

Abstract
We have conducted a phase 1 study of intravenous vvDD, a Western Reserve strain oncolytic vaccinia virus, on 11 patients with standard treatment-refractory advanced colorectal or other solid cancers. The primary endpoints were maximum tolerated dose and associated toxicity while secondary endpoints were pharmacokinetics, pharmacodynamics, immune responses, and antitumor activity. No dose-limiting toxicities and treatment related severe adverse events were observed. The most common adverse events were grades 1/2 flu-like symptoms. Virus genomes were detectable in the blood 15–30 minutes after virus administration in a dose-dependent manner. There was evidence of a prolonged virus replication in tumor tissues in two patients, but no evidence of virus replication in non-tumor tissues, except a healed injury site and an oral thrush. Over 100-fold of anti-viral antibodies were induced in patients' sera. A strong induction of inflammatory and Th1, but not Th2 cytokines, suggested a potent Th1-mediated immunity against the virus and possibly the cancer. One patient showed a mixed response on PET-CT with resolution of some liver metastases, and another patient with cutaneous melanoma demonstrated clinical regression of some lesions. Given the confirmed safety, further trials evaluating intravenous vvDD in combination with therapeutic transgenes, immune checkpoint blockade or complement inhibitors, are warranted.
Introduction
Oncolytic viruses (OVs) are tumor-selective live agents that work to kill cancer and associated stromal cells via multiple mechanisms of action. Many studies have demonstrated that OVs act through three pronged mechanisms of action: induction of direct oncolysis, inhibition of angiogenesis, and eliciting antitumor immunity.1,2,3 The use of tumor-selective OVs for the treatment of advanced cancer is a promising alternative or adjunct to existing therapies. The successful phase 3 trial of T-VEC (Imlygic) in melanoma patients and the recent approval of T-VEC as the first drug of this class by the Food and Drug Administration (FDA) have validated the potential of this novel class of anticancer drugs.4
The first clinically applicable OVs were developed over 20 years ago. The first one tested in human cancer patients was ONYX-015, an E1B-55kD gene-deleted adenovirus.5 It has been demonstrated not only that tumor cells can be infected, but that replication and spread to cancer cells occurs in humans.6 Since then, over 1,000 patients have been treated in phase 1–3 clinical trials with various OVs.2 While most of the clinical trials were conducted with intralesional injection of the OVs, quite a few trials have been conducted with OVs delivered intravenously, including adenoviruses (Ad) ONYX-015 and CG7870 (refs. 6,7), Newcastle disease viruses PV701 and NDV-HUJ,8,9 Herpes simplex virus (HSV (NV1020) (refs. 10,11), Reolysin (reovirus type 3 Dearing),12 picornavirus called Seneca Valley Virus (SVV-001) (ref. 13), and poxvirus Pexa-Vec (pexastimogene devacirepvec, JX-594) which was derived from Wyeth strain vaccinia virus (VV).14,15 In addition, measles virus MV-NIS has been used to treat two patients with relapsing drug-refractory myeloma by intravenous infusion, with one patient experienced durable complete remission at all disease sites.16 These studies have demonstrated the feasibility and safety to deliver a variety of OVs intravenously into human cancer patients.
Several advanced phases of clinical trials with OVs have been conducted. Thus far, the most successful trial has been with T-VEC as an intratumoral injection in unresectable melanoma. The phase 3 trial demonstrated improvements in durable response rate and a trend toward improved overall survival compared to GM-CSF alone, which led to the approval by the FDA of its use in melanoma patients.4 Pexa-Vec has undergone multiple clinical trials in multiple types of cancer.14,15,17,18,19,20 In one of those trials in human patients with liver cancer, treatment with Pexa-Vec demonstrated a 15% response by RECIST criteria and improved survival when high-dose intralesional treatment was compared to low-dose treatment for hepatocellular carcinoma.18 However, TRAVERSE trial, a randomized phase 2b study of Pexa-Vec in second-line, advanced liver cancer patients did not reach its primary survival endpoint for this population of patients. Nevertheless, a global phase 3 trial to evaluate Pexa-Vec in combination with sorafenib versus sorafenib alone in patients with advanced unresectable hepatocellular carcinoma, called PHOCUS trial, was initiated at the end of 2015.
VV, a prototype poxvirus, is ideal for oncolytic therapy due to its efficiency of replication and spread and its immune evading properties. It possesses intrinsic tumor tropism, and TK-mutated VV, such as Pexa-Vec, selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers.21 Through deletion of viral genes encoding vaccinia growth factor (VGF) and thymidine kinase (TK), we have genetically engineered a tumor-selective, oncolysis-potent Western Reserve (WR) strain VV named vvDD.22,23 Both vgf and tk genes are essential for viral replication in normal cells but not in cancer cells. Studies in non-human primates demonstrated the virulence of wild type WR VV and verified the safety of the tumor-selective, genetically engineered vvDD.24 Finally, the safety and tumor-selectivity of vvDD (also called JX-929) has been proven in humans in our recently published phase 1 trial of intratumoral injection.25
However, intravenous delivery of OVs for metastatic cancer is the goal given the disseminated nature of the disease and the inherent limitations of intratumoral delivery. Due to its proven safety and toxicity profile and evidence of antitumor activity as an intratumoral injection in humans, we proceeded with a phase 1 trial of intravenous delivery of vvDD for metastatic cancers with the goal of developing a systemic therapy for metastatic cancer. Here, we present the data on this study in human cancer patients.
Results
Patient population
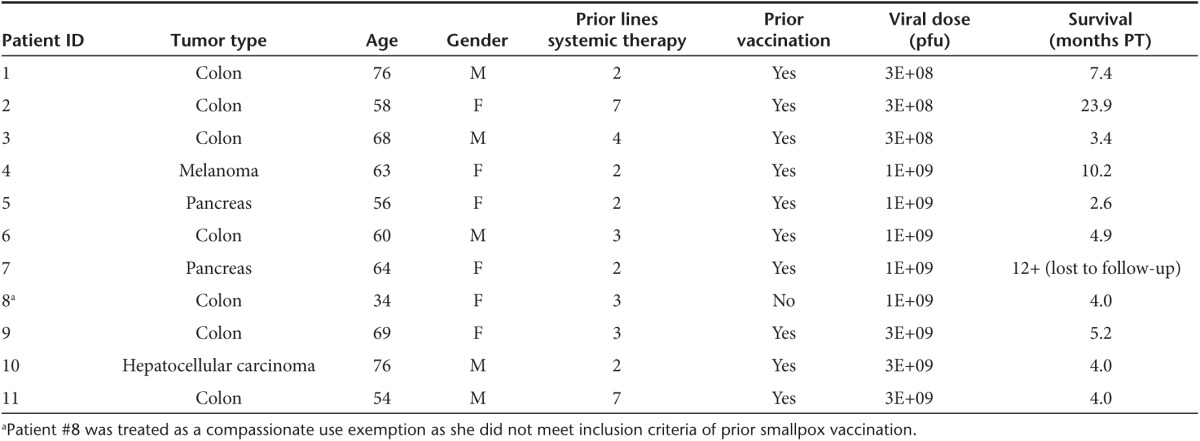
Eleven patients were screened, enrolled, and treated in this clinical trial (Table 1). Patient #8 was enrolled as a compassionate use exemption since she had not received prior smallpox vaccination. All other patients were previously vaccinated against smallpox. All patients were white, six were female and five were male with a median age of 63.1 years (range 34–76 years). Prior to enrollment, patients were treated with a median of three lines of systemic therapy (range 2–7) and five of eleven patients underwent surgical therapy for their disease. All patients progressed through standard treatment regimens. Diagnoses included colon cancer (n = 7), pancreatic cancer (n = 2), hepatocellular carcinoma (n = 1), and melanoma (n = 1) (Table 1).
Table 1. Patients at baseline.
Treatment and safety
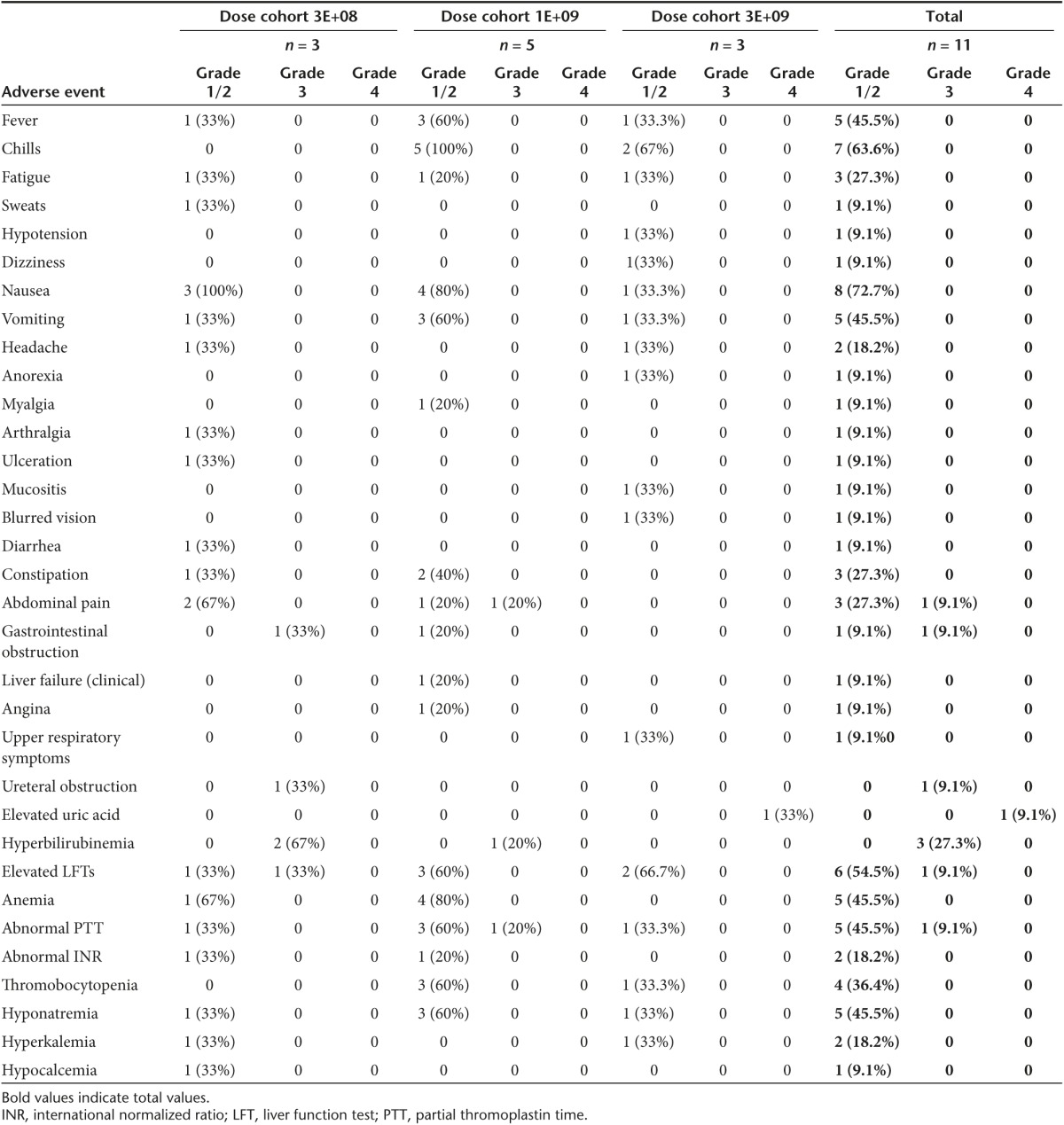
Eleven patients were treated in three dose cohorts (3 × 108 pfu, 1 × 109 pfu, or 3 × 109 pfu). vvDD was infused in 250 ml of bicarbonate-buffered saline over 1 hour. There were no dose-limiting toxicities. There were no treatment-related severe adverse events (SAEs) (Table 2). Treatment-related toxicities (all Grades 1 and 2) included fever and/or chills in 11 patients. Other treatment-related toxicities include abdominal pain, nausea, vomiting, fatigue, and headache and ulceration. There were nine nontreatment-related SAEs reported which included Grade 3 abdominal pain, intestinal obstruction (related to progression of disease), ureteral obstruction, abnormal partial thromboplastin time, and transiently elevated liver function tests (LFTs). There was one Grade 4 elevation in uric acid in a patient with elevated uric acid at baseline. These significant adverse events (SAEs) were determined to be unrelated to treatment after independent review.
Table 2. Adverse events.
Laboratory findings
While there were no particular temporal patterns in laboratory data abnormalities, the most common aberrations were in measures of liver function. Several subjects had mild elevations in their liver transaminases, which could be related to viral delivery. Three patients (27%) had mild elevation in ALT, eight patients (72%) had mild elevation in AST, and seven (76%) patients had mild elevation in alkaline phosphatase. Three patients (27%) had elevation in total bilirubin. All of these patients had disease burden in their liver or pancreas causing biliary obstruction, which explains these abnormalities. Early swelling of tumor from inflammation caused by viral infection could lead to this effect but it is impossible to differentiate from tumor progression and all patients were treated with stents. Four patients (36%) experienced a one-time elevation in LDH. Two patients (18%) experienced mild leukocytosis and two patients (18%) experienced leukopenia. Three patients (27%) experienced thrombocytosis and four patients (36%) experienced thrombocytopenia. No patients had abnormalities in serum creatinine after treatment.
Acute pharmacokinetics
Blood draws were performed 15 minutes, 30 minutes, and 4 hours post-treatment with vvDD to assess the acute pharmacokinetics. All patients (n = 11) had viral genomes detected in their blood 15 minutes after injection. Genome copies/ml were significantly higher in patients treated at the highest dose cohort (3 × 109) (Figure 1a). This finding was also true at 30 minutes post-treatment wherein all patients had detectable vvDD genomes in their blood by quantitative polymerase chain reaction (qPCR) and the quantity was significantly greater in patients treated at the highest dose cohort (Figure 1b). At 4 hours post-treatment, 4 of 11 patients still had detectable vvDD viral genomes in their blood by qPCR (2 in the lowest dose cohort, 1 in the intermediate dose cohort, and 1 in the highest dose cohort). The maximum genome concentrations were detected in the first 15 minutes after treatment and declined with time (Figure 1c). The majority of viral genomes were cleared by 4 hours.
Figure 1.

Acute pharmacokinetics after virus treatment. The virus genomic DNA in the sera was quantified by qPCR at 15 minutes, 30 minutes, and 4 hours post-virus infusion. Shown are data from patients who received the virus doses at 3.0e8, 1.0e9, and 3.0e9 pfu. For the virus dose at 3.0e9 pfu, the quantities of the virus genome in the blood are statistically higher than those at two lower doses at 15 and 30 minutes post-treatment (**P < 0.01).
Anti-VV antibody kinetics
Antibodies to VV in the sera were analyzed before treatment and at day 22 post-treatment by an enzyme-linked immunosorbent assay (ELISA) in all analyzed patients (Figure 2). All patients demonstrated the induction of anti-VV antibodies at 22 days. The difference of 1.0 OD value represents the difference of ~100-fold antibody induction. Thus, the data showed that there was a 100-fold or more induction of anti-VV antibodies at day 22 post-treatment compared to pretreatment. Of note, our healthy volunteer vaccinated with smallpox vaccine 5 years ago had served as a “vaccinated control” and her antibody titer against VV was comparable to the baseline levels (D0) from those patients who were vaccinated 54–76 years ago (Figure 2a,b).
Figure 2.

The antibodies against vaccinia virus in the sera of patients right before treatment and 22 days PT. The anti-VV antibodies as measured by enzyme-linked immunosorbent assays (ELISA). (a) The data were collected on sera from patients who received the virus dose at 1.0E9 PFU (n = 3). The “vaccinated control” indicated a healthy volunteer from our laboratory who was vaccinated with smallpox vaccine 5 years ago and served as a control to patients who had been vaccinated 54–76 years ago. (b) The data were collected on sera from patients who received the virus dose at 3.0E9 PFU (n = 3). (c) Shown are another presentation on the mean values when the sera were diluted at 1: 5,120. Data on D0 (day 0) provided the baseline and the data on D22 (day 22) post-treatment showed increased titers of antisera as determined by ELISA assays. The data are mean ± SD from six patients treated at two higher doses of the virus.
Evidence of vvDD infection and replication in blood and tumor
Blood draws performed at late time points (days 3, 5, 8, and 22 post-treatment) demonstrated evidence of re-emergence of vvDD genomes by qPCR in one patient at day 3 (after a negative qPCR at day 1 hour 4) and one patient at day 8 (after a negative qPCR at day 1 hour 4, day 3, and day 5) post-treatment (Table 3). Of the eight patients who had evaluation of the viral genomes from biopsied tumor samples, two had evidence of vvDD at late time points (days 8 and 22 post-treatment), providing further evidence of prolonged survival and replication of the virus in tumor tissues. The first patient had no evidence of vvDD genome in a biopsied liver tumor specimen at day 8, but tested positive from a biopsied liver tumor specimen biopsied at day 22. A second patient also had evidence of vvDD genome by qPCR in a biopsied liver tumor 8 days after treatment (Table 3).
Table 3. Evidence of vvDD replication and infection.
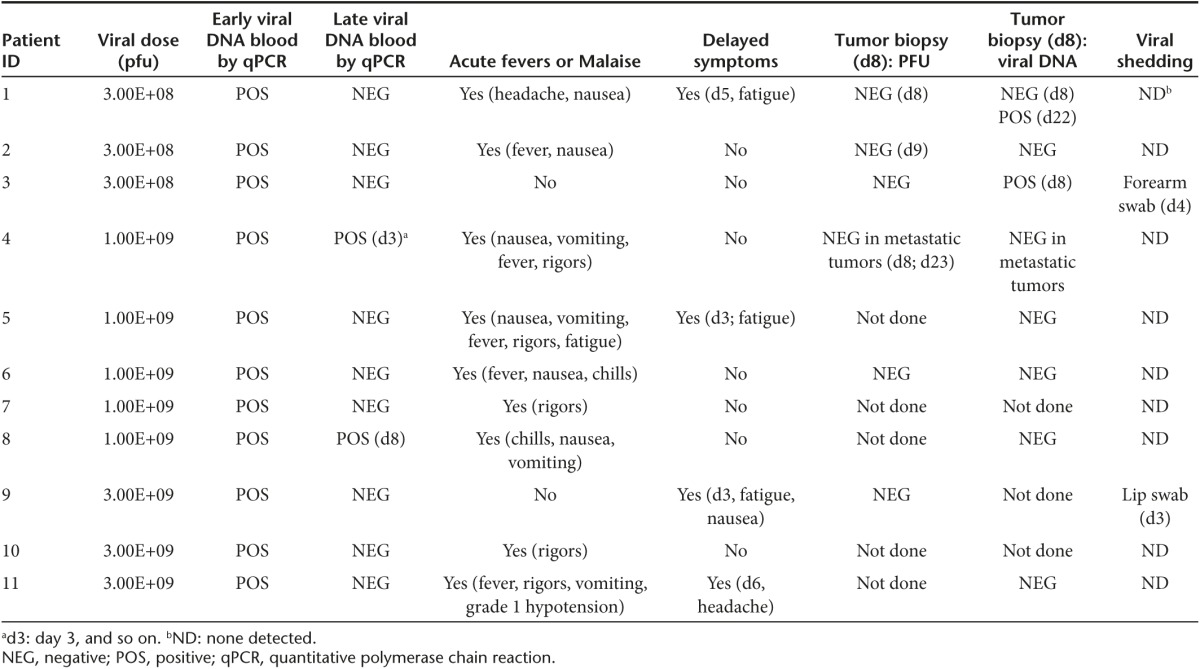
Nine of eleven patients experienced acute systemic symptoms (fevers/malaise/rigors/nausea) within 24 hours of treatment. Four of 11 patients experienced late symptoms (days 3–6) consistent with an ongoing inflammatory response against the virus (Table 3). The presence of ongoing symptoms may indicate continued viral replication in patients.14 Predetermined blood draw times and tumor biopsies may also result in missed evidence of vvDD replication by qPCR.
vvDD shedding, recovery, and tumor specificity
vvDD shedding was analyzed by plaque assay from the blood, plasma, urine, and saliva on days 3, 5, 8, 15, 22, and 28 post-treatment. Patient #3 developed an exudative ulcer on the forearm (Figure 3a) 4 days after treatment and 2.5 × 103 pfu vvDD were recovered from this lesion. This patient had a completely healed injury at this site from 2 weeks prior to vvDD injection, and the scar opened and developed an exudate on day 4, with vvDD cultured from the exudate. Patient #9 had oral thrush at the time of virus administration (Figure 3b). On day 3, a lip swab was performed and a total of 10 pfu of virus was detected. There was no other evidence of vvDD shedding by plaque assay at any time points in any additional patients. There was no other clinical evidence of vvDD replication in normal tissues.
Figure 3.

Detection of the virus at non-tumor sites in two patients. (a) Patient #3 developed an exuative ulcer on the forearm on day 4 post-treatment. This lesion was biopsied on day 4 and the exudate was subjected to analysis of the infectious virus. (b). Patient #9 had oral thrush at the time of virus administration. Lip swab was performed on day 3 to detect infectious virus.
Induction of inflammatory, Th1 but not Th2 cytokines
Patient plasma samples collected pre- and post-treatment (4 hour and day 22 post-treatment) were evaluated for a series of cytokines by Luminex assay. Analysis was performed after log-transformation of most cytokines (except IL-10 and IL-12p70) and the P values were then adjusted using step-down Bonferroni method of Holm to control the familywise error rate. IL-2, IL-7, IL-8, IL-10, IFN-γ, GM-CSF, TNF-α, and IL-6 demonstrated significant increases in patient plasma samples collected 4 hours after treatment (Figure 4a). The most impressive increases were IL-6 and IL-8, two key inflammatory cytokines (Figure 4b). The infection-triggered induction of these inflammatory cytokines might have brought the acute flu-like symptoms to the patients. We also observed significant increases in Th1 cytokines (IL-2, IFN-γ, and TNF-α) and Th1-related cytokines (IL-7 and GM-CSF) (Figure 4c). In contrast, Th2 cytokines (IL-4, IL-5, and IL-13) were not enhanced after treatment (Figure 4d). We have performed the same assays on sera collected on day 22, and the majority of cytokines went back to basal levels (data not shown). In summary, at 4 hours, inflammatory cytokines (IL-6, IL-8) and Th1 cytokines (IL-2, IFN-α, TNF-γ; (IL-1β, IL-7, and GM-CSF are Th1 related)) were upregulated, while Th2 cytokines (IL-4, IL-5, and IL-13) were unchanged. These data strongly suggest that virus infection elicited a Th1 immune response against the virus and possibly the cancer in the patient.
Figure 4.

Cytokine levels in plasma. Cytokine concentrations in the plasma collected before and 4 hours after vvDD treatment were determined via Luminex assays. (a) The whole panel of cytokines assayed. All cytokines were analyzed on a logarithmic scale except those denoted by * were analyzed using the Box-Cox transformation method. * indicated adjusted P values based on log-transformed values. (b) Key inflammatory cytokines (IL-6 and IL-8). (c) Th1 (IL-2, IFN-γ, and TNF-α) and Th1-related (IL-7 and GM-CSF) cytokines. (d) Th2 cytokines (IL-4, IL-5, and IL-13). Pre-T: pretreat; Post-T: 4 hours post-treatment. The P values in the graphs are indicated, *P < 0.05; **P < 0.01; ns: no significant.
Antitumor activity
All but one patient (compassionate use patient #8) were assessed for clinical response to treatment by sequential PET-CT scanning. The scan at 3 weeks was to determine any immediate effect of the viral infection, but no necrosis or change in PET signal intensity was noted. Three patients had documented stable disease at 12 week post-treatment and one of these had a mixed response.
One patient with cutaneous lesions (melanoma, #4) was followed with photographs. This patient demonstrated clinical regression of some lesions shown by necrosis of an in-transit metastasis at 23 days (Figure 5a). Patient #2, with metastatic colorectal cancer in the liver, demonstrated a mixed response (Figure 5b). The patient had been previously treated with isolated hepatic perfusion 2 years prior to vaccinia therapy and had multiple lesions that became calcified and necrotic and never progressed after liver perfusion. However, she ultimately developed new PET-avid lesions in her liver and was subsequently enrolled in this trial. Six weeks after treatment, PET-CT imaging demonstrated complete resolution of these new hepatic metastases and only residual FDG-avid calcium is visualized at the previously treated sites (presumed not to represent viable tumor after 2 years as stable disease). An enlarged retroperitoneal lymph node did not respond to treatment with vvDD however this lesion remained stable for 6 months after treatment before regrowing. This patient survived 23.9 months after treatment.
Figure 5.

Clinical activity of vvDD in cancer patients after systemic delivery. (a). One patient with cutaneous lesions (patient #4, with melanoma) was photographed before and 23 days after treatment. The clinical regression of lesions is shown by necrosis of an in-transit metastasis at 23 days post-treatment. (b). The patient #2 with metastatic colorectal cancer in the liver demonstrated a mixed response 6 weeks after treatment on PET-CT with resolution of some of the liver metastases.
The median survival of all patients was 4.8 months (range 2.6–23.9 months) (Table 1). One patient (#10) was lost to follow-up and survival data is unknown.
Discussion
We have previously reported the first-in-human phase 1 trial of intratumoral injection of vvDD and demonstrated both the safety and feasibility of this route of vvDD delivery as well as some promising antitumor activity.25 In that report, we evaluated the capacity of vvDD for tumor infectivity in preimmunized patients. The viral administration was tolerated well and there were no dose-limiting toxicities. We defined a maximum feasible dose of 3 × 109 pfu in preimmunized human patients.
In the current study, we have described the results of the first trial of intravenous delivery of vvDD in human patients. All enrolled patients demonstrated evidence of vvDD genomes in their blood acutely and two patients had vvDD persistence in blood at later time points. Two patients demonstrated evidence of prolonged vvDD replication in the biopsied tumors (days 8 and 22). Nearly all patients demonstrated clinical symptoms consistent with acute viral infection and 4 of 11 had symptoms consistent with an ongoing inflammatory response to vvDD at 3 to 6 days post-treatment (Table 3). One patient demonstrated a mixed response to treatment with resolution of some metastatic hepatic lesions from colon cancer post-treatment as measured by PET-CT scan. Another demonstrated a response in some of the melanoma cutaneous lesions. Unfortunately, we were unable to correlate circulating viral genomes with positive biopsy results and tumor response in this small study. This is most likely due to sampling errors in terms of timing of the blood draws, the sensitivity of our assays, and the fact that tumor biopsies are only representative of a fraction of the tumor. In future studies, we believe post-treatment day 4 would be a better time to biopsy tumors, as in our animal models, the virus is mostly eliminated in 8 days by the immune system. Unlike in our intratumoral injection trial, in this trial, we had two patients with evidence of vvDD infection of normal tissues, including one patient with oral thrush and another patient with a 2-week-old arm wound which became infected with the virus. The recovery of replicating virus in a healing wound and inflamed oral cavity suggests that systemic delivery of the virus to tissues is possible, and that the selective mutations of the vaccinia do not differentiate tumor tissue from healing or inflamed tissue. It has been previously noted that active psoriatic skin rashes supported vaccinia replication, and it is therefore a contraindication to smallpox vaccination. While virus recovery in our study was not associated with any significant toxicity, actively healing wounds, and acute inflammatory conditions of the skin or oral mucosa should be an exclusion criterion in systemic delivery of WR strain-derived oncolytic virus.
While all patients demonstrated acute evidence of vvDD viral genomes in their blood, most patients appeared to clear the virus quickly, limiting its ability to infect sites of metastases. When serum samples were evaluated by Luminex assay, there was strong evidence for an acute inflammatory response, with induction of inflammatory cytokines (IL-6 and IL-8) and Th1 cytokines (IL-2, IFN-γ, and TNF-α), that may result in the elimination of vvDD before it has a chance to exert significant anti-tumor effects.
vvDD is unique from other VVs used in clinical trials, in that it is based on an aggressive backbone strain (WR), which was found to be markedly more potent than other strains of VV.22,24 It is also the only VV in clinical trials with a deletion in the vaccinia growth factor gene in combination with the thymidine kinase gene. This combined deleted virus was shown to be more attenuated in normal tissues than the TK-deleted virus, while maintaining its potent phenotype in tumor tissue.22,24 Also, vvDD does not have a working therapeutic transgene as seen in other oncolytic VVs. It is logical to compare phase 1 studies of vvDD with other OVs including other poxvirus, herpes simplex virus and adenovirus. First let us compare vvDD with Pexa-Vec when both delivered intravenously.14,15 In both cases, no dose-limiting toxicities were reported, and the maximum tolerable dose was not reached. The most common adverse events were grades 1/2 flu-like symptoms. The pharmacokinetics and pharmacodynamics of the two recombinant OVs were similar. Pexa-Vec infusion was associated with skin-pustules (pox lesions) in normal skin in two of nine patients treated at a dose of ≥1.0e9 pfu. With vvDD, we did not see any pox lesions develop in normal skin in any dose level (up to 3 × 109 pfu). In both cases, antivirus antibodies were induced significantly in all of tested patients. As for efficacy, neither showed objective clinical responses. Comparing the results of intravenous and intralesional deliveries of the same virus vvDD,25 in both cases, we have observed a high degree of safety. Viral replication was tumor tissue-selective as we observed no viral replication in normal tissues when delivered intralesionally, and only low amounts of infectious virus recovered from nontumor, but pathological tissues in 2 out of 11 patients when delivered systemically. The acute pharmacokinetics was similar with most viral genomes cleared from the blood by 4 hours. Pharmacodynamic studies indicated re-emergence of viral genomes at days 3 and 8 post-treatment in blood in two patients, suggesting persistent viral replication in tissues (perhaps tumor). In both cases, a highly significant induction of antiviral antibodies was observed. In terms of antitumor activities, we have seen clinical responses in a small fraction of patients. It is interesting that treatments of two melanoma patients (one by intravenous delivery and the other by intratumoral injection) resulted in significant clinical responses in some tumor nodules. This is reminiscent of the fact that the FDA-approved drug Imlygic (T-VEC) was effective in melanoma patients. The therapeutic efficacy of OVs may be most effective with immunogenic cancers.
Clinical results with three OVs armed with GM-CSF, but derived from VV, HSV, and Ad, highlight the progress of this rapidly moving field. Pexa-Vec via intravenous delivery showed that 67% (7/11) of patients with advanced colorectal cancer had radiographically stable disease.15 For Ad5/3-E2F-Δ24-GMCSF, its multiple treatments were well tolerated, and tumor- and adenovirus-specific T-cell immunity was frequently observed in the patients. Overall, antitumor responses were seen in 9/12 evaluable patients (75%). The radiological disease control rate was 83% while the response rate (including minor responses) was about 50%.26 However, these patients were also treated with low-dose cyclophosphamide to reduce Treg, and some patients also received low-dose pulse temozolomide for enhanced autophagy. Despite these additional treatments, the overall survival (OS) of all virus-treated patients was not increased over controls.27 As for T-VEC (Imlygic) in the phase 2 trial, the overall response rate by RECIST was 26%, and regression of both injected and distant (including visceral) lesions occurred.28 In the phase 3 trial, overall response rate was at 26.4% and median overall survival was 23.3 months in melanoma patients.4 Given the high immunogenicity of melanoma, it is not too surprising to see better objective clinical responses. In summary, our results on safety and efficacy of vvDD via systemic delivery are in line with other unarmed OVs via systemic delivery (JX-594, NV1020 and Reolysin).10,11,12,15 However, arming an OV with an immunostimulatory gene such as GM-CSF (as found in T-VEC) may improve the overall efficacy.
The immune responses have played yin-yang roles in OV-mediated antitumor activity. Oncolytic virotherapy can be considered a form of immunotherapy.1,2 To enhance this antitumor immunity, OVs are often armed with immunostimulatory genes. T-VEC, Ad5/3-E2F-Δ24-GMCSF, and Pexa-Vec are armed with the GM-CSF gene. We and others have studied oncolytic VVs in preclinical models which are armed with cytokines or chemokines to stimulate antitumor immunity.29,30 Further modulation of the tumor microenvironment may promote the functions of the antitumor immune cells.31 On the other hand, the innate immunity and/or elicited antiviral adaptive immunity often eliminates the OV prematurely, thus diminishing the oncolytic potency. We and others have studied various strategies to overcome this hurdle. OVs can be delivered in conjunction with immune evasion or immune inhibition.32,33,34 Similarly, the pretreatment with an immunosuppressive cytokine, TGF-β, enhances HSV-mediated virotherapy of glioblastoma by inhibiting the innate immune response.35
The lack of clinical response with systemic delivery may be secondary to poor delivery to the tumors. We only recovered viral genomes in two of the patients biopsied. This may be due to timing, as in animal models the virus is cleared by day 8, but it may also be due to premature viral clearance and poor delivery to the tumor. The complement system acts as a rapid and efficient immune surveillance mechanism that has distinct effects on altered host cells and viruses, directly destroying the virus, sending a ‘danger signal' to antigen-presenting cells, and orchestrating immune responses.36 In the case of VV, the complement system limits VV infection as it enhances the neutralizing capacity of smallpox vaccine-induced antibodies.37,38 Neutralization by anti-vaccinia antibodies is ineffective in the absence of complement.39 In addition, there is antibody-independent triggering of the classic complement pathways.40 All of these mechanisms inhibit the trafficking to and infection of the cancer cells by VV in the tumor tissue. Investigators have designed strategies to overcome this hurdle. Complement depletion using Compstatin in primates enhances viral stability in primates.41 Complement depletion with a protein inhibitor (cobra venom factor) enhanced viral delivery to tumors in immunized animals and CP40 complement inhibition in immunized human blood samples inhibits antibody-mediated virus neutralization in vitro.41 We have demonstrated that complement inhibition using either protein inhibitors or a monoclonal antibody to C5 markedly enhanced vvDD infectivity and oncolysis of cancer cells in vitro.42 The complement depletion has also been used for facilitation of delivery of an oncolytic HSV in a tumor model.43 This approach should be considered for future systemic oncolytic virus trials.
Immune checkpoint blockade is a highly effective approach to stimulate antitumor immunity in a number of cancers.44 The rational combination of an OV and immune checkpoint blockade is highly efficacious in tumor models.45,46,47,48,49 This could be performed with either physical delivery of antibodies such as anti-PD-1, anti-CTLA-4, or all in one approach with the OV armed with genes encoding recombinant antibodies against PD-1, CTLA-4 or other immune checkpoint molecules.45,46 In this aspect, it is exciting to know that an ongoing phase 1–3 clinical study combining T-VEC and anti-PD-1 antibody has achieved significant clinical responses in melanoma patients.50
In summary, we have demonstrated feasibility, safety, infectivity, and some limited antitumor effects in patients with intravenous delivery of vvDD. The lack of uniform antitumor activity may be explained by inadequate viral delivery to the tumor, the need for a therapeutic immunogenic transgene, or the need for additional alterations of the tumor microenvironment using checkpoint blockade. These results indicate the next step is to consider two combining complement inhibition with intravenous vvDD treatment, and combining vvDD with immune checkpoint blockade and an immunogenic transgene.
Materials and Methods
Clinical trial
Patients. Eleven patients were enrolled in this phase 1 dose-escalation trial over 17 months between October 2011 and February 2013. One patient (#7) left the trial after 8 days and limited follow-up was obtained for this patient. Patient #8 was treated as a compassionate use exemption as she did not have prior smallpox vaccination. Patients were eligible for enrollment if they were over 18 year of age with a Karnofsky performance status of ≥80 or an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 with a histologically-confirmed cancer diagnosis that was not surgically curable and had advanced despite standard therapy. Bone marrow function was also assessed for adequacy prior to enrollment (WBC > 3,500, absolute neutrophil count > 1,500 cells/mm3, CD4 T-cell count > 350 per µl of blood, hemoglobin > 10 g/dl, and platelet count > 150,000 cells/mm3 and patients were also required to have normal renal function (serum creatinine ≤1.2 times upper limit of normal) and coagulation (international normalized ratio <1.1 times upper limit of normal). Unlike the intratumoral trial of vvDD, prior smallpox vaccination was required for enrollment in this study. Patients with previous side effects from smallpox vaccination were excluded. Due to the risk of skin infection after VV administration, any patients who ever required systemic therapy for eczema were excluded. Additionally, pregnant or nursing patients, those with active viral infection (HIV, Hepatitis C or B), those with systemic corticosteroid or immunosuppressive medication use (including chemotherapy or radiation) within 4 weeks of screening, or those with household contacts with significant immunodeficiency were excluded. Finally, patients with unstable cardiac disease or rapidly accumulating or clinically significant ascites, pericardial or pleural effusions were excluded from enrollment.
Prior to study initiation, the protocol and consent forms were approved by the University of Pittsburgh Institutional Review Board, Institutional Protocol Review Committee, and Institutional Biosafety Committee as well as the Recombinant Advisory Committee and US Food and Drug Administration (Institutional protocol #UPCI-06-041; ClinicalTrials.gov Identifier: NCT00574977). All patients gave written informed consent prior to participation and the Declaration of Helsinki protocols were fulfilled. An independent data-safety monitoring committee reviewed data before all dose escalations as well as adverse events.
vvDD Production. The production of vvDD (also called JX-929 or vvDD-CDSR) has been described elsewhere.23,25 The vgf gene has been previously deleted by our group by insertion of a lacZ gene into the VGF gene locus by homologous recombination and color selection after X-gal staining of plaques for a nonfunctional β-galactosidase. For the creation of vvDD, homologous recombination of the cytosine deaminase and somatostain receptor genes in the TK-locus of vSC20 (vgf-deleted WR strain VV) was performed. While vvDD allows imaging via somatostatin receptor scintigraphy (octreotide scanning) as well as prodrug treatment with 5-fluorocytosine, these techniques were not utilized in this trial.
Production of vvDD intended for use in the clinical trial followed Good Manufacturing Practice guidelines. The virus vvDD was created using Vero cells (American Type Culture Collection (ATCC), Manassas, VA) and purified through sucrose-gradient centrifugation by Novavax (Rockville, MD). The tk and vgf deletions were verified in the final product and lacZ mutation was also confirmed by sequencing and the product was tested for sterility, endotoxin and assayed for potency. Genome-to-pfu ratio was 50:1 and the final product was created in phosphate-buffered saline with 10% glycerol, 138 mmol/l sodium chloride with a pH of 7.4. The virus stock at a concentration of 2 × 109 pfu/ml was diluted to a total of 250 ml of bicarbonate-buffered saline prior to administration.
Treatment and monitoring. We followed a phase 1, open-label, dose-escalation, single-dose group, sequential dose-escalation format. Patients were treated at the following doses: 3 × 108 pfu (n = 3), 1 × 109 pfu (n = 5), and 3 × 109 (n = 3) with 4 weeks between each dose escalation.
Patients were admitted to the Clinical and Translational Research Center Montefiore University Hospital for 24 hours (or more) on the day of treatment for physical examination and vital signs monitoring. vvDD at the selected dose was infused over 1 hour in a total volume of 250 ml of sodium bicarbonate-buffered saline. Blood draws for pharmacokinetic monitoring were performed pretreatment and 15 minutes, 30 minutes, 1 hour, and 4 hours PT. Additional blood draws were performed at days 3, 5, 8, 15, 22, and 28 PT. Consenting patients underwent biopsy of selected lesions at day 8 PT. Treatment response was evaluated by standard RECIST criteria at weeks 3 and 8 using CT scanning.
Laboratory analysis
Quantitation analysis for vvDD genomes. Quantitative PCR (Q-PCR) was employed as a method of pharmacokinetic monitoring of plasma especially because of its ability to detect vvDD genome even in the presence of antibody and/or complement neutralization. Q-PCR was performed as described elsewhere.17,18,25 Given the potential public health consequences of vvDD shedding during treatment, in many cases, the infectious vvDD particles in patient saliva, urine, and biopsy samples were also tittered by plaque assays. Samples underwent three freeze-thaw cycles prior to analysis to release infectious particles. For a similar reason, biopsy samples were homogenized prior to analysis using a FastPrep Cell Disrupter (Model FP120; Qbiogene, Carlsbad, CA). Samples or cell lysates (from biopsy samples) were used to infect CV-1 cells and titres were determined using standard, previously described plaque assay techniques.22
ELISA for antibodies against VV. ELISA plates were incubated with 1 × 105 pfu of vvDD and patient serum samples were serially diluted twofold at a starting dilution of 1:10 and incubated on the same plate. A vaccinated nontreated subject's serum was used a positive control and wells with no serum added were used at negative control. Detection antibody (Goat anti-human IgG (H+L) horseradish peroxidase (HRP) from Chemicon (Temecula, CA) was used at a dilution of 1:5,000 and after developing the samples, results were read on a spectrophotometer at 450 nm after the addition of stop solution.
Luminex assay. Luminex assay on pretreatment and 4-hours post-treatment patient plasma samples for the following cytokines was performed in triplicate: IL-2, IL-1β, IL-4, IL-5, IL-13, IL-7, IL-8, IL-10, IL12p70, IFN-γ, CM-CSF, TNF-α, and IL-6 using the MILLIPLEX MAP Human High Sensitivity T Cell Panel- Immunology Multiplex Assay Kit (EMD Millipore, Billerica, MA). This assay was performed by the UPCI Cancer Biomarkers Facility Luminex Core Laboratory using multianalyte profiling (LabMAP) system (Luminex Corporation, Austin, TX). Statistical analysis was performed using SAS v9.4 (SAS Institute, Cary, NC). A significance level was set at 0.05 and all P values reported were two-sided. Raw means and P values were calculated, however, the P values were then adjusted using step-down Bonferroni method of Holm to control the familywise error rate after log transformation of all biomarkers (except that IL-10 and IL-12p70 were not log transformed). A similar analysis was performed for pretreatment and 22 days post-treatment patient serum samples for the same cytokines. Statistical analysis was performed in a similar manner and most biomarkers were analyzed after log transformation or by Box-Cox transformation (IL-2 and IL-7). Again to control for familywise error rate due to testing done in multiple patients in triplicate, the step-down Bonferroni method of Holm was used to calculate adjusted P values.
Statistical analysis. The primary objectives of this study were to assess the safety and maximum tolerated dose of vvDD used intravenously for metastatic cancer. The sample size was chosen to assess these primary outcomes and the expected sample size was 9 to 18 patients, based on a single-dose group sequential dose-escalating design, with three to six patients treated at each of three dose levels.
For Luminex assays, statistical analysis was performed using SAS v9.4 (SAS Institute). For other data, statistical analyses were performed with SPSS Statistics Software version 18 (IBM, NY), or Prism (GraphPad Software, La Jolla, CA). An α value (P) of 0.05 was considered as statistically significant. The standard symbols are used in the figures: * indicates P < 0.05; **P < 0.01; ***P < 0.001; and “ns” means not significant.
Acknowledgments
This project was supported by grant R01CA155925 and training grant T32CA113263 from the National Cancer Institute of NIH. This project used the University of Pittsburgh Cancer Institute (UPCI) shared facilities that are supported in part by the NIH grant award P30CA047904. This work was also partially funded by generous support from Valerie Koch and the New Era Cap Company. D.L.B. served as a consult to Jennerex Biotherapeutics from 2001–2013, and holds a patent on vvDD. A.M. was a consult to and CJB was an employees of SillaJen Biotherapeutics, Inc.
References
- Bartlett, DL, Liu, Z, Sathaiah, M, Ravindranathan, R, Guo, Z, He, Y et al. (2013). Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer 12: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichty, BD, Breitbach, CJ, Stojdl, DF and Bell, JC (2014). Going viral with cancer immunotherapy. Nat Rev Cancer 14: 559–567. [DOI] [PubMed] [Google Scholar]
- Kaufman, HL, Kohlhapp, FJ and Zloza, A (2015). Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 14: 642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andtbacka, RH, Kaufman, HL, Collichio, F, Amatruda, T, Senzer, N, Chesney, J et al. (2015). Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 33: 2780–2788. [DOI] [PubMed] [Google Scholar]
- Ganly, I, Kirn, D, Eckhardt, G, Rodriguez, GI, Soutar, DS, Otto, R et al. (2000). A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res 6: 798–806. [PubMed] [Google Scholar]
- Nemunaitis, J, Cunningham, C, Buchanan, A, Blackburn, A, Edelman, G, Maples, P et al. (2001). Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther 8: 746–759. [DOI] [PubMed] [Google Scholar]
- Small, EJ, Carducci, MA, Burke, JM, Rodriguez, R, Fong, L, van Ummersen, L et al. (2006). A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther 14: 107–117. [DOI] [PubMed] [Google Scholar]
- Pecora, AL, Rizvi, N, Cohen, GI, Meropol, NJ, Sterman, D, Marshall, JL et al. (2002). Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol 20: 2251–2266. [DOI] [PubMed] [Google Scholar]
- Freeman, AI, Zakay-Rones, Z, Gomori, JM, Linetsky, E, Rasooly, L, Greenbaum, E et al. (2006). Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther 13: 221–228. [DOI] [PubMed] [Google Scholar]
- Kemeny, N, Brown, K, Covey, A, Kim, T, Bhargava, A, Brody, L et al. (2006). Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther 17: 1214–1224. [DOI] [PubMed] [Google Scholar]
- Geevarghese, SK, Geller, DA, de Haan, HA, Hörer, M, Knoll, AE, Mescheder, A et al. (2010). Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther 21: 1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal, L, Pandha, HS, Yap, TA, White, CL, Twigger, K, Vile, RG et al. (2008). A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res 14: 7127–7137. [DOI] [PubMed] [Google Scholar]
- Rudin, CM, Poirier, JT, Senzer, NN, Stephenson, J Jr, Loesch, D, Burroughs, KD et al. (2011). Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res 17: 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach, CJ, Burke, J, Jonker, D, Stephenson, J, Haas, AR, Chow, LQ et al. (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477: 99–102. [DOI] [PubMed] [Google Scholar]
- Park, SH, Breitbach, CJ, Lee, J, Park, JO, Lim, HY, Kang, WK et al. (2015). Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol Ther 23: 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ, Federspiel, MJ, Peng, KW, Tong, C, Dingli, D, Morice, WG et al. (2014). Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc 89: 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, BH, Hwang, T, Liu, TC, Sze, DY, Kim, JS, Kwon, HC et al. (2008). Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 9: 533–542. [DOI] [PubMed] [Google Scholar]
- Heo, J, Reid, T, Ruo, L, Breitbach, CJ, Rose, S, Bloomston, M et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, MK, Breitbach, CJ, Moon, A, Heo, J, Lee, YK, Cho, M et al. (2013). Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med 5: 185ra63. [DOI] [PubMed] [Google Scholar]
- Cripe, TP, Ngo, MC, Geller, JI, Louis, CU, Currier, MA, Racadio, JM et al. (2015). Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther 23: 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parato, KA, Breitbach, CJ, Le Boeuf, F, Wang, J, Storbeck, C, Ilkow, C et al. (2012). The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther 20: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCart, JA, Ward, JM, Lee, J, Hu, Y, Alexander, HR, Libutti, SK et al. (2001). Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 61: 8751–8757. [PubMed] [Google Scholar]
- Chalikonda, S, Kivlen, MH, O'Malley, ME, Eric Dong, XD, McCart, JA, Gorry, MC et al. (2008). Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer Gene Ther 15: 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik, AM, Chalikonda, S, McCart, JA, Xu, H, Guo, ZS, Langham, G et al. (2006). Intravenous and isolated limb perfusion delivery of wild type and a tumor-selective replicating mutant vaccinia virus in nonhuman primates. Hum Gene Ther 17: 31–45. [DOI] [PubMed] [Google Scholar]
- Zeh, HJ, Downs-Canner, S, McCart, JA, Guo, ZS, Rao, UN, Ramalingam, L et al. (2015). First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther 23: 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki, O, Parviainen, S, Juhila, J, Turkki, R, Linder, N, Lundin, J et al. (2015). Immunological data from cancer patients treated with Ad5/3-E2F-Δ24-GMCSF suggests utility for tumor immunotherapy. Oncotarget 6: 4467–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanerva, A, Koski, A, Liikanen, I, Oksanen, M, Joensuu, T, Hemminki, O et al. (2015). Case-control estimation of the impact of oncolytic adenovirus on the survival of patients with refractory solid tumors. Mol Ther 23: 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzer, NN, Kaufman, HL, Amatruda, T, Nemunaitis, M, Reid, T, Daniels, G et al. (2009). Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27: 5763–5771. [DOI] [PubMed] [Google Scholar]
- Chard, LS, Maniati, E, Wang, P, Zhang, Z, Gao, D, Wang, J et al. (2015). A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res 21: 405–416. [DOI] [PubMed] [Google Scholar]
- Liu, Z, Ravindranathan, R, Li, J, Kalinski, P, Guo, ZS and Bartlett, DL (2016). CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology 5: e1091554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis L, Guo ZS, Liu Z, Ravindranathan R, Urban JA, Sathaiah M, Kalinski P, Bartlett DL (2016). Modulation of chemokines in the tumor microenvirnment enhances oncolytic virotherapy for colorectal cancer. Oncotarget 7: 22174–22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, K, Ichikawa, T, Wakimoto, H, Silver, JS, Deisboeck, TS, Finkelstein, D et al. (1999). Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med 5: 881–887. [DOI] [PubMed] [Google Scholar]
- Thomas, MA, Spencer, JF, Toth, K, Sagartz, JE, Phillips, NJ and Wold, WS (2008). Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther 16: 1665–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, ZS, Parimi, V, O'Malley, ME, Thirunavukarasu, P, Sathaiah, M, Austin, F et al. (2010). The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther 17: 1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J, Chen, X, Chu, J, Xu, B, Meisen, WH, Chen, L et al. (2015). TGFβ Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res 75: 5273–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin, D, Hajishengallis, G, Yang, K and Lambris, JD (2010). Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, C, Nomura, M, Kitaoka, M, Takeuchi, Y and Kimura, M (1968). Complement requirement of the neutralizing antibody appearing after immunization with smallpox vaccine. Jpn J Microbiol 12: 256–259. [DOI] [PubMed] [Google Scholar]
- Takabayashi, K and McIntosh, K (1973). Effect of heat-labile factors on the neutralization of vaccinia virus by human. Infect Immun 8: 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhnia, MR, McCausland, MM, Moyron, J, Laudenslager, J, Granger, S, Rickert, S et al. (2009). Vaccinia virus extracellular enveloped virion neutralization in vitro and protection in vivo depend on complement. J Virol 83: 1201–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, NR, Jensen, FC, Welsh, RM Jr and Oldstone, MB (1976). Lysis of RNA tumor viruses by human serum: direct antibody-independent triggering of the classical complement pathway. J Exp Med 144: 970–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgin, L, Acuna, SA, Tanese de Souza, C, Marguerie, M, Lemay, CG, Ilkow, CS et al. (2015). Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther 23: 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magge, D, Guo, ZS, O'Malley, ME, Francis, L, Ravindranathan, R and Bartlett, DL (2013). Inhibitors of C5 complement enhance vaccinia virus oncolysis. Cancer Gene Ther 20: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, K, Wakimoto, H, Ichikawa, T, Jhung, S, Hochberg, FH, Louis, DN et al. (2000). Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol 74: 4765–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow, MA, Callahan, MK and Wolchok, JD (2015). Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 33: 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias, JD, Hemminki, O, Diaconu, I, Hirvinen, M, Bonetti, A, Guse, K et al. (2012). Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther 19: 988–998. [DOI] [PubMed] [Google Scholar]
- Engeland, CE, Grossardt, C, Veinalde, R, Bossow, S, Lutz, D, Kaufmann, JK et al. (2014). CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther 22: 1949–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin, D, Holmgaard, RB, Subudhi, SK, Park, JS, Mansour, M, Palese, P et al. (2014). Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 6: 226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quetglas, JI, Labiano, S, Aznar, MÁ, Bolaños, E, Azpilikueta, A, Rodriguez, I et al. (2015). Virotherapy with a Semliki Forest virus-based vector encoding IL12 synergizes with PD-1/PD-L1 blockade. Cancer Immunol Res 3: 449–454. [DOI] [PubMed] [Google Scholar]
- Rajani, K, Parrish, C, Kottke, T, Thompson, J, Zaidi, S, Ilett, L et al. (2016). Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol Ther 24: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke K (2016). A new age for melanoma via immune Rx. http://wwwclinicaloncologycom/Melanoma/Article/02-16/A-New-Age-for-Melanoma-Via-Immune%C2%A0Rx/35301.