myeloid cells as essential for ACT efficacy, we next investigated changes in the number and types of intra-tumoral myeloid cells after ACT. Following the gating strategy shown in Figure 2A, we observed that ACT induced the expansion of Ly6C+ class II MHC (MHCII+) CD11b+ cells within the tumor (Figure 2B). Moreover, inside the CD11b+NOS2+ cell fraction (comprising the whole NOS2+ cell population), which expanded after ACT, the number of Ly6C+MHCII+ cells increased whereas F4/80high mature macrophages declined in number (Figure 2C, D, S2A), suggesting that Ly6C+MHCII+ cells but not F4/80high macrophages contributed to the NOS2 activity required for cancer elimination. Importantly, in agreement with other observations (Korrer and Routes, 2014; Kratochvill et al., 2015), ARG1 expression is linked to the F4/80high mature macrophages, indicating that intra-tumoral ARG1+ cells described above are macrophages (Figure S2B). Compared to control mice, Ly6G−CD11b+ cells isolated from the tumor bearing mice adoptively transferred with tumor-specific CD8+ T cells had enhanced cytostatic activity when co-cultured with tumor cells (Figure S2C). This anti-tumor action was absent in cells isolated from Nos2−/− mice (Figure S2C) and was accompanied by increased NOS2 expression in cells isolated from WT mice (Figure S2D), suggesting that ACT-induced NOS2 activity in myeloid cells contributed to the antitumor activity. ACT also enhanced the ability of Ly6C+MHCII+CD11b+NOS2+ cells but not F4/80hi macrophages to produce TNF ex vivo, another hallmark of activated myeloid cells (Figure 2E).
Figure 2. Adoptive transfer of tumor-specific CD8+ T cells induces Tip-DC expansion.

(A) Dot plots indicating the overall gating strategy to define the myeloid sub-populations in EG7 tumor mass.
(B) Quantification of tumor-infiltrating myeloid subpopulations in the CD11b+ gate in mice treated (CD8) or untreated (w/o CD8) with ACT, n=18, pooled from 3 experiments
(C, D) Frequency (C, n=18 pooled from 3 experiments) and representative flow cytometry plots (D, percentages of different populations are indicated in each quadrant.) for tumor infiltrating myeloid populations in CD11b+NOS2+ gate (red boxes) in WT mice untreated (w/o CD8) or treated with (CD8) ACT.
(E) Frequency of different myeloid subpopulations in tumor cell suspensions stimulated or not with LPS in CD11b+ gate, n=6 representative experiment of 3.
(F) Sorted myeloid subpopulations were cultured with naive CD4+ lymphocytes to evaluate IFN-γ released in supernatants by ELISA, representative experiment of 3.
(B, C, E, F) Mean ± s.d., ***p ≤ 0.001, **p ≤ 0.01, by using One Way ANOVA.
See also figure S2.
In addition to tumor killing, we found that ACT triggered the ability of intra-tumoral Ly6C+MHCII+ cells to stimulate naive, OVA-specific CD4+ T cells from OT-II transgenic mice (Figure 2F). The TNF and NOS2 up-regulation coupled with the antigen stimulating cell capacity and the cell surface marker profile (Figure 2A–D and Figure S2E) suggested that ACT was linked to the accumulation of an anti-tumor population of myeloid cells most similar to TNF- and NOS2-producing inflammatory dendritic cells, often termed Tip-DCs (Aldridge et al., 2009; Serbina et al., 2003). We define these cells as NOS2+Ly6C+MHCII+ and refer to them as Tip-DCs hereafter.
Tip-DCs revert ACT therapeutic effect
Transcriptome characterization showed that Tip-DCs cells sorted from the tumor mass following ACT are more similar to tissue DCs (DCs) than to monocytic myeloid-derived suppressor cells (M-MDSCs), or tissue macrophages (MFs) (Figure S3 A–B and table S1 and S2). This similarity emerged when we used subsets of most variable genes across tissue MFs, tissue DCs, M-MDSCs and Tip-DCs (Figure S3A), as well as a set of previously described gene signatures specific for DCs (Miller et al., 2012), macrophages (Gautier et al., 2012) and M-MDSCs (Figure S3B) (Conde et al., 2015).
In order to demonstrate the relevance of NO production in the anti-tumor activity of Tip-DCs and to confirm their therapeutic benefit, we injected EG7 tumors grown in Nos2−/− mice with Tip-DCs sorted from WT or Nos2−/− EG7-tumor bearing mice previously treated with ACT (Figure 3A). After ACT, survival of Nos2−/− mice was increased only after intra-tumoral transfer of Tip-DCs from WT mice (Figure 3B).
Figure 3. Tip-DCs are functionally active in tumor environment.

(A) A schematic representation of the experiment.
(B) Survival of EG7-tumor-bearing NOS KO mice treated with ACT and with sorted Tip-DCs from WT or NOS KO mice; n=6. **p ≤ 0.01, Log-rank test.
(C) In vitro generated human Tip-DCs were analyzed by flow cytometry for the indicated molecules.
(D) NOG mice were injected s.c. with MDA-MB-231 and intratumorally with CD14 monocytes, Tip-DCs, or PBS and either treated or untreated i.v. with anti-hTERT specific CD8+ T cells. Survival percentages of n=15, pooled from 3 independent experiments, are reported. ***p ≤ 0.001, logrank test.
See also figure S3.
To test the ability of human Tip-DCs to improve ACT we generate these cells by incubating human CD14+ monocytes with the supernatants collected from the co-culture of CD8+ T cells specific for human (h)TERT with peripheral blood mononuclear cells (PBMCs) pulsed with the HLA-A2-restricted, hTERT peptide. Human cells consistent with the phenotype of Tip-DCs differentiated in vitro expressed high levels of MHCI, MHCII, CD86 and TNF and up-regulated NOS2, compared to monocytes from which they were derived (Figure 3C). NOG mice bearing the MDA-MB-231 mammary carcinoma were treated by ACT with hTERT–specific CD8+ T cells, in association with injection of either human monocytes or hTip-DCs (Figure S3C). Only the supply of Tip-DCs significantly improved the ACT (Figure 3D).
Tip-DC differentiation is dependent on the affinity between TCR and MHC-peptide complexes and can be in vitro generated
To further evaluate the ability of activated CD8+ T cells to induce Tip-DCs expansion and activity in response to antigen expression, we transferred the OVA-specific CD8+ T cells into B16 melanoma-bearing mice (Figure 4A). We selected two melanoma lines expressing high or low levels of OVA antigen, as assessed by the staining with a mAb recognizing the surface expression of the complex between MHCI molecule and OVA peptide and by the amount of IFN-γ released by OVA-specific T cells after co-incubation with the tumor cell lines (Figure S4A, B). We found that the in vivo expansion of NOS2+Ly6C+MHCII+ cells and the efficacy of the immunotherapy mediated by ACT were dependent on the tumor antigenicity, being evident only in B16 melanoma expressing high amounts of OVA (Figure 4A).
Figure 4. Affinity between TCR and MHC-peptide complexes regulates Tip-DC generation and ACT effectiveness.

(A) Percentage of survival (*p ≤ 0.05, logrank test) and percent of different tumor infiltrating myeloid subpopulations in CD11b+NOS2+ gate (Mean ± s.d., representative of 3 independent experiments, **p ≤ 0.01, by using One Way ANOVA) for WT mice bearing B16-OVAhigh (n=7) or B16-OVAlow (n=6) tumor treated or not with ACT.
(B) Splenic CD11b+ cells were cultured in the presence of cell culture supernatant of antigen-stimulated OT-I, TRP2high or TRP2low CD8+ T cells and anti-CD40 antibody for 48 hr then analyzed for Tip-DC differentiation. Representative dot plots are shown.
(C) Percentage of CD11b+Ly6C+MHCII+ cells generated in vitro by splenic CD11b+ cells in different culture conditions is shown.
(D) As in D showing MFI of NOS2 and TNF for CD11b+Ly6C+MHCII+ cells.
(C, D) Mean ± s.d.; n=4 representative experiment of 3, ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05, by using One Way ANOVA.
(E) B16 tumor-bearing mice were adoptively transferred with CD8+ T cells from mice expressing the TCR specific for TRP-2 antigen either with TRP-2high or TRP-2low. As negative control, anti-OVA CD8+ T cells were transferred. Survival of mice untreated or treated with ACT is reported; n=10 mice for group, 3 independent experiments. **p ≤ 0.01, logrank test.
See also figure S4.
We next evaluated whether Tip-DC generation from circulating precursors could be affected by factors produced from activated T cells. To test this, CD11b+ cells isolated from the spleens of naive mice were cultured in the presence of supernatants from in vitro stimulated CD8+ T cells, with or without the addition of CD40 agonist, to induce the generation of Tip-DCs in vitro. We used CD8+ T cells from OT-I transgenic mice or transgenic mice bearing a TCR recognizing an H-2Kb–restricted epitope of the melanoma antigen tyrosinase-related protein-2 (TRP-2180–188) with high or low affinities (Zhu et al., 2013), indicated hereafter as TRP-2high and TRP-2low respectively. Supernatant from antigen-stimulated CD8+ T cells from OT-I and TRP-2high but not TRP-2low mice induced the in vitro differentiation of Tip-DCs whereas stimulation with anti-CD3 and anti-CD28 mAbs was poorly effective (Figure 4B, C). NOS2 expression was significantly higher with supernatant from OT-I and in the presence of CD40 agonist, TNF expression was not significantly affected (Figure 4D). Similarly, human Tip-DCs induced in vitro, from CD14+ monocytes stimulated with the supernatant of the in vitro co-culture between the high affinity hTERT specific TCR-engineered T cells and HLA-A2-restricted hTERT peptide-pulsed PBMCs, expressed a significant amount of NOS2 and TNF (Figure S4C).
Collectively, degree of antigen expression, TCR avidity, and the efficient antigen recognition by the CD8+ T lymphocytes contributed to Tip-DC generation. Consistent with these results, mice bearing the B16 melanoma, naturally expressing the TRP-2180–188 epitope, showed a significant survival benefit only following transfer of CD8+ T cells with high affinity towards TRP-2 antigen (Figure 4E and Figure S4D).
Ligation of CD40 stimulates expression of NOS2 in macrophages (Weiss et al., 2010) and microglia (Jana et al., 2001) via an IFN-γ-dependent mechanism and is therefore potentially important in activating NOS2 in the tumor microenvironment. The in vitro generation of Tip-DCs was dependent on anti-CD40 stimulation (Figure 4D, E) but only partially dependent by IFN-γ whereas the expression of NOS2 and TNF was not affected by IFN-γ blockade (Figure S4E, F).
CD40-CD40L axis is required for tumor rejection mediated by CD8+ T cells
To evaluate whether CD40 engagement stimulated a prompt NO release following CD8+ T cell recognition of tumor, we stained living tissue slices of tumors grown in WT or different KO mice to assess NO following CD40 activation (Figure S5A, B). Tumor-antigen specific (OVA) CD8+ T cells from OT-I mice but not cells recognizing an irrelevant antigen (gp100) induced NO release in WT tumor slices, which was abrogated in Nos2−/− tissue (Figure 5A), confirming specificity of NO production by NOS2 and not by the non-immune NOS isoforms (NOS1, NOS3). Slices from tumors grown in either CD40 or CD40L KO mice had reduced NO release following interaction with tumor-specific CD8+ T cells from OT-1, suggesting a role of CD40/CD40L axis on tissue-infiltrating cells to sustain NOS2 activation (Figure 5A).
Figure 5. CD40-CD40L is required for ACT effectiveness.

(A) CFSE-labeled, CD8+ T cells specific for either OVA or gp100 antigens were incubated with EG7 tumor slices from either WT or different KO mice as indicated. NO was detected by confocal microscopy of slices loaded with DAR-4M AM. NO-release levels were measured as mean of fluorescence and expressed as fold induction over control (without CD8). Mean ± s.e.m.; n=12 slices pooled from 3 independent experiments, ***p ≤0.001, **p ≤0.01, by using One Way ANOVA.
(B) CFSE-labeled, polyclonal CD8+ T cells specific for OVA were derived from immunized WT or E8Icre x Cd40lgflox/flox mice and were incubated with EG7 tumor slices from either WT or CD40L KO mice, as indicated. NO was detected as in A. Error bars, mean ± s.e.m.; n=12 slices pooled from 3 independent experiments, ***p ≤ 0.001, **p ≤ 0.01, by using One Way ANOVA and the Holm–Sidak method of correction for all pairwise multiple comparison.
(C) Survival percentages of WT, CD40 KO, and CD40L KO EG7 tumor-bearing mice untreated or treated with ACT (n=10). *p ≤ 0.05 *** p ≤ 0.001, logrank test.
(D) EG7 tumor-bearing RAG-deficient mice were reconstituted with CD8+ T lymphocytes isolated from spleens and lymph nodes of WT and CD40L KO, either EG7 tumor-bearing or tumor-free, mice. After 2 days, ACT with OVA-specific CD8+CD45.1+ T lymphocytes was performed. Tumor area at days 9, and 13 following ACT are reported. Horizontal lines represent means of n=7. **p ≤ 0.01, unpaired Student t-test.
See also figure S5.
To investigate the role of CD40L expression by CD8+ T cells in the induction of the NO burst, we isolated polyclonal CD8+ T cells expanded from the spleens of WT or CD40L KO mice previously vaccinated with OVA peptide and tested their ability to induce NO release in contact with slices of tumors grown in WT or CD40L KO mice. Even though lack of CD40L in CD8+ T cells specific for OVA decreased the NO release in WT tumors, the highest reduction was observed in tumors grown in CD40L KO mice (Figure 5B), suggesting CD40L signaling is ‘upstream’ of NO release by NOS2 following ACT.
To test the specificity of the CD40/CD40L axis in tumor rejection, we monitored the survival of WT, CD40 (encoded by Tnfrsf5) KO, and CD40L (encoded by Cd40lg) KO mice bearing EG7 tumors and treated with tumor-specific CD8+ T cells. We found that lack of CD40L abrogated therapeutic activity while lack of CD40 had a partial effect (Figure 5C). To understand how CD40 was involved in the NO-dependent therapeutic activity induced by CD8+ T cells, we isolated Ly6G−CD11b+ cells from tumors grown in CD40-deficient mice. Different from WT mice (Figure S2C), ACT did not increase the cytostatic effect of Ly6G−CD11b+ cells against EG7 tumor cells (Figure S5C) and did not increase the expression of NOS2 (Figure S5D) in these cells from Tnfrsf5−/− mice. These data indicate NOS2-induced tumor killing requires activation of CD40 within intra-tumoral myeloid cells.
To identify which cells in the recipient provided the endogenous CD40L necessary for the therapeutic efficacy of ACT, we quantified Cd40lg mRNA in different cells isolated from the tumors of WT mice undergoing ACT. Apart from the transferred CD8+CD45.1+ T lymphocytes, the other cells expressing Cd40lg mRNA among tumor-infiltrating populations were the recipient, host-derived CD4+ and CD8+ T cells (Figure S5E). However, OVA-specific CD8+ T cells did not require help from CD4+ T cells to induce tumor regression, since the depletion of CD4+ T cells had no effect on ACT efficacy (Figure S5F).
To interrogate the mechanism of how CD40L expression on endogenous CD8+ T cells contributes to tumor rejection, we transferred CD8+ T cells isolated from secondary lymphoid organs of EG7 tumor-bearing WT or Cd40lg−/− mice to immunodeficient EG7 tumor-bearing RAG mice. With this strategy, we reconstituted the endogenous antitumor CD8+ T cell repertoire pre-ACT with either normal or CD40L-deficient CD8+ T lymphocytes. As an additional control, CD8+ T cells were enriched from tumor-free mice. After reconstitution, mice were adoptively transferred with OVA-specific CD8+ T cells and tumor growth was analyzed (Figure S5G). Following ACT, CD40L competent, endogenous CD8+ T cells from tumor-bearing WT mice contributed to control tumor growth more effectively than CD8+ T lymphocytes isolated from either tumor-free or Cd40lg−/− mice (Figure 5D). These data demonstrate that the CD8+ T cell repertoire activated by tumors contributes to tumor eradication following ACT by providing endogenous CD40L help.
Enhanced ACT efficacy based on supporting Tip-DC function
F4/80high mature macrophages express high amounts of ARG1 (Kratochvill et al., 2015) and are dependent on CSF-1 (Noy and Pollard, 2014). Like Tip-DCs, these cells originate from inflammatory monocytes recruited to the tumor and increase in number over time during tumor growth (Franklin et al., 2014; Kratochvill et al., 2015; Movahedi et al., 2010). Therefore, interfering with CSF-1-dependent cells could enhance NO-dependent ACT by reducing the number of ARG1+ cells. EG7 tumor-bearing WT mice were acutely treated with a single dose of anti-CSF-1R mAb and then injected with tumor-specific CD8+ T cells (Figure S6A). While ACT sustained and enhanced the decrease in F4/80high macrophages following treatment with the anti-CSF-1R mAb for 6 days, macrophage depletion did not alter the expansion of NOS2+Ly6C+MHCII+ cells, suggesting a CSF-1R-independent step for their accrual or differentiation following ACT (Figure 6A).
Figure 6. Improving ACT effectiveness by favoring intra-tumoral myeloid balance towards Tip-DC accumulation.

(A) EG7 tumor-bearing WT mice were treated with either anti-CSF-1R or Ctrl antibody, followed or not by ACT with OVA-specific CD8+CD45.1+ T lymphocytes. Percent of different myeloid subpopulations in CD11b+NOS2+ gate within the tumor mass is shown. n=12, pooled from 2 independent experiments.
(B–C) WT (B) or NOS KO (C) MCA203 tumor-bearing mice were treated weekly with either anti-CSF-1R or Ctrl antibody and injected or not with mTERT-specific CD8+ T lymphocytes. Tumor volume at indicated days is shown as mean ± s.d., (n=15).
(D) Percent of different immune subpopulations in tumors from mice treated with anti-CSF-1R or Ctrl antibody with or without ACT, (n=8).
(A, D) Error bars indicate mean ± s.d., ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05, by using One Way ANOVA.
See also figure S6.
Considering the activity of anti-CSF-1R on F4/80high mature macrophages but not on Tip-DCs, we speculated that reprogramming tumor microenvironment with anti-CSF-1R mAb could also improve the efficacy of ACT with limited reactivity towards tumor-antigens. To test this we used a model where CD8+ T cells have weak reactivity to their cognate antigen and fail to mediate anti-tumor effects in normal mice. We used mouse telomerase (mTERT)-specific CD8+ T cells, which lack anti-tumor activity against the TERT+ MCA203 fibrosarcoma when used alone (Ugel et al., 2012; Ugel et al., 2010). Repeated administrations of anti-CSF-1R mAb with mTERT-specific CD8+ T cells on WT mice induced an additive delay in tumor growth (Figure S6B and Figures 6B). The combined treatment had no effect when administered to Nos2−/− mice (Figure 6C). ACT plus anti-CSF-1R therapy caused a reduction in intra-tumoral macrophages and monocytes while the numbers of granulocytes, Tip-DCs, total CD8+ T cells and, in particular, activated CD8+ T cells were expanded (Figure 6D).
CD40L, NOS2 and TNF within the intratumoral immune network linked with outcome of colorectal cancer patients
The most complete picture of the immune reaction in human cancer has been described in colorectal cancer (CRC) (Bindea et al., 2013b). In CRC tissues, we correlated CD40LG, NOS2 and TNF expression with markers specific for immune cell subpopulations using ClueGO (Bindea et al., 2009) and CluePedia (Bindea et al., 2013a). We found an association between the intratumoral expression of these genes with markers of cytotoxic CD8+ T cells, T cells, Th1 and activated DCs (Figure 7A). B cells, Th2, Th17 and macrophages were included in the network but had weaker correlations with the expression of CD40LG, NOS2 and TNF compared to cytotoxic CD8+ T cells, T cells, Th1, and activated DCs. Other immune cell populations did not correlate with the three target genes (Figures 7A, S7A). The expression of cytotoxic CD8+ T cells, T cells, and Th1 markers was higher in tumors with high expression of CD40LG, NOS2 and TNF compared to tumors with either heterogeneous or low levels (Figure S7B). Similar results were observed for MHCII genes and genes related with antigen presenting machinery (Figure S7B). Results obtained by comparison of gene expression profiles were validated by immunohistochemistry. A significantly higher density of CD3+ T cells was observed in tumors with high CD40LG (Figure S7C) and TNF (Figure S7D), with a similar trend observed for NOS2 (Figure S7E). Tumors with high expression of CD40LG, TNF or NOS2 showed an increased density of CD8+, CD4+, T-Bet+ T cell infiltrate (Figure S7C–E). Patients with tumors having high (Hi) expression of all three markers CD40LG, NOS2 and TNF (HiHiHi) had a significant higher intratumoral density of CD3+ T cells, CD4+ T cells and T-Bet+ positive Th1 cells (Figure 7B). In addition, CD8+ T cells were significantly increased in the stroma of HiHiHi patients, whereas Th17 cells and macrophages showed no differences (Figure 7B). We investigated the effect of CD40LG, NOS2 and TNF on disease free survival (DFS, Figure 7C). Patients having a high intratumoral expression of all three markers had longer DFS compared to patients with low levels of those markers [hazard ratio=4.35, 95%CI=1.66–11.75, log rank p=0.033].
Figure 7. CD40LG, NOS2 and TNF correlate with survival in human CRC.

(A) Correlations between CD40LG, NOS2 and TNF expression (qPCR) in CRC (n=125 patients) and immunome markers visualized in a ClueGO-CluePedia network. Markers associated to specific immune cell populations share the color of the node. The lines between nodes (edges) represent Spearman’s rank correlation values and are colored in red (CD40LG, ρ >0.5), blue (NOS2, ρ >0.2) and green (TNF, ρ >0.5), respectively. Negative correlation is shown with a sinusoidal line. The lower graphs show pair-wise correlation plots corresponding to the network edges.
(B) Density of immune cells (number of positive cells per mm2 of tissue surface area) infiltrating the center of the tumor (CT) in patients with (HiHiHi), heterogeneous (Htg) or low (LoLoLo) expression of CD40LG, NOS2 and TNF. A parametric or nonparametric test was applied. Error bars, mean ± s.e.m.; n=107. The median cell count/mm2 is shown in blue, ** p ≤0.01, and * p ≤0.05.
(C) Disease free survival (DFS) for patients having high, heterogeneous, and low expression of CD40LG, NOS2 and TNF is shown.
See also figure S7.
DISCUSSION
We describe an interaction between anti-tumor CD8+ T cells and Tip-DCs causing tumor growth control dependent on NO production. ACT-based therapy requires the triggering of a ‘virtuous circle’ where CD8+ T cell recognition of tumor antigen is necessary to recruit and activate Tip-DCs which in turn present tumor antigens promoting T cell expansion, and tumor killing activity by TNF and NO production. We found NO release is dominant for the antitumor activity of ACT and restrained by ARG1 expression in F4/80hi macrophages. NO produced by NOS2 in the tumor environment can restrain T cell ability to produce IFN-γ. Although we did not investigate the source of the immune suppressive NO, it is likely that F4/80+ cells in tumor might contribute in some situations (Vicetti Miguel et al., 2010). That study also highlighted that some tissues, such as the anterior chamber of the eye, can negatively regulate the NOS2 activity induced by ACT in tumor-infiltrating F4/80+ cells by restraining NO production (Vicetti Miguel et al., 2010).
We define a myeloid population expanded after ACT as Tip-DCs; these cells have an inflammatory DC profile based on their transcriptome and immune phenotype, functional activity as antigen presenting cells, and the ability to secrete NO and TNF. The number of Tip-DCs varies between 0.01% and 0.15% in mouse tumors of different origins (data not shown) and their number increases between 3.5 and 12 fold following ACT. In all tumors, NOS2+ macrophages were either unchanged or reduced. These data suggest further studies are needed to evaluate whether clinical efficacy of TCR- or Chimeric Antigen Receptor (CAR)-transduced T lymphocytes correlates with the intra-tumoral expansion of human Tip-DCs.
Importantly, Tip-DCs did not require CSF-1R signaling for their differentiation. However, CD40 engagement on Tip-DCs by CD8+ T cells is one of the key molecular events underlying both NO production and the antitumor effect. Tumors of CRC patients with prolonged disease-free survival shared a signature comprising CD8+ T cells, activated DCs, as well as CD40LG, TNF, and NOS2 expression, suggesting anti-tumor functions of Tip-DCs and their activation and effector mechanisms might overlap with the cognate events in the mouse (Bindea et al., 2013b; Galon et al., 2006).
A basic requirement for T cells to eradicate tumors is the high affinity of the targeted peptides for MHC class I molecules (Engels et al., 2013). We found that when tumor cells express a sufficient amount of MHCI-peptide complexes, tumor-specific CD8+ T cells with sufficient TCR avidity expand and activate Tip-DCs. Tip-DCs originate from bone marrow monocytes and their main roles are the rapid and local activation of T cell response (Aldridge et al., 2009; Bosschaerts et al., 2010; Serbina et al., 2003) and the pro-inflammatory activity in psoriatic lesions of patients skin (Chong et al., 2011; Lowes et al., 2005). A key feature of Tip-DCs is their ability to prime naive T cells (Chong et al., 2011). Our data indicate that the immune functions of Tip-DCs extend beyond control of infections, are necessary for tumor growth control and might be part of a broader mechanism of tissue homeostasis, by locally coupling the adaptive immunity to cell growth control.
Mok and colleagues reported an improvement of immunotherapy by ACT using the treatment interfering with CSF-1R signaling (Mok et al., 2014). Moreover, anti-CSF-1R blocking antibody is currently being tested in pre-clinical and early phase clinical trials as a means to deplete pro-tumor macrophages (Ries et al., 2014). We demonstrate here that it is possible to improve ACT towards a poorly recognized tumor-associated antigen by titling the balance between immunosuppressive cells like macrophages and Tip-DCs, without altering Tip-DC generation from precursors. The use of anti-CSF-1R antibodies in ACT could be modified to deplete F4/80+ macrophages at the time where Tip-DCs are most active in killing tumors. Alternatively, Tip-DCs might be prepared in vitro and targeted to tumor environment to aid both locally distributed and incoming anti-tumor T lymphocytes. The antithetical relationship between in-dwelling tumor macrophages and Tip-DCs is therefore a further point of attack in designing rational immunotherapies.
CD40-CD40L interplay controls an array of pathways at the base of both initiation and progression of cellular and humoral adaptive immunity (Gommerman and Summers deLuca, 2011). Moreover, CD40 agonists induce T cell-dependent and independent tumor regression in both mice and patients with pancreatic cancer (Beatty et al., 2011). Contrary to our initial model, CD40L was not provided by tumor-infiltrating CD4+ but rather CD8+ T lymphocytes. A proportion (around 25% in average) of central and effector memory CD8+ T cells express CD40L and can display helper functions, induce maturation of monocyte-derived DCs and in vivo antigen presenting cell (APC) activation (Frentsch et al., 2013). The present study assigns an important role on expression of CD40L on endogenous CD8+ T cells. However, under some circumstances, CD40L might be provided by CD4+ T lymphocytes recognizing mutated tumor epitopes, which are frequently detected in mouse and human tumors (Gubin et al., 2014; Kreiter et al., 2015; Linnemann et al., 2015).
Clinical evidence suggests that ACT efficacy can be increased by chemotherapy-induced lymphodepletion of the host (Restifo et al., 2012), but our data open alternatives. The CD8+ T cells recognizing the self-antigen mTERT can treat established melanomas as effectively as T cells derived from Pmel-1 TCR transgenic mice, which recognize the hgp-100 melanoma antigen (Ugel et al., 2010). However, melanoma growth control following ACT with both CD8+ T cells was achieved only in mice completely lacking adaptive immunity, lymphodepleted by nonmyeloablative irradiation, or pretreated with low doses of the chemotherapeutic drug 5- flourouracil (Ugel et al., 2012; Ugel et al., 2010). In normal hosts, mTERT-specific CD8+ T lymphocytes lack therapeutic efficacy and do not induce the appearance of Tip-DCs unless coupled with CSF-1R blockade.
In conclusion, we have dissected mechanisms governing the interplay between therapeutic anti-cancer T cells and the local immunosuppressive microenvironment. Most significantly, we have elucidated distinct nodes in the interplay between activated T cells and myeloid cells that can be exogenously manipulated to enhance cancer immunotherapy.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 (WT) and congenic CD45.1 (Ly5+), OT-I mice (C57BL/6-Tg (TcraTcrb)1100Mjb/Crl) and OT-II mice (C57BL/6-Tg (TcraTcrb)425Cbn/Crl) were from Charles River Laboratories. Pmel-1 transgenic mice with a Vα1Vβ13 H-2b restricted TCR specific for murine and human melanoma peptide gp10025-33 by CD8+ T lymphocytes, were provided by N. Restifo (NIH, Bethesda, MD). Rag2−/−γc−/− (RAG), Tnfrsf5−/− mice (B6.129P2-Cd40tm1Kik/J), Cd40lg−/− mice (B6.129S2-Cd40lgtm1Imx/J) and Nos2−/− mice were from Jackson Laboratories. Tie2cre+, Arg1flox/flox, Nos2−/− Arg1flox/flox and Nos2−/−; Arg1flox/flox; Tie2cre+ mice have been previously described (Duque-Correa et al., 2014; El Kasmi et al., 2008; Pesce et al., 2009). NOG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) from Taconic; the TCR Tg mouse strains 37B7 (TCRlow mice) and 24H9 (TCRhigh mice), that bear a TCR transgene that recognizes an H-2Kb–restricted epitope of TRP-2180–188, were a gift from A. Hurwitz (NCI, NIH). Females of 8 weeks were used for all experiments. Mice were maintained in specific-pathogen-free conditions at Veneto Institute of Oncology and were randomized before the first treatment.
Animal Studies
All animal experiments were approved by our local animal ethics committee at the University of Padova and were executed in accordance with governing Italian law and EU directives and guidelines. Mice were monitored daily and euthanized when displaying excessive discomfort.
Cell lines, Synthetic peptides and Reagents
Cell lines, Synthetic peptides and reagents are detailed in the Supplemental Experimental procedures.
Cytofluorimetric analysis and sorting
Antibodies used, staining procedures and cell sorting procedures are detailed in the Supplemental Experimental procedures.
Adoptive cells therapy (ACT)
Mice were s.c. injected with 0.5×106 EG7 cells. After 7 days, mice were injected i.v. with 0.5×106 antigen stimulated T lymphocytes specific for OVA class I epitope. Tumor volume was monitored every two days with a caliper and mice were euthanized when tumors reached 200 mm2. OVA specific CD8+CD45.1+ T lymphocytes were prepared from OT-I splenocytes and plated in 24-well plates in presence of 1 μg/ml OVA257-264 (SIINFEKL) peptide. Cultures were maintained for 7 days in complete medium with 20 IU/ml IL-2. Data are presented as the percentage of survival after ACT. In MCA203 tumor-bearing mice, ACT was performed as described in Figure S6B and in Figure 6B, C, D, by using mTERT-specific CD8+ T lymphocytes, as previously described (Ugel et al., 2012).
Ex vivo antigen stimulation assays
Myeloid cells sorted from the tumor masses of EG7-tumor bearing, WT mice either treated or untreated with ACT were co-cultured with 5×104 naive CD4+ T lymphocytes isolated by immunomagnetic sorting from OT-II mice, in a final volume of 200 μl of complete DMEM in a U-bottom-96 well plates for 72hr. Supernatants from co-cultures were collected and assessed for the concentration of mouse IFN-γ.
ELISA
ELISA for mouse IFN-γ (R&D, Minneapolis, MN) was performed according to manufacturer’s instructions.
NO detection within viable tumor slices
C57BL/6, NOS2 KO, CD40L KO and CD40KO mice were inoculated s.c with 0.5 × 106 EG7-OVA cells. At day 7, tumors were dissected, embedded in 6% agarose (Invitrogen) and cut on vibratome (Leica, VT1000S) to obtain thick viable tumor slices (250 μm). Slices were loaded with the NO fluorescent probe (diaminorhodamine-4M AM; Sigma) for 1 hr at 37°C. CFSE-labeled CD8 + T cells (1.2 × 106) were added on the top of tumor slices. After 2 hr, tumor slices were fixed with 4% PFA for 30 min at RT. Nuclei were counterstained with 1 μg/ml ToPRO (Invitrogen). Slides were mounted with ProLong (Invitrogen) and analyzed by confocal microscopy (TCS SP5, Leica). For quantitative analysis of NO staining, different and noncontiguous regions of interest (50 ROIs) were randomly selected, and DAR-4M AM mean intensity was quantified for each ROI. Results were expressed as fold induction over control (no CD8+ T cells).
Human Tip-DC differentiation
Human CD14+ monocytes, immunomagnetically separated from buffy coat of HLA-A2+ healthy donors, were co-cultured for 48 hr in vitro using the supernatant of a 24 hr co-culture between specific anti h-TERT865-873 CD8+ T cells (obtained as reported in Supplemantal Experimental Procedures) and HLA-A2 restricted PBMCs pulsed with the specific human hTERT865-873 (RLVDDFLLV) peptide.
The study was approved by local Ethics Committee (AOUI of Verona, n. 1496) and the donors were enrolled after signing informed consent.
Combined ACT and human Tip-DC treatment
For in vivo testing of the therapeutic activity of hTERT-specific T cells, 1×106 MDA-MB231 cells were inoculated s.c. in the left flank of NOG mice. After 10 days, mice were inoculated i.v. with hTERT-specific CD8+ T cells (2.5×106 cells/mouse) in association with intra-tumoral injection of either monocytes or hTip-DCs (both at 1×106 cells/mouse). The treatment was repeated three times every 5 days. Tumor volume was calculated according to the following equation: V (mm3) = (d2 x D)/2, where d (mm) and D (mm) are the smallest and largest perpendicular tumor diameters, respectively, as assessed by caliper measurement.
Combined ACT and murine Tip-DC treatment
Murine Tip-DCs were sorted from tumor mass of WT or Nos2−/− EG7 tumor bearing mice after 3 days from ACT with 0.5×106 antigen stimulated T lymphocytes specific for OVA class I peptide as indicated in supplemental experimental procedures. Nos2−/− EG7 tumor bearing mice, after 7 days, were inoculated i.v. with OVA specific CD8+ T cells (0.5×106 cells/mouse) in association with intra-tumoral injection of either Tip-DCs from WT or Nos2−/− mice (both at 1×106 cells/mouse) the same day and two days after. Data are presented as the percentage of survival as described in Figure 3A, B.
Vaccination protocol to obtain polyclonal CD8+ T cells recognizing OVA
WT and CD40L KO mice were intraperitoneally injected with 100 μg of FGK45.5 antibody against CD40 (BioXcell, NH 03784-1671 USA) and then rubbed with imiquimod (Meda, Sweden) after the administration of OVA257-264 (SIINFEKL) peptide (200 μg/mouse) at the tail base. Peptide injection with repeated twice every 7 days.
Statistical analysis
Values are reported as mean ± standard deviation (s.d.). We performed statistical analysis by Student’s t-test, or by One Way ANOVA and the Holm–Sidak method of correction for all pairwise multiple comparisons. We assumed normality and equal distribution of variance between the different groups analyzed. Survival in mouse experiments are reported as Kaplan-Meyer curves and significance was determined by logrank test. Values were considered significant with p≤0.05 and are indicated as *p ≤ 0.05; **p ≤ 0.01 and ***p ≤ 0.001. All analyses were performed using SigmaPlot Software.
Colorectal cancer patients
CRC patients who underwent a primary resection of their tumor at the Laennec-HEGP Hospitals between 1996 and 2004 were reviewed and previously described (Galon et al., 2006). Ethical, Legal and Social Implications were approved by ethical review board from CPP Hotel Dieu, Ile de France. All experiments were performed according to the Helsinki guidelines. Informed consent was obtained from all subjects at the collection time.
Low density array (LDA) real-time Taqman qPCR analysis
The human tissue samples for RT qPCR were treated and processed as reported in detail in the Supplemental Experimental procedures section.
Tissue microarray (TMA) immunohistochemistry analysis
Tissue microarray (TMA) from the center (CT) and invasive margin (IM) of colorectal tumors were constructed (Galon et al., 2006). Procedures for immunohistochemistry analysis are detailed in the Supplemental Experimental procedures.
Statistical analysis for studies with human samples
All bar plots are shown as mean ± standard error mean (SEM). For pairwise comparisons of parametric and non-parametric data the Student’s t-test and Mann–Whitney–Wilcoxon rank-sum test were used, respectively. The hazard ratio (Cox proportional hazards model) and the Log rank test were used to compare disease-free and overall survival between patients in different groups. To avoid over-fitting, hazard ratios and logrank p values obtained by the “minimum p value” approach were corrected. A p value<0.05 was considered statistically significant. Analyses were performed with the statistical software R implemented as a statistical module in TME.db. Functional and correlation analyses were performed using CluePedia and ClueGO (Bindea et al., 2013a; Bindea et al., 2009) within the Cytoscape framework. The normality of the data was tested using the Shapiro-Wilk test. Correction of the hazard ratios and logrank p values were done as suggested by Hollander et al. and by Altman et al., respectively, as previously reported (Mlecnik et al., 2011).
Supplementary Material
SIGNIFICANCE.
Overcoming immunosuppressive tumor environment is fundamental for increasing the efficacy of adoptive cell therapy (ACT). We demonstrate that Tip-DCs accumulating within the tumor after ACT fuel an intra-tumoral “virtuous circle” by cross-presenting tumor antigens and activating adoptively transferred and endogenous CD8+ T-cells. Through a pathway requiring CD40-CD40L, Tip-DCs produce anti-tumor NO and TNF. Our data show that colorectal cancer patients with good survival share a tumor signature comprising the key molecules identified in the study (CD40L, TNF, and NOS2). Blocking CSF-1R signaling before ACT reduced immunosuppressive macrophage accumulation while preserving Tip-DC activation and enhancing anti-tumor activity of poorly active CD8+ T-cells. These observations provide a rational to switch the balance between pro- and anti-tumor myeloid cells in tumor microenvironment.
Highlights.
Effective anti-tumor CD8+ T cell therapy requires expansion of Tip-DCs
Tip-DC differentiation depends on the affinity between TCR and MHC-peptide complexes
CD40/CD40L axis is necessary for tumor rejection mediated by CD8+ T cells
Favoring Tip-DC function in tumor environment enhances ACT efficacy
Acknowledgments
We thank Kevin Leone for the artwork. This work was supported by grants from the Italian Ministry of Health (FINALIZZATA RF-2011-02348435 cup: E35G1400019001); Italian Ministry of Education Universities, and Research (FIRB cup: B31J11000420001 and RBAP11T3WB); EPIGEN - Italian Flagship Project Epigenomics; Italian Association for Cancer Research (AIRC, grants 6599, 12182, 14103 and Special Program Molecular Clinical Oncology 5 per mille); PRIN 2009 (2009NREAT2_003); the National Cancer Institute of France (INCa), INSERM, Qatar National Research Fund under its National Priorities Research Program award number NPRP09-1174-3-291; the Cancer research for personalized medicine (CARPEM); Paris Alliance of Cancer Research Institutes (PACRI); the LabEx Immuno-oncology; NIH grants CA189990 (PJM) and CA138064 (JEQ); by Alex’s Lemonade Stand Foundation and the Hartwell Foundation (PJM); Cancer Center Core Grant P30 CA21765, the Austrian Science Fund J3309-B19 (FK), and the American Lebanese Syrian Associated Charities. A. HR. A was supported by Cariparo Fundation Fellowship.
Footnotes
ACCESSION NUMBER
The GEO accession number for the microarray data reported in this paper is GSE74427.
Supplemental information includes Supplemental Experimental Procedures, seven figures and two tables.
AUTHOR CONTRIBUTIONS
Conceptualization, I.M. and V.B.; Methodology, P.J.M., F.K. and B.Mo.; Formal Analysis, B.M., G.B., J.G., S.B., E.M.C.M.; Investigation, I.M., S.Z., A.H.R.A., M.S.S., G.D., S.U., J.E.Q. and A.M.B.; Resources, C.H.R., V.R. and S.H.; Writing-Original Draft, I.M., G.B., E.M.C.M., P.Z., C.H.R.; Writing –Review & Editing, I.M., J.G., S.B., P.J.M. and V.B.; Supervision, J.G., S.B., P.J.M. and V.B.; Funding acquisition, V.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldridge JR, Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arina A, Bronte V. Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Curr Opin Immunol. 2015;33:120–125. doi: 10.1016/j.coi.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Galon J, Mlecnik B. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics. 2013a;29:661–663. doi: 10.1093/bioinformatics/btt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013b;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, Herin M, De Baetselier P, Beschin A. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog. 2010;6:e1001045. doi: 10.1371/journal.ppat.1001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Chong SZ, Wong KL, Lin G, Yang CM, Wong SC, Angeli V, Macary PA, Kemeny DM. Human CD8(+) T cells drive Th1 responses through the differentiation of TNF/iNOS-producing dendritic cells. Eur J Immunol. 2011;41:1639–1651. doi: 10.1002/eji.201041022. [DOI] [PubMed] [Google Scholar]
- Colegio OR, Chu N, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Borowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumor-associated macrophages by tumor-derived lactic acid. Nature. 2014 doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde P, Rodriguez M, van der Touw W, Jimenez A, Burns M, Miller J, Brahmachary M, Chen HM, Boros P, Rausell-Palamos F, et al. DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance. Immunity. 2015;42:1143–1158. doi: 10.1016/j.immuni.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. The Journal of experimental medicine. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque-Correa MA, Kuhl AA, Rodriguez PC, Zedler U, Schommer-Leitner S, Rao M, Weiner J, 3rd, Hurwitz R, Qualls JE, Kosmiadi GA, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4024–4032. doi: 10.1073/pnas.1408839111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nature immunology. 2008;9:1399–1406. doi: 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, Kranz DM, Meredith SC, Rowley DA, Schreiber H. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell. 2013;23:516–526. doi: 10.1016/j.ccr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frentsch M, Stark R, Matzmohr N, Meier S, Durlanik S, Schulz AR, Stervbo U, Jurchott K, Gebhardt F, Heine G, et al. CD40L expression permits CD8+ T cells to execute immunologic helper functions. Blood. 2013;122:405–412. doi: 10.1182/blood-2013-02-483586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nature immunology. 2015;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Glynn SA, Boersma BJ, Dorsey TH, Yi M, Yfantis HG, Ridnour LA, Martin DN, Switzer CH, Hudson RS, Wink DA, et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. The Journal of clinical investigation. 2010;120:3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommerman JL, Summers deLuca L. LTbetaR and CD40: working together in dendritic cells to optimize immune responses. Immunol Rev. 2011;244:85–98. doi: 10.1111/j.1600-065X.2011.01056.x. [DOI] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL. In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. Eur J Immunol. 2011;41:749–759. doi: 10.1002/eji.201041069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602. doi: 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Korrer MJ, Routes JM. Possible role of arginase-1 in concomitant tumor immunity. PloS one. 2014;9:e91370. doi: 10.1371/journal.pone.0091370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, McEvoy J, Roussel MF, Dyer MA, Qualls JE, Murray PJ. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell reports. 2015;12:1902–1914. doi: 10.1016/j.celrep.2015.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nature medicine. 2015;21:81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, Novitskaya I, Carbonaro H, Cardinale I, Kikuchi T, et al. Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a) Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19057–19062. doi: 10.1073/pnas.0509736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nature immunology. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, Bruneval P, Trajanoski Z, Fridman WH, Pages F, Galon J. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29:610–618. doi: 10.1200/JCO.2010.30.5425. [DOI] [PubMed] [Google Scholar]
- Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber TG, West BL, Bollag G, Ribas A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer research. 2014;74:153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer research. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, Thompson RW, Cheever AW, Murray PJ, Wynn TA. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371. doi: 10.1371/journal.ppat.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nature medicine. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125:3365–3376. doi: 10.1172/JCI80006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S, Peranzoni E, Desantis G, Chioda M, Walter S, Weinschenk T, Ochando JC, Cabrelle A, Mandruzzato S, Bronte V. Immune Tolerance to Tumor Antigens Occurs in a Specialized Environment of the Spleen. Cell reports. 2012 doi: 10.1016/j.celrep.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Ugel S, Scarselli E, Iezzi M, Mennuni C, Pannellini T, Calvaruso F, Cipriani B, De Palma R, Ricci-Vitiani L, Peranzoni E, et al. Autoimmune B-cell lymphopenia after successful adoptive therapy with telomerase-specific T lymphocytes. Blood. 2010;115:1374–1384. doi: 10.1182/blood-2009-07-233270. [DOI] [PubMed] [Google Scholar]
- Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol. 2010;185:6706–6718. doi: 10.4049/jimmunol.0903411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Ridnour LA, Back T, Hussain SP, He P, Maciag AE, Keefer LK, Murphy WJ, Harris CC, Wink DA, Wiltrout RH. Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy. The Journal of experimental medicine. 2010;207:2455–2467. doi: 10.1084/jem.20100670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Singh V, Watkins SK, Bronte V, Shoe JL, Feigenbaum L, Hurwitz AA. High-avidity T cells are preferentially tolerized in the tumor microenvironment. Cancer research. 2013;73:595–604. doi: 10.1158/0008-5472.CAN-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.