

Abstract
We examined the efficiency, specificity, and mutational signatures of zinc finger nucleases (ZFNs), transcriptional activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 systems designed to target the gene encoding the transcriptional repressor BCL11A, in human K562 cells and human CD34+ progenitor cells. ZFNs and TALENs were delivered as in vitro transcribed mRNA through electroporation; CRISPR/Cas9 was codelivered by Cas9 mRNA with plasmid-encoded guideRNA (gRNA) (pU6.g1) or in vitro transcribed gRNA (gR.1). Analyses of efficacy revealed that for these specific reagents and the delivery methods used, the ZFNs gave rise to more allelic disruption in the targeted locus compared to the TALENs and CRISPR/Cas9, which was associated with increased levels of fetal hemoglobin in erythroid cells produced in vitro from nuclease-treated CD34+ cells. Genome-wide analysis to evaluate the specificity of the nucleases revealed high specificity of this specific ZFN to the target site, while specific TALENs and CRISPRs evaluated showed off-target cleavage activity. ZFN gene-edited CD34+ cells had the capacity to engraft in NOD-PrkdcSCID-IL2Rγnull mice, while retaining multi-lineage potential, in contrast to TALEN gene-edited CD34+ cells. CRISPR engraftment levels mirrored the increased relative plasmid-mediated toxicity of pU6.g1/Cas9 in hematopoietic stem/progenitor cells (HSPCs), highlighting the value for the further improvements of CRISPR/Cas9 delivery in primary human HSPCs.
Keywords: zinc finger nucleases, TALENs, CRISPR/Cas9, nonhomologous end joining, BCL11A, fetal hemoglobin
Introduction
The ability to edit DNA sequences is critical to a wide range of basic and applied sciences. Detailed comparison studies of genome engineering strategies are imperative to evaluate the characteristics and effects of these technologies. Zinc finger nucleases (ZFNs) and transcriptional activator-like effector nucleases (TALENs) have been firmly established as efficient gene-editing technologies over the past decade. In clinical trials for human immunodeficiency virus infection, ZFN-mediated disruption of the C-C chemokine receptor 5 locus in T lymphocytes has produced salutary effects, and ZFNs are currently being evaluated in clinical trials for treatment of glioblastoma and hemoglobinopathies.1,2 The recent development of clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 has enabled widespread application of genome engineering and virtually constitutes a paradigm shift in the research field due to the simplicity of designing a guideRNA (gRNA) to any target. Nevertheless, although high site-specific nuclease activity of the CRISPR/Cas9 has been demonstrated in cell lines, there has, with the exception of two recent publications(3, 4), been to date a paucity of studies examining the efficiency of CRISPR/Cas9 in primary human hematopoietic stem and progenitor cells (HSPCs).
Inherent nuclease properties determine their efficacy to facilitate permanent site-specific genome engineering by induction of DNA double-strand breaks (DSBs) that result in insertions and deletions (indels) through the error-prone nonhomologous end-joining (NHEJ) DNA repair pathway. ZFNs and TALENs rely on protein-based systems with designable DNA binding specificities. The ZFNs consist of an endonucleolytic domain, FokI, that is tethered to engineered Cys2His2 zinc-finger DNA-binding polypeptides5,6; two ZFNs are required to bind opposite DNA strands at a set distance for the FokI domains to dimerize and create a DSB. TALENs also depend on two FokI nuclease domains to dimerize in order to induce site-specific DNA cleavage but consist of assembled TAL effector DNA-binding motifs, each motif consisting of tandem repeats that bind to a single base pair.7,8 The Type II RNA-guided CRISPR/Cas9 consists of a short gRNA sequence (20 variable bases target sequence that encodes crRNA) that targets the Cas9 endonuclease through Watson–Crick RNA–DNA base pairing to cleave the DNA.9,10
Efficient non-cytotoxic delivery is a prerequisite to achieve maximum genome engineering potential of nucleases, especially in delicate primary cells that cannot be effectively expanded ex vivo, such as HSPCs. Introduction of DNA to cells is associated with higher cytotoxicity compared to in vitro transcribed mRNA and introduced DNA might potentially undergo integration into the host genome.3,11,12,13 Conversely, RNA instability is challenging for the process of delivery, since RNA can undergo rapid intracellular degradation.
The zinc-finger transcriptional factor BCL11A has been shown to silence embryonic and fetal globin genes in both mouse models and human cells during development and thus directly regulates fetal hemoglobin (HbF) switching.14,15,16,17,18,19 BCL11A silences HbF by associating with other known γ-globin transcriptional repressors and binding to the locus control region as well as other intergenic sites, which prevents the interaction between the locus control region and the HbF globin gene required for fetal globin expression.17,20,21,22 Individuals with incomplete hemoglobin switching and elevated HbF levels in adult life (hereditary persistence of HbF) exhibit milder symptomatology of sickle cell disease and β-thalassemia. Genome-wide association studies have revealed that variations in HbF levels are frequently caused by common polymorphisms in the BCL11A gene and the β-globin cluster.15,18,19,23 Knockdown of the BCL11A gene using precise artificially engineered nucleases that enable permanent site-specific genome engineering through the NHEJ DNA repair pathway is an attractive strategy to reactivate HbF and restore functional erythrocytes. In this study, one ZFN pair, two TALEN pairs, and four CRISPR gRNAs, all designed to target exon 2 of the transcriptional repressor of HbF—the BCL11A gene, were evaluated for efficacy, specificity, and toxicity in human cell lines and primary CD34+ cells. We compared the frequencies of indels induced in the BCL11A locus for the three systems in CD34+ cells, and functional reactivation of HbF by high-performance liquid chromatography (HPLC) after in vitro erythroid differentiation. The nuclease specificity was analyzed by investigating predicted off-target events in addition to a genome-wide screen to identify DSBs. Nuclease-treated CD34+ cells were tested in vivo by intravenous transplantation into immune-deficient NOD-PrkdcSCID-IL2Rγnull (NSG) mice. This study demonstrates that component delivery optimization is still required for CRISPR/Cas9 to achieve high genome editing efficiency in CD34+ cells.
Results
Delivery of plasmid and mRNA transcripts of the nucleases results in different toxicity responses and genome editing efficiencies
One ZFN pair, two TALENs, and four CRISPR/Cas9 gRNAs were designed to target exon 2 of the human BCL11A locus to induce HbF expression (Figure 1a, Table 1). To confirm functionality, the seven nucleases were initially evaluated for gene disruption efficiencies of the BCL11A locus after electroporation of the nucleases into the K562 human hematopoietic cell line (Figure 1b). The ZFNs were delivered as in vitro transcribed mRNA (2 μg) transcripts, and the TALENs (2 μg) and CRISPR/Cas9 (4 μg) were delivered as expression plasmids. Administration of all nucleases resulted in high levels of cleavage at the BCL11A locus, as determined by the Surveyor nuclease assay (Figure 1b).
Figure 1.

ZFNs, TALENs, and CRISPR/Cas9 nucleases targeting the human BCL11A gene delivered to human hematopoietic cells via plasmids and RNA. (a) Schematic of engineered nucleases targeting the BCL11A gene. Top: the human BCL11A gene; bottom: exon 2 with the binding recognition sites of the nucleases (zinc finger nuclease (Z), TALENs (T1, T2), and CRISPR/Cas9 guide sequence targets (g1–4)). The nucleases were delivered through electroporation. (b) Representative figure of CEL-1 surveyor assay used to measure the functional activity of the nucleases in the K562 cell line. The type of nuclease used is indicated; negative control (−) was not exposed to a nuclease and the positive control (+) was from a genomic DNA sample previously nuclease-treated and known to have insertions and deletions (indels). The nucleases were delivered at the following concentrations: ZFNs, 2 μg; T1–2, 2 μg; pX330.g1–4, 4 μg. (c) Assessment of expression and toxicity after electroporation of GFP expression plasmid and mRNA by flow cytometry (7-AAD) in CD34+ cells, 1 day post-electroporation, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001. CD34+ cells were either suspended in electroporation buffer but not electroporated (untreated), electroporated without added nucleic acid (Mock (E)) or were electroporated with the indicated amounts of GFP plasmid (black bars) or GFP mRNA (gray bars). (d) Surveyor assay displaying CD34+ cells electroporated with RNA transcripts of ZFN (5 μg), TALENs (5 μg), and gR.1/Cas9 (5 μg/5 μg). CRISPR, clustered regularly interspaced short palindromic repeat; TALEN, transcriptional activator-like effector nuclease; ZFN, zinc finger nuclease.
Table 1. Nuclease types and expression templates.
Initially, we assessed the efficiency and toxicity of electroporation of a GFP expression plasmid versus in vitro transcribed green fluorescent protein (GFP) mRNA into CD34+ cells, determined by flow cytometry (GFP fluorescence and 7-AAD uptake) (Figure 1c). The cytotoxicity of the GFP plasmid was higher (and concentration dependent) compared to the GFP mRNA. Furthermore, expression from the in vitro transcribed mRNA was more efficient than expression from the plasmid. The TALENs and the Cas9 were cloned into a pGEM plasmid and thereafter in vitro transcribed to mRNA; gRNA + scaffold sequences were transcribed to RNA (gR.1) or delivered as a plasmid (pU6.g1) (Table 1). All nucleases were able to induce cleavage of the target locus in CD34+ cells with differing levels of efficiency. The ZFNs induced the highest percentage indels at 27%, the TALENs reached 12% indels, and the gR.1/Cas9 induced only 4% indels (Figure 1d).
CRISPR/Cas9-mediated genome editing in CD34+ cells
Electroporation of the CRISPR/Cas9 plasmid (pX330.g1) that expresses both the gRNA (from a U6 promoter) and the Cas9 mRNA (from a CBh promoter) was examined at the concentrations 1, 2, 5, and 10 μg and compared to codelivery of gRNA expression plasmid (pU6.g1) and Cas9 mRNA. Electroporation of the pX330.g1 plasmid did not result in any cleavage at any of the concentrations tested in bulk CD34+ cells (Figure 2a), which is in concordance with previous results showing that this delivery method may require selection of the nuclease-treated CD34+ cells to detect activity.4 However, codelivery by electroporation of the U6-driven gRNA expression plasmid (pU6.g1) and in vitro transcribed Cas9 mRNA resulted in 10–13% cleavage of the target locus when delivered at the concentrations: pU6.g1(5 μg): Cas9 (5 μg) and pU6.g1(10 μg): Cas9 (10 μg) (Figure 2a). The cytotoxicity induced in CD34+ cells after delivery of plasmid alone (pX330) was assessed by flow cytometry after 1 and 5 days (Supplementary Figure S1a). Electroporation of pX330.g1 or pU6.g1/Cas9 induced a similar percentage of cell death (Supplementary Figures S1a and S3c). Furthermore, electroporation of the in vitro transcribed gR.1 with Cas9 mRNA resulted only in 4–6% cleavage of the target locus, delivered at the concentrations: gR.1(5 μg): Cas9(5 μg) and gR.1(10 μg): Cas9(10 μg) to CD34+ cells (Figure 2a). On the other hand, the toxicity associated with CRISPR/Cas9 delivered as RNA was much lower (17–35% cell death) compared to the pX330 and pU6.g1/Cas9 (Supplementary Figure S3b). Despite the higher toxicity from the pU6.g1/Cas9 compared to gR.1/Cas9, combinatorial delivery of plasmid and mRNA was the most effective mode of delivery for CD34+ cells for disrupting the BCL11A locus.
Figure 2.

Delivery of CRISPR/Cas9 as plasmid, RNA or a combination of Cas9 mRNA and a plasmid expressing gRNA to CD34+ cells. (a) Electroporation of CRISPR/Cas9 plasmid (pX330.g1 at the concentrations: 1, 2, 5, 10 μg), or codelivery of pU6.g1 and Cas9 mRNA at the concentrations: pU6.g1(5 μg): Cas9 (5 μg) and pU6.g1(10 μg): Cas9(10 μg), or electroporation of gR.1 and Cas9 mRNA at the concentrations: gR.1(5 μg): Cas9(5 μg) and gR.1(10 μg): Cas9(10 μg) to CD34+ cells. Representative Surveyor assay showing efficiency of Cas9-mediated cleavage in CD34+ cells. (b) Efficiency of gRNA multiplexing mediated cleavage in CD34+ cells. gRNA multiplexing by expressing two to three copies of the same gRNA target sequence from one plasmid pU6.g2/Cas9 (one copy), pU6.g2.2/Cas9 (two copies), or pU6.g2.2.2/Cas9 (three copies) (n = 3–5). (c) Multiplexing mediated cleavage in CD34+ cells using different gRNAs; pU6.g1/Cas9 (one copy), pU6.g1.2/Cas9 (two copies), or pU6.g1.2.3/Cas9 (three copies) (n = 3–4). CRISPR, clustered regularly interspaced short palindromic repeat; gRNA, guideRNA.
In an attempt to increase the cleavage efficiencies further, we increased the number of gRNA expression cassettes in the plasmid, delivered with the Cas9 mRNA. Because transfection of multiple plasmids can increase cytotoxicity and lower targeting efficiency, we instead produced four additional gRNA expression plasmids containing two or three copies of the same gRNA targeting the BCL11A sequence (pU6.g2.2, pU6.g2.2.2) and two or three gRNA targeting different BCL11A sequences (pU6.g1.2, pU6.g1.2.3). While the cytotoxicity was comparable to the gRNA plasmid expressing one single gRNA (Supplementary Figure S1b,c), none of the multiplexed gRNA plasmids resulted in significant increased cleavage efficiency over the plasmid with the single gRNA (Figure 2b,c and Supplementary Figure S2a–c).
ZFN-mediated cleavage results in higher levels of allelic disruption in CD34+ cells compared to the other nucleases
After demonstrating successful cleavage at the targeted BCL11A locus in CD34+ cells, we titrated the ZFNs (Z1), TALENs (T1), and the two forms of CRISPR/Cas9 (gR.1/Cas9 and pU6.g1/Cas9) to determine the optimal concentration resulting in the highest cleavage efficiency for each of the nucleases. At a concentration of 5 μg/arm, the ZFNs and TALENs produced ~25% and 15% cleavage, respectively (Figure 3a), and resulted in average acute cytotoxicity levels of 47% (range 40–60%) (ZFNs) and 38% (range 35–40%) (TALENs) (Supplementary Figure S3a).
Figure 3.

Determination of nuclease efficiencies and mutation signatures. (a) Electroporation of ZFNs and TALENs to CD34+ cells at various concentrations (n = 4). (b) Electroporation of gR.1 and Cas9 mRNA (n = 3). (c) Electroporation of pU6.g1 and Cas9 mRNA (n = 3–5). (d) (%) Indel frequencies and sizes induced by the respective nucleases (Z: n = 41, T(1): n = 45, T(2): n = 41, pX330.g1: n = 42, pX330.g2: n = 42), pX330.g3: n = 41, pX330.g4: n = 43. (e) (%) Mutations resulting in reading-frame shift. TALEN, transcriptional activator-like effector nuclease; ZFN, zinc finger nuclease.
Delivery of the gR.1/Cas9 and pU6.g1/Cas9 resulted in an average of 5% and 12% cleavage at the targeted locus in CD34+ cells when delivered at the concentrations of 5 μg gRNA and 5 μg Cas9 mRNA (Figure 3b,c). The cytotoxicity induced in CD34+ cells after delivery of gRNA expression plasmid (pU6.g1) or RNA (gR.1) together with Cas9 mRNA was assessed by flow cytometry after 1 day. RNA delivery of gRNA was clearly associated with limited toxicity compared to the gRNA expression plasmid (Supplementary Figure S3b,c).
To compare the DNA repair signatures created by these specific nucleases, we measured the frequencies of different sizes of indels at the target sites in K562 cells. After treating cells with the nucleases, the target region of the BCL11A locus was amplified by polymerase chain reaction (PCR) and the products TOPO cloned and sequenced. We found that the ZFNs were associated with 38% deletions (0–5 bp), 28% deletions (11–20 bp) and also had 10% small insertions (0–5 bp), in contrast to the other nucleases that produced no insertions. The TALENs gave rise to larger lengths of deletions: T(1) 28% (6–10 bp) and 23% (21–30 bp) deletions and T(2) 35% (6–10 bp) and 24% (21–30 bp) deletions. The four CRISPR/Cas9 combinations studied created mostly small deletions (0–5 bp), 69% (pX330.g1), 50% (pX330.g2), 54% (pX330.g3), and 35% (pX330.g4), respectively, although the second CRISPR/Cas9 (pX330.g2) also yielded 25% deletions with the size range 11–20 bp and the fourth CRISPR/Cas9 (pX330.g4) gave rise to 42% deletions with the range of 6–10 bp (Figure 3d). The overall mutations resulting in a reading frameshift ranged between 90% and 53%, where the pX330.g1 resulted in the highest percentage of reading frame shift (90%), followed by the pX330.g2 and the ZFNs (81%). The lowest reading frame shift was caused by the second TALEN pair (T2) (Figure 3e).
Assessment of nuclease specificity
To investigate the specificity of the nucleases, we began by analyzing the predicted off-target effects using the PROGNOS online tool for the ZFNs and TALENs24 (both obligate heterodimers) and the MIT-design web page for the CRISPRs. We designed primers and amplified the top five predicted regions with high homology to the on-target site from the bulk CD34+ population and analyzed cleavage by Surveyor assay. None of the predicted off-target sites for ZFNs and TALENs showed any cleavage activity. For the CRISPR/Cas9, only one potential off-target site was detected when using pX330.g4, displaying 12% cleavage at the top predicted site (Supplementary Table S1).
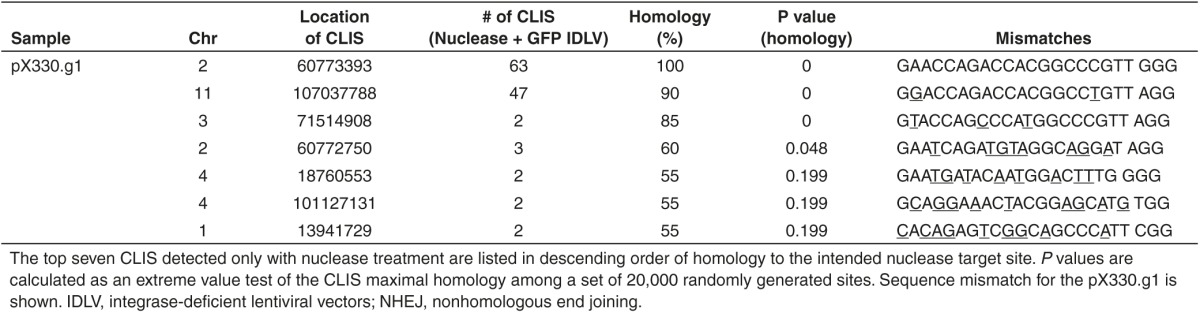
We also investigated off-target cleavage with a less biased technique using NHEJ-mediated trapping of integrase-deficient lentiviral vectors (IDLVs) to find unknown loci with recurrent DSBs.25 Sites where IDLVs were trapped were mapped using nonrestrictive linear amplification-mediated PCR followed by Illumina high-throughput sequencing.26 In the resulting sets of IDLV trapping sites, we looked for 500 bp windows containing more than one unique trapped IDLV, and then searched 1 kb of the hg19 human genome reference sequence centered on the center of mass of the IDLV integrations for the sequence with maximal homology to the nuclease recognition site. The ZFNs displayed high specificity for the BCL11A target locus, as all clustered integration sites (CLIS) identified had maximal homologies that were not significantly higher than those from a set of 20,000 random cluster sites generated in silico (all CLIS P > 0.05, Table 2). This is consistent with the identified CLIS being nearby trapping events by chance, rather than the result of off target nuclease activity. The TALEN pairs yielded five off-target CLIS with maximal homologies significantly higher than those from randomly generated sites (P < 0.05, Table 2). Two of these significant off-target CLIS observed for the TALENs were confirmed by Surveyor nuclease assay, measuring the allelic disruption (Supplementary Figure S4a). pX330.g1 yielded three off-target CLIS with significant maximal homology, and one of these comprised 47 independent trapped IDLV on chromosome 11, a site that was not predicted as a potential off-target site by the MIT CRISPR Design Tool web resource (Table 3). The Surveyor assay revealed 29% allelic disruption at this off-target site, which is a similar level of cleavage as observed at the on-target site (Supplementary Figure S4b).
Table 2. Determination of nuclease specificity through genome-wide off-target site analysis; clustered integration sites (CLIS) within a 500 bp window resulting from NHEJ-mediated trapping of a GFP IDLV were analyzed for ZFNs and TALENs.
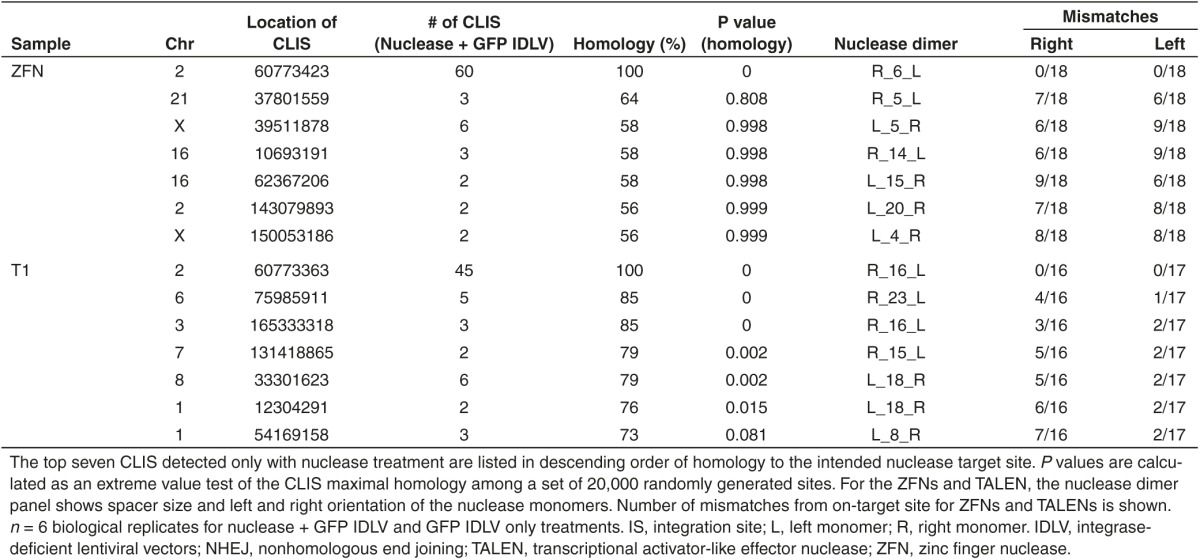
Table 3. Determination of nuclease specificity through genome-wide off-target site analysis; clustered integration sites (CLIS) within a 500 bp window resulting from NHEJ-mediated trapping of a GFP IDLV were analyzed for pX330.g1.
In vitro erythroid differentiation of BCL11A-edited CD34+ cells results in significantly increased HbF with the ZFNs
To assess the functional effects of the disruption created by the nucleases at the targeted BCL11A locus, we differentiated the nuclease-treated CD34+ cells using an established two-phase erythroid differentiation culture model that generates enucleated erythroid cells27,28 (Figure 4a). Surveyor nuclease assay was performed on day 4 of the erythroid differentiation (Figure 4b), revealing indel rates of 25% for ZFNs, 11% for TALENs, 9% for pU6.g1/Cas9, and 5% for gR.1/Cas9.
Figure 4.

In vitro erythroid differentiation of BCL11A-edited CD34+ cells. (a) Experimental design. Nuclease-treated CD34+ cells were subjected to in vitro erythroid differentiation. Samples were taken at the indicated days and stages for end-point testing. Basophilic erythroid (Baso-E), orthochromatic erythroid (Ortho-E). (b) Cleavage frequencies were assessed by the surveyor assay 4 days after electroporation of the ZFNs (Z), the TALENs (T1) nuclease, or the CRISPR/Cas9 using the gRNA expression plasmid (pU6.g1/Cas9) or RNA with Cas9 mRNA (gR.1/Cas9). Mean (±SD), *P < 0.05, **P < 0.01. (c) Flow cytometry measuring lineage markers at day 18–20 of erythroid differentiation. (d) On day 12 of the erythroid differentiation, the cells were lysed and HPLC was performed to determine proportions of HbA and HbF. (e) Quantification of the percentage of HbA and HbF determined by HPLC (n = 3). (f) Flow cytometric analysis of HbF and HbA in erythroid progenitors at day 14 of the erythroid differentiation (n = 3). CRISPR, clustered regularly interspaced short palindromic repeat; HPLC, high-performance liquid chromatography; TALEN, transcriptional activator-like effector nuclease; ZFN, zinc finger nuclease.
To determine whether the erythrocyte differentiation capacity had been altered, surface markers (including: CD45, CD34, CD71 (transferrin receptor), CD235a (Glycophorin A)) and Draq5 staining to verify enucleation of red blood cells were used to monitor the cells appearance and contribution to the populations. Compared to unmodified controls from nontransfected samples, no differences were seen in the cultures from nuclease-treated CD34+ cells (Figure 4c).
Erythroblasts were isolated and globin expression (HbA and HbF) was quantified by HPLC at day 12 and by flow cytometry at day 14. The ZFN nuclease-treated cells displayed a significant increase in HbF, resulting in a significant 4.3-fold increase of HbF expression measured by HPLC and a significant 3.4-fold increase in γ-globin measured by flow cytometry (Figure 4d–f). The TALENs and CRISPR/Cas9 were less effective and revealed no significant increase when measured by HPLC or flow cytometry.
Transplantation of modified CD34+ cells into immune-deficient NSG mice
Finally, we transplanted nuclease-treated CD34+ cells in vivo intravenously into immune-deficient NSG mice. The mice received one million unmodified CD34+ cells (n = 6), or one million viable nuclease-treated CD34+ cells treated with ZFNs (n = 4), TALENs (n = 5), or CRISPR/Cas9 (pU6.g1/Cas9) (n = 4). Cell counts of nuclease-treated cells were assessed by trypan blue on the day after electroporation, showing more cell death induced by the ZFNs and CRISPR/Cas9 compared to the TALENs (Figure 5b), and cleavage efficiency was determined by Surveyor nuclease assay, after 4 days of in vitro culture (Figure 5a).
Figure 5.

Transplantation of nuclease-modified CD34+ cells into immune-deficient NSG mice. NSG mice were irradiated (250 cGy) and transplanted with 1 × 106 unmodified CD34+ cells Mock (n = 6) or 1 × 106 viable gene-modified CD34+ cells treated with ZFNs (Z) (n = 4), TALENs (1) (n = 5), or CRISPR/Cas9 (pU6.g1/Cas9) (n = 4). (a) Surveyor assay determining the frequencies of cleavage in nuclease-treated CD34+ cells after 4 days of in vitro culture. The remainder of the cells were transplanted into irradiated (250 cGy) NSG mice (1E6 viable cells from each nuclease-treated group). (b) Cell counts 1 day post-electroporation (starting cell count number 9 × 106 cells). (c) Indel frequencies associated with the nucleases in peripheral blood (PB) 2 months after transplantation. (d) Engraftment analysis (%hCD45/(%hCD45+%mCD45)) of human cells in PB, 3 months after transplantation. (e) Lineage analysis of human cells in PB, 3 months after transplantation. Myeloid cells (CD33), B-lymphocytes (CD19). (f) Engraftment analysis (%hCD45/(%hCD45+%mCD45)) of human cells in PB, bone marrow (BM), and spleen, 5 months after transplantation. (g) Lineage analysis of human cells in PB, BM, and spleen, 5 months after transplantation. Myeloid cells (CD33), B-lymphocytes (CD19), T-lymphocytes (CD3), and CD34+ cells. (h) Determination of (%) indels measured by the surveyor assay, in PB, BM, and spleen 5 months after transplantation. CRISPR, clustered regularly interspaced short palindromic repeat; TALEN, transcriptional activator-like effector nuclease; ZFN, zinc finger nuclease.
Two months after transplantation, we analyzed the cleavage percentage created by the nucleases in human cells in the peripheral blood (PB) from the transplanted mice (Figure 5c). We detected cleavage only in the human cells that had been treated by ZFNs, ranging from 2% to 10%. No cleavage of the BCL11A gene was detected in the human cells treated by CRISPR/Cas9 and TALENs. Engraftment (%hCD45) and lineage analysis of human cells performed 3 months post-transplantation in PB showed no significant differences between the ZFN-treated group compared to the untreated group. Strikingly, the mice transplanted with pU6.g1/Cas9-treated cells showed no human engraftment in PB at 3 months (Figure 5d,e). Five months post-transplantation, measurement of engraftment levels as the percentage of hCD45+ cells in PB, bone marrow (BM), and spleen and lineage analyses of the human cells were conducted. Once again we observed failure of engraftment of the pU6.g1/Cas9-treated CD34+ cells measured in all three organs (Figure 5f).
The engraftment potential of the ZFN- and TALEN-treated CD34+ cells showed no significant difference compared to untreated CD34+ cells (Figure 5f), and the lineage analysis of these treatment groups showed no skewing in differentiation potential (Figure 5g). Surveyor assay of PB, BM, and spleen was performed 5 months post-transplantation and showed cleavage of the BCL11A gene in ZFN-treated CD34+ cells in BM (3.3–4.3% cleavage) and spleen (1.5–2.1% cleavage) (Figure 5h, Supplementary Figure S5a–c), but no cleavage was observed in PB. Neither of the other two nucleases (TALENs or CRISPR/Cas9) were able to induce cleavage of the BCL11A locus in CD34+ cells capable of engrafting in vivo (Figure 5h, Supplementary Figure S5a–c).
Discussion
Manipulation of the physiologic fetal-to-adult transition of hemoglobin expression via induction of knockdown of HbF repressors may ameliorate clinical manifestations of β-hemoglobinopathies. BCL11A, a direct regulator of HbF switching, was until recently considered a potential clinical target.18,19,20,21,22,23 Emerging evidence indicates that BCL11A knockdown may lead to HSC impairment, as observed in competitive transplantations,29 thus the erythroid-specific BCL11A enhancer might constitute a more suitable clinical target.24,30 Nevertheless, the main focus of this study was not to determine the optimal clinical target, but to investigate and characterize the technical aspects of targeted nucleases directed at the same locus, the BCL11A gene. Whereas mRNA delivery systems, i.e., ZFNs and TALENs, produce high frequencies of nonhomologous chromosomal DNA indels in primary CD34+ cells, the CRISPR/Cas9 has only recently been reported to yield relatively efficient gene modification in human CD34+ cells.3 Previous studies have demonstrated high efficacy of ZFNs and TALENs after electroporation of in vitro transcribed mRNA to CD34+ cells.1,31,32 Data from Mandal et al.4 indicate that delivery of the CRISPR/Cas9 from the expression plasmid pX330 through electroporation to CD34+ cells requires selection by fluorescence-activated cell sorting (FACS) to detect the nuclease-treated cells, due to low gene-editing efficiency in primary HSPCs. Using the same approach of plasmid-mediated CRISPR/Cas9 delivery and expression, but without selection, we were unable to detect any cleavage despite delivery of high concentrations of the plasmid, which confirms low efficiency associated with this strategy. Moreover, while the study by Mandal et al. did not address cytotoxicity induced by DNA delivery to HSPCs, we conducted such analyses and found that acute cytotoxicity induced cell death in ~50% of the cells. Due to the absence of cleavage, we investigated codelivery of plasmid gRNA and Cas9 mRNA, which proved to be a more efficient mode of delivery, as cleavage was detected, however, similar acute cytotoxicity was comparable to plasmid delivery alone. The limited pool of RNA pol III that expresses the gRNA in human cells may explain why we were not able to improve cleavage efficiency by multiplexing gRNA sequences to the BCL11A locus. Multiplexing of CRISPR/Cas9 might require further development of a system where the gRNA is expressed from RNA pol II.33,34 Delivery of gR.1/Cas9 mRNA resulted in ~5% allelic disruption, the lowest of all the nucleases. Nonetheless, RNA delivery of the CRISPR exhibited the lowest toxicity to CD34+ cells. A recent publication by Hendel et al.3 showed increased cleavage efficiency in CD34+ cells as well as T cells after chemically modifying the gRNA in three different ways (2́-O-methyl, 2́-O-methyl 3́́phosphorothioate, 2́-O-methyl 3́thioPACE). Furthermore, Osborn et al.35 also investigated chemical modifications of the gRNA (2́-O-methyl) in Jurkat cells and human T cells and observed higher cleavage efficiency after the chemical modification of the gRNA. While recognizing that efficacy and specificity may vary depending on target sequence, cell type, and delivery method, chemical alterations of the gRNA, protecting the sequence from degradation, could be a promising strategy to increase cleavage efficiencies further in primary CD34+ cells.
In contrast to TALENs and CRISPRs, which only created deletions in this study, the mutation signature of the ZFNs was characterized by the presence of indels. The ZFNs and TALENs used in this study were obligate heterodimers, and thus required both proteins to heterodimerize in order to induce cleavage. Whereas the smaller spacer size of ZFNs (5–6 bp) might avert further cleavage after an indel has been created due to prevention of heterodimerization, the TALENs, with a larger spacer size (12–21 bp), may recleave the DNA despite being farther apart. It has been suggested that ZFNs induce more defined overhangs that get filled prior to NHEJ and this produces small insertions.36 The TALENs gave rise to larger deletions compared to ZFNs and CRISPRs, which most commonly induced small deletions. The different mutation signatures were associated with differences in the frequency of reading-frame shifts. The nuclease pX330.g1 had the highest frequency of small deletions and gave rise to 90% frameshift mutations. The pX330.g2 and the ZFNs, both nucleases displaying similar mutation signatures (large proportion of 0–5 bp and 11–20 bp), both gave rise to 81% frameshift mutations. Furthermore, the cleavage efficiencies differed among the nucleases; the studied ZFN pair had the highest cleavage efficiency, as confirmed by functional assessment in erythroid differentiation. At day 12 of differentiation, we observed a fourfold increase in HbF expression as shown by HPLC and flow cytometry. The TALENs and CRISPR-modified populations resulted in a modest, nonsignificant increase in HbF expression, measured by HPLC.
ZFN and TALEN gene-edited CD34+ cells had the capacity to engraft in NSG mice, while retaining multi-lineage potential after 5 months. However, only ZFN gene-edited CD34+ cells exhibited gene modification after engraftment in vivo. Despite BCL11A being essential for lymphoid development,37,38 the observed knockdown of BCL11A in bulk BM cells (4%) (Figure 5b) did not alter levels of B lymphoid cells in the ZFN-treated group. This could be due to the low-to-absent gene modification levels of the sorted B cells from the ZFN-treated BM cells (0–3.7%) (Supplementary Figure S6). Strikingly, when the plasmid-mediated CRISPR nuclease-treated CD34+ cells were transplanted into NSG mice, they failed to successfully engraft; the toxicity caused by plasmid delivery of the gRNA prevented engraftment of CRISPR-treated HSPCs in the mice, indicating that such a delivery method is not appropriate for HSPCs.13,39
Previously, the specificity of target site recognition has been studied extensively in ZFN-based technology25 and CRISPR-based systems,40,41,42 but less so for TALEN-based approaches. Overall, evidence suggests that every targeted locus is different and requires individual evaluation for off-target cleavage. While it is natural to commence with analyses of predicted off-targets to guide nuclease selection using web-tools, previous studies illustrate the need for unbiased genome-wide analyses to determine the true specificity.42 In this comparison study, we examined both in silico predicted off-targets and genome-wide analysis. The ZFN pair showed no significant off-target activity in either of these analyses. For the TALEN pair and the CRISPR, the less biased IDLV trapping based approach revealed significant off-target cleavage, while the top five off-targets predicted in silico incurred no DSBs upon nuclease treatment. For the CRISPR, the IDLV trapping analysis found 47 integration sites at chromosome 11, a target site that was not predicted by the CRISPR Design Tool web-based software. Hence, global unbiased identification of DSBs is warranted to properly evaluate nuclease safety.
The ZFNs used for this study were identified via screening of a small panel of related designs for gene modification activity at the target shown in Figure 1a, as well as binding specificity as gauged via SELEX. In contrast, our TALENs and Cas9 nucleases were generated via assembly and testing of single reagents, since at the time of these studies the standard engineering principles for these platforms did not provide for alternative designs. More recently, variations to these platforms have been developed that afford greater design complexity, including alternative Cas9 sequences as well as an expanded repertoire of repeat variable biresidue (RVDs) for improved TALEN function.43,44,45 It is possible that the use of these platform variations, coupled with a small-scale screening effort, could have yielded TALENs or CRISPR/Cas9 nucleases with improved properties.
In conclusion, our data contributes to an emerging appreciation of the need to further investigate ways to optimize delivery of the CRISPR/Cas9 to CD34+ cells and shows that careful assessment of different gRNA sequences are required due to high variability of cleavage efficiencies in different loci and different types of cells.
Materials and Methods
Cell lines and culture. K562 cells (ATCC, Manassas, VA) were cultured in RPMI 1640 (Cellgro, Manassas, VA) supplemented with Penicillin/Streptomycin/l-glutamine (100 units/ml) (Gemini Bio-products, West Sacramento, CA) and 10% fetal bovine serum (FBS) (Gemini Bio-products). HEK293T cells (ATCC) were cultured in Dulbecco's Modified Eagle's Medium (Thermo Fisher, Grand Island, NY) supplemented with 1% Penicillin/Streptomycin/l-glutamine (100 units/ml) and 10% FBS. Adult human mobilized PB apheresis was purchased from the Division of Experimental Hematology and Cancer Biology, Cincinnati Children's Hospital Medical Center (Cincinnati, OH). CD34+ cells were isolated using the MACS CD34+ enrichment kit (Miltenyi Biotec, Auburn, CA) and the CliniMACS system (Miltenyi Biotec), resulting in a purity of 96–98%. Human PB CD34+ cells were routinely cultured in medium containing X-VIVO 15 (Lonza, Walkersville, MD) supplemented with Penicillin/Streptomycin/l-glutamine and the cytokines recombinant human Stem Cell Factor (50 ng/µl), recombinant human thrombopoietin (TPO) (50 ng/µl), and Recombinant Human Flt3-Ligand (50 ng/µl) (Peprotech, Rocky Hill, NJ). The cells were cultured for 1–3 days prior to electroporation.
Transfection. Nucleofection of K562 cells was performed using a Nucleofector 4D (Lonza). 2 × 105 K562 cells (alternatively 1 × 106) were centrifuged (90g) for 15 minutes and cell pellets resuspended in 100 μl of SF solution (Lonza) together with the DNA or RNA encoding the nuclease. The cells were thereafter nucleofected using the recommended program FF120 and transferred into a 24-well (alternatively a 6-well) tissue culture plate (Corning, Corning, MA) containing culture medium (RPMI 1640) supplemented with Penicillin/Streptomycin/l-glutamine (100 units/ml) and 10% FBS and moved into the 37 °C incubator (NuAire, Plymouth, MN). Electroporation of PB CD34+ cells was performed using the ECM 830 Electroporation System (Harvard Apparatus, Holliston, MA). 2 × 105 cells were centrifuged (90g) for 15 minutes and cell pellets resuspended in 100 μl of BTX solution (Harvard Apparatus) together with the DNA or mRNA encoding the nuclease. The cells were thereafter electroporated using 250 V, 5 ms, 1 pulse, and transferred into a 24-well tissue culture plate (Corning) containing X-VIVO 15 (Lonza) supplemented with Penicillin/Streptomycin/l-glutamine and the cytokines recombinant human SCF (50 ng/μl), recombinant human TPO (50 ng/μl), and Recombinant Human Flt3-Ligand (50 ng/μl) (Peprotech).
Surveyor nuclease assay. Genomic DNA was isolated using the PureLink Genomic DNA mini kit (Life Technologies). To determine allelic disruption, a 410 bp region surrounding the binding sites of the nucleases in BCL11A was amplified using PCR (Applied Biosystems/Life Technologies, Carlsbad, CA). The PCR reaction was performed using Accuprime Taq HiFi (Life Technologies) and the forward primer (5́-GGTGGGATGGCATGGGGTTGAGA-3́) and reverse primer (5́-CAGGTCACGCCAGAGGATGACG-3́) (94 °C for 2 minutes, then 30 cycles of 94 °C for 30 seconds, 62 °C for 30 seconds, and 68 °C for 1 minute, 68 °C for 5 minutes and finally 4 °C). PCR products were diluted 1:2 in 1× Accuprime buffer and thereafter denatured and reannealed (94 °C for 10 minutes, 85 °C (cooling at −2 °C per second) and cooling to 25 °C (−0.1 °C per second)). Finally, the PCR product was digested with 1 μl of Surveyor Nuclease (Transgenomic, Omaha, NE) at 42 °C for 15 minutes and run on an 8% TBE polyacrylamide gel (Life Technologies) at 120 V. The gel was stained with Gel Green (Phenix Research Products, Candler, NC) (1:10) in 1× TBE for 20 minutes and imaged on a Kodak Molecular Imaging Station (Kodak, Rochester, NY). Allelic disruption was determined by densitometry using the formula: % Allelic disruption = 100 × (1−√(intensity of the uncut band/total intensity of cut and uncut bands)).
In vitro erythroid differentiation. The erythroid differentiation protocol is adapted from Giarratana et al.28 as described in Romero et al.27 The PB CD34+ cells were prestimulated for 3 days in X-VIVO 15 (Lonza) supplemented with Penicillin/Streptomycin/l-glutamine and the cytokines recombinant human Stem Cell Factor (50 ng/μl), recombinant human TPO (50 ng/μl), and Recombinant Human Flt3-Ligand (50 ng/μl) (Peprotech) prior to electroporation and thereafter transferred into erythroid medium (Iscove's Modified Dulbecco's Medium (Life Technologies) (1× glutamine, penicillin, and streptomycin)) supplemented with 10% bovine serum albumin, 40 μg/ml inositol, 10 μg/ml folic acid, 1.6 μmol/l monothioglycerol, 120 μg/ml transferrin, and 10 μg/ml insulin (Sigma-Aldrich, St. Louis, MO). The cells were cultured with 10−6 mol/l hydrocortisone (Sigma-Aldrich), 100 ng/ml hSCF, 5 ng/ml hIL-3, and 3 IU/ml erythropoietin (Epo) (Janssen Pharmaceuticals, Titusville, NJ) for the first 6 days. The following 3 days, the cells were cocultured with stromal cells (MS-5, murine stromal cell line46) and only Epo (3 IU/ml) was added to the erythroid medium. From day 11 to day 21, all the cytokines were removed from the basic erythroid medium. HPLC of hemoglobin tetramers in erythroblasts at day 12 of culture was performed as described.31
Chromosomal indel analysis. Genomic DNA was collected from K562 cells electroporated with the respective nucleases after 3 days, and PCR was performed to amplify the region surrounding the cut site in BCL11A (Fwd 5́-GGTGGGATGGCATGGGGTTGAGA-3́ and Rev 5́-CAGGTCACGCCAGAGGATGACG-3́). The PCR product was cloned into a Topo TA cloning vector (TOPO TA cloning kit from Invitrogen, Carlsbad, CA) and transformed into Top10 cells. The colonies were screened using X-gal to enrich for clones with the PCR product. Plasmid isolation was performed using the Purelink Quick Plasmid Miniprep Kit (Invitrogen) and sent for sequencing analysis to Laragen (Culver City, CA). A plasmid Editor (Ape) by M. Wayne Davis was used for sequence alignments.
Predicted and genome-wide off-target analysis. The predicted off-target sequences for the four CRISPR/Cas9 were assessed using the Zhang CRISPR Design online Tool.40 The off-target sequences for TALENs and ZFNs were assessed using the Bao lab online PROGNOS (Predicted Report of Genome-wide Nuclease Off-target Sites) tool.24 Primers were designed for regions with the highest likelihood of having off-target cleavage for each nuclease tested, and Cel-1 assays were performed of these sites on genomic DNA previously isolated from electroporation experiments in K562 cells and CD34+ PBSCs.
The IDLV trapping protocol was adapted from Gabriel et al.25 A titration of the nucleases with an IDLV expressing GFP (1.55E + 09 TU/ml) was performed at an multiplicity of infection (MOI) of 3, 30, and 100 to determine the optimal amount of IDLV to be trapped by NHEJ at nuclease-induced DSB with minimal background capture in control samples. Six independent replicates of K562 (2 × 105) cells were electroporated with optimized amount of nuclease and GFP IDLV was added at an MOI of 30 to the post-electroporation recovery medium: RPMI 1640 (Cellgro, Tewksbury, MA) supplemented with 1% Penicillin/Streptomycin/l-glutamine (100 units/ml) (Gemini Bio-products) and 10% FBS (Gemini Bio-products). Six control replicates were performed with only IDLV (MOI 30) added to determine the background capture rate and to distinguish between DSBs caused by nuclease activity and DSBs occurring at fragile sites in the genome. Five days post-electroporation, we confirmed that cleavage occurred at the target sites in the nuclease + IDLV samples by the Surveyor nuclease assay, but not in the control samples that received no nuclease. The cells were subsequently cultured for 7 weeks. Each week, the percentage and mean fluorescent intensity of GFP-positive cells were analyzed by flow cytometry. Additionally, the vector copies per cell (VC/cell) were determined by digital PCR to ensure elimination of nontrapped GFP IDLV. Genomic DNA for digital PCR was isolated using the PureLink Genomic DNA Mini Kit (Invitrogen). The average VC/cell was determined by duplex droplet digital PCR with normalization to the autosomal locus uc378 to calculate the average VC/cell. After 50 days, nontrapped IDLV was sufficiently diluted out of the sample and GFP-positive cells were sorted by FACS for genomic DNA extraction. A biotinylated primer designed to the U5 region of the IDLV LTR was used to selectively amplify 3ʹ vector–genome junctions by nonrestrictive linear amplification-mediated PCR.26,47 Libraries were then prepared for high-throughput Illumina paired-end 100 sequencing. Vector integration sites were analyzed using a custom Python wrapper for the Bowtie2 aligner, and CLIS were defined as two or more integration sites in a sliding 500 bp window. Any CLIS found in nuclease + IDLV samples that did not overlap CLIS in IDLV only control samples were considered to be potential off-target sites for further analysis.
Mice. NOD/SCID IL2R gamma−/− (NSG) mice obtained from the Jackson Laboratory (Bar Harbor, ME) were maintained by the UCLA Department of Laboratory Animal Medicine under protocols reviewed and approved by the UCLA Chancellor's Animal Research Committee (ARC# 2008–167).
1 × 106 CD34+ cells were electroporated with the respective nucleases, as previously described, and transplanted intravenously by tail vein injection, 1-day post-electroporation, into irradiated (250 cGy) NSG recipient mice. Engraftment and lineage analyses of PB, BM, and spleen were performed. Antibodies directed against hCD45-V450 and mCD45-FITC were used for engraftment analysis and CD19-PeCy7, CD3-PerCP, CD33-PE, and CD34-APC were used for lineage analysis. Genomic DNA was prepared as previously described to assess the BCL11A disruption.
Statistical analysis. For measurements related to efficiency and toxicity, descriptive statistics such as mean, standard deviation, count, and percentage were summarized and presented in figures and tables. Statistical significance of comparison between two experimental arms was determined by two-sided unpaired t-test. P < 0.05 was considered statistically significant. Statistical analyses were carried out using GraphPad Prism version 6.00 for Mac OS X (GraphPad Software, San Diego, CA, www.graphpad.com). In the bioinformatic analysis of nuclease specificity assessment, an extreme value test was used for testing maximal homology. Specifically, a set of 20,000 random sites was generated in silico and 1 kb windows for these sites were searched for maximal homologies to the recognition site of the nuclease being considered. P values were calculated by determining the percentile within the 20,000 maximal homologies of random sites at which the CLIS maximal homology fell. For example, if a CLIS maximal homology would rank 200th in the list of random site homologies sorted in descending order, it would receive a P value of 0.01 (200/20,000).
SUPPLEMENTARY MATERIAL Figure S1. Short-term cytotoxicity of CRISPR/Cas9 delivered as plasmid alone or plasmid and RNA codelivery in CD34+ cells. Figure S2. Multiplex genome editing in CD34+ cells. Figure S3. Short-term cytotoxicity of the nucleases Z, T1, and CRISPR/Cas9 in CD34+ cells. Figure S4. Off-target allelic disruption for the top significant CLIS induced by T1 and CRISPR/Cas9 in K562 cells. Figure S5. Allelic disruption of PB, BM, and spleen from transplanted NSG mice. Figure S6. Frequency of cleavage in sorted subpopulations in BM from transplanted NSG mice. Table S1. Predicted off-target activity of the nucleases.
Acknowledgments
The authors thank Sangamo Biosciences for the ZFNs targeting the BCL11A locus. This work has been supported by an Innovations in Clinical Research Award from the Doris Duke Charitable Foundation (2013158) (not used for the animal work), NIH (PO1 HL73104) and a generous donation by the Shaffer Family Foundation, and the Swedish Tegger Foundation. The authors have no financial conflicts of interest.
Supplementary Material
References
- Holt, N, Wang, J, Kim, K, Friedman, G, Wang, X, Taupin, V et al. (2010). Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28: 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel, A, Bak, RO, Clark, JT, Kennedy, AB, Ryan, DE, Roy, S et al. (2015). Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol 33: 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, PK, Ferreira, LM, Collins, R, Meissner, TB, Boutwell, CL, Friesen, M et al. (2014). Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15: 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov, FD, Miller, JC, Lee, YL, Beausejour, CM, Rock, JM, Augustus, S et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651. [DOI] [PubMed] [Google Scholar]
- Kim, HJ, Lee, HJ, Kim, H, Cho, SW and Kim, JS (2009). Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19: 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak, T, Doyle, EL, Christian, M, Wang, L, Zhang, Y, Schmidt, C et al. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, JC, Tan, S, Qiao, G, Barlow, KA, Wang, J, Xia, DF et al. (2011). A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148. [DOI] [PubMed] [Google Scholar]
- Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE et al. (2013). RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z, Troilo, PJ, Wang, X, Griffiths, TG, Pacchione, SJ, Barnum, AB et al. (2004). Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther 11: 711–721. [DOI] [PubMed] [Google Scholar]
- Gurunathan, S, Klinman, DM and Seder, RA (2000). DNA vaccines: immunology, application, and optimization*. Annu Rev Immunol 18: 927–974. [DOI] [PubMed] [Google Scholar]
- Shimokawa, T, Okumura, K and Ra, C (2000). DNA induces apoptosis in electroporated human promonocytic cell line U937. Biochem Biophys Res Commun 270: 94–99. [DOI] [PubMed] [Google Scholar]
- Xu, J, Peng, C, Sankaran, VG, Shao, Z, Esrick, EB, Chong, BG et al. (2011). Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 334: 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda, M, Galanello, R, Sanna, S, Lettre, G, Sankaran, VG, Chen, W et al. (2008). Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA 105: 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran, VG, Xu, J, Ragoczy, T, Ippolito, GC, Walkley, CR, Maika, SD et al. (2009). Developmental and species-divergent globin switching are driven by BCL11A. Nature 460: 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran, VG, Menne, TF, Xu, J, Akie, TE, Lettre, G, Van Handel, B et al. (2008). Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322: 1839–1842. [DOI] [PubMed] [Google Scholar]
- Menzel, S, Garner, C, Gut, I, Matsuda, F, Yamaguchi, M, Heath, S et al. (2007). A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 39: 1197–1199. [DOI] [PubMed] [Google Scholar]
- Lettre, G, Sankaran, VG, Bezerra, MA, Araújo, AS, Uda, M, Sanna, S et al. (2008). DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA 105: 11869–11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J, Sankaran, VG, Ni, M, Menne, TF, Puram, RV, Kim, W et al. (2010). Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev 24: 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawaid, K, Wahlberg, K, Thein, SL and Best, S (2010). Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells Mol Dis 45: 140–146. [DOI] [PubMed] [Google Scholar]
- Tolhuis, B, Palstra, RJ, Splinter, E, Grosveld, F and de Laat, W (2002). Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 10: 1453–1465. [DOI] [PubMed] [Google Scholar]
- Sedgewick, AE, Timofeev, N, Sebastiani, P, So, JC, Ma, ES, Chan, LC et al. (2008). BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis 41: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine, EJ, Cradick, TJ, Zhao, CL, Lin, Y and Bao, G (2014). An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res 42: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823. [DOI] [PubMed] [Google Scholar]
- Paruzynski, A, Arens, A, Gabriel, R, Bartholomae, CC, Scholz, S, Wang, W et al. (2010). Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat Protoc 5: 1379–1395. [DOI] [PubMed] [Google Scholar]
- Romero, Z, Urbinati, F, Geiger, S, Cooper, AR, Wherley, J, Kaufman, ML et al. (2013). β-globin gene transfer to human bone marrow for sickle cell disease. J Clin Invest (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Giarratana, MC, Kobari, L, Lapillonne, H, Chalmers, D, Kiger, L, Cynober, T et al. (2005). Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat Biotechnol 23: 69–74. [DOI] [PubMed] [Google Scholar]
- Guda, S, Brendel, C, Renella, R, Du, P, Bauer, DE, Canver, MC et al. (2015). miRNA-embedded shRNAs for lineage-specific BCL11A knockdown and hemoglobin F induction. Mol Ther 23: 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver, MC, Smith, EC, Sher, F, Pinello, L, Sanjana, NE, Shalem, O et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, MD, Cost, GJ, Mendel, MC, Romero, Z, Kaufman, ML, Joglekar, AV et al. (2015). Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 125: 2597–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese, P, Schiroli, G, Escobar, G, Di Tomaso, T, Firrito, C, Calabria, A et al. (2014). Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510: 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim, L, Perli, SD, Fridkin, A, Perez-Pinera, P and Lu, TK (2014). Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell 54: 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma, T, Nishikawa, A, Kume, S, Chayama, K and Yamamoto, T (2014). Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep 4: 5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn, MJ, Webber, BR, Knipping, F, Lonetree, CL, Tennis, N, DeFeo, AP et al. (2016). Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther 24: 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y, Kweon, J and Kim, JS (2013). TALENs and ZFNs are associated with different mutation signatures. Nat Methods 10: 185. [DOI] [PubMed] [Google Scholar]
- Liu, P, Keller, JR, Ortiz, M, Tessarollo, L, Rachel, RA, Nakamura, T et al. (2003). Bcl11a is essential for normal lymphoid development. Nat Immunol 4: 525–532. [DOI] [PubMed] [Google Scholar]
- Yu, Y, Wang, J, Khaled, W, Burke, S, Li, P, Chen, X et al. (2012). Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med 209: 2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiyoshi, T, Holt, NG, Hollis, RP, Ge, S, Cannon, PM, Crooks, GM et al. (2009). Stable transgene expression in primitive human CD34+ hematopoietic stem/progenitor cells, using the Sleeping Beauty transposon system. Hum Gene Ther 20: 1607–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, PD, Scott, DA, Weinstein, JA, Ran, FA, Konermann, S, Agarwala, V et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Foden, JA, Khayter, C, Maeder, ML, Reyon, D, Joung, JK et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, JC, Zhang, L, Xia, DF, Campo, JJ, Ankoudinova, IV, Guschin, DY et al. (2015). Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat Methods 12: 465–471. [DOI] [PubMed] [Google Scholar]
- Zetsche, B, Gootenberg, JS, Abudayyeh, OO, Slaymaker, IM, Makarova, KS, Essletzbichler, P et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163: 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver, BP, Pattanayak, V, Prew, MS, Tsai, SQ, Nguyen, NT, Zheng, Z et al. (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529: 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, J, Fujita, J, Taniguchi, S, Sugimoto, K and Mori, KJ (1992). Characterization of murine hemopoietic-supportive (MS-1 and MS-5) and non-supportive (MS-K) cell lines. Leukemia 6: 452–458. [PubMed] [Google Scholar]
- Candotti, F, Shaw, KL, Muul, L, Carbonaro, D, Sokolic, R, Choi, C et al. (2012). Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood 120: 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.