

Abstract
Development of RNA and DNA aptamers for diagnostic and therapeutic applications is a rapidly growing field. Aptamers are identified through iterative rounds of selection in a process termed SELEX (Systematic Evolution of Ligands by EXponential enrichment). High-throughput sequencing (HTS) revolutionized the modern SELEX process by identifying millions of aptamer sequences across multiple rounds of aptamer selection. However, these vast aptamer HTS datasets necessitated bioinformatics techniques. Herein, we describe a semiautomated approach to analyze aptamer HTS datasets using the Galaxy Project, a web-based open source collection of bioinformatics tools that were originally developed to analyze genome, exome, and transcriptome HTS data. Using a series of Workflows created in the Galaxy webserver, we demonstrate efficient processing of aptamer HTS data and compilation of a database of unique aptamer sequences. Additional Workflows were created to characterize the abundance and persistence of aptamer sequences within a selection and to filter sequences based on these parameters. A key advantage of this approach is that the online nature of the Galaxy webserver and its graphical interface allow for the analysis of HTS data without the need to compile code or install multiple programs.
Keywords: aptamer, high-throughput sequencing, next-generation sequencing, SELEX
Introduction
Aptamers are small (20–100 nucleotides) DNA or RNA oligonucleotides that specifically bind target molecules with high affinity. Current aptamer selections utilize the SELEX (Systematic Evolution of Ligands by EXponential enrichment) process1,2 to enrich a library of 1012–1036 aptamers for sequences that are highly specific for a target of interest (Figure 1). Enrichment of aptamers is accomplished through iterative rounds of positive and negative selection pressure, leading to the accumulation and persistence of selected aptamers. A major advance in the SELEX process, high-throughput SELEX, is the application of high-throughput sequencing (HTS) to identify millions of sequences across multiple rounds of selection.3,4 HTS is particularly well suited for aptamer selections because the entire sequence of an aptamer is contained within each HTS read. However, the large datasets associated with aptamer HTS necessitated advanced bioinformatics methods in order to narrow down the millions of sequences to a selective few candidate aptamers to test experimentally.
Figure 1.

Schematic of SELEX process and the pipeline using Galaxy Workflows to analyze aptamer high-throughput sequencing (HTS) data. The SELEX process is used to enrich an aptamer library for aptamers sequences specific to a target using a combination of negative and positive selection pressure. Selection rounds may be multiplexed and sequenced using HTS methods. The HTS aptamer HTS datasets is first preprocessed then used to build a nonredundant database (NrD) of aptamer sequences. The aptamers within the NrD may then be analyzed for abundance and persistence to filter selected from nonselected aptamers. See Supplementary Materials for screenshots of Galaxy Workflows along with step-by-step examples of using the Workflows within Galaxy.
The Galaxy Project Webserver,5,6,7 originally created for analysis of genomic HTS data, is a collection of free bioinformatics tools that are powerful, flexible, dynamic, easy to use, and accessible using any web browser, including mobile devices. A visual scripting language within the Galaxy Webserver allows for the creation of an automated analysis process, or “Workflow.” A Galaxy Workflow can be shared and executed with user-defined data and parameters. We previously reported the use of individual Galaxy tools to analyze aptamer HTS datasets.8 However, use of these Galaxy tools for analysis of aptamer HTS data requires a sophisticated understanding the individual tools within Galaxy, and in what order to run the various tools to properly manipulate the data files. To allow the average researcher with no training in bioinformatics to analyze aptamer HTS data, we have now created Galaxy Workflows that adapt existing Galaxy Webserver tools to achieve semiautomated aptamer HTS data analysis (Figure 1).
We developed seven Galaxy Workflows to accomplish three steps of aptamer HTS data analysis: preprocessing, nonredundant database, and abundance/persistence analyses (Figure 1). The first step, preprocessing, utilized two Galaxy Workflows (Preprocess Workflow 1, Preprocess Workflow 2) to separate multiplexed HTS FASTA data, remove adapter sequences, remove constant region sequences, isolate variable region sequences of a defined nucleotide length, and collapse duplicate reads. Next, the NrD Workflow was used to compile multiple rounds of selection into a single database of unique aptamer sequences, termed a “Nonredundant Database” or “NrD.” For each aptamer sequence, the NrD included a measure of the aptamer persistence throughout the selection (round representation) and a measure of aptamer abundance from each selection round (read count). Two additional Galaxy Workflows (Abundance NrD Analysis Workflow, Persistence NrD Analysis Workflow) were used to analyze the persistence and abundance of aptamers within the NrD. Lastly, Abundance NrD Filter Workflow and Persistence NrD Filter Workflow were used to filter selected aptamers from nonselected aptamers within the NrD. Together, this pipeline of Galaxy Workflows will allow users with limited bioinformatics experience to analyze aptamer HTS data and obtain a manageable, organized database of candidate aptamer sequences.
Results
Our objective was to validate that Galaxy Workflows can be used for the semiautomated analysis aptamer HTS selection data (Figure 1). We used an example HTS Aptamer Dataset that consists of 2,076,187 reads representing six rounds of aptamer selection (Rounds 1, 3, 5, 6, 7, and 8) and the nonselected starting aptamer library (Round 0).8 After running Preprocess Workflow 1, Preprocess Workflow 2, and NrD Workflow: 7 round, we identified 913,347 unique aptamers. The sequences were compiled within the NrD with read counts ranging from 1 read to 272,799 reads and round representation from 1 round to 7 rounds. Read count refers to the number of duplicate reads of an aptamer sequence within each round of selection, and round representation refers to the total number of rounds in which an aptamer sequence is observed.
We next separated selected from nonselected sequences by analyzing the abundance and persistence of the aptamers contained within the NrD. Aptamers that have been selected will accumulate during the SELEX process. This increase in abundance is reflected in the read count of an aptamer across selection rounds. Additionally, aptamers that have been selected will exhibit persistence across multiple rounds of selection, which is measured by round representation (i.e., the number of rounds in which an aptamer appears). HTS data from the starting aptamer library, Round 0, served as a control for nonselected aptamers. We compared aptamer abundance and persistence in Rounds 1, 3, 5, and 8 to Round 0 using the Abundance NrD Analysis Workflow: Compare 5 rounds and the Persistence NrD Analysis Workflow: Compare 5 rounds (Figure 2).
Figure 2.

Abundance and persistence analysis of nonredundant database (NrD). (a) Plotting data from the Abundance NrD Analysis Workflow and the (b) Persistence NrD Analysis Workflow. Read count indicates the number of duplicate reads of an aptamer sequence within a selection round, and round representation refers to the number of rounds in which the aptamer sequence is observed.
The abundance analysis indicated 99.9% aptamers (282,486 of 282,849) contained within the nonselected Round 0 had a single read count (Figure 2a and Supplementary Figure S9C). The nonselected Round 0 only had a few hundred aptamers with read counts of 2, <100 aptamers with a read count of 3, and <10 aptamers with a read count of ≥4. HTS data from the selected rounds (Rounds 1, 3, 5, and 8) exhibited a significant increase in the number of aptamer sequences with read counts of ≥4. From these data we concluded that a selected aptamer sequence must have a read count >2.
With the persistence analysis, the nonselected Round 0 data exhibited a striking trend, with <10 nonselected aptamers having a round representation of ≥2 (Figure 2b and Supplementary Figure S10C). These data indicate that <10 aptamer sequences of the 282,849 aptamer sequences in Round 0 were observed in any of the subsequent selection rounds. Alternatively, the selected rounds contained a significant number of aptamers that persisted throughout most of the other rounds of selection. From these data, we determined that a selected aptamer required a round representation of ≥2.
We next filtered the NrD based upon the abundance and persistence analysis using the Abundance NrD Filter Workflow (>2 reads) and the Persistence NrD Filter Workflow (≥2 rounds), respectively. After running both Workflows, the NrD was reduced from 913,347 unique sequences (representing 2,076,187 reads) to 1,284 selected sequences (representing 1,103,172 reads). Thus, these Galaxy Workflows filtered 99.86% of the nonselected aptamer sequences from the NrD. This analysis and filtering was sufficient to allow for importing data into spreadsheet software.
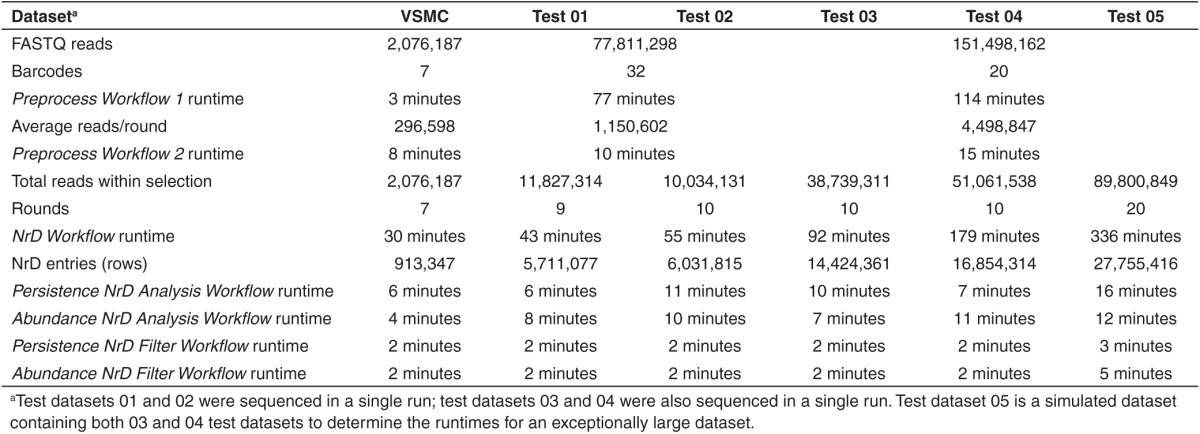
Since the example dataset contains ~2 million reads and recent advances in HTS yield >50 million reads, we examined the scalability and runtimes of the Galaxy Workflows with increasingly larger FASTQ datasets of up to ~151 million reads (Table 1). Preprocess Workflow 1 runtime for these FASTQ dataset was 77–114 minutes, yielding an average of 1.1–4.5 million reads per selection round. These selection rounds were then processed by Preprocess Workflow 2 in 10–15 minutes per round. The NrD Workflows processed selections containing 7–20 rounds into NrDs in 43–336 minutes. The resultant NrDs were comprised of 5.7–27.7 million entries (rows). Analysis of these NrDs by the Persistence Analysis NrD Workflow and Abundance NrD Analysis Workflow took 6–16 minutes and 7–12 minutes, respectively. The Persistence NrD Filter Workflow and Abundance NrD Filter Workflow filtered the NrDs in 2–5 minutes and 2–3 minutes, respectively. All Galaxy Workflows runtimes exhibited a linear relationship with the number of reads analyzed (Supplementary Figure S12). These data support that the Galaxy Workflows for aptamer HTS analysis have both efficient runtimes and are scalable with FASTQ datasets containing hundreds of millions of reads.
Table 1. Galaxy Workflow runtimes.
Discussion
Aptamer HTS bioinformatics is a growing and expanding field9 addressing an important need to facilitate the development of aptamers as diagnostic and therapeutic molecules. Key advances in aptamer bioinformatics include the separation of selected aptamers from nonselected aptamers,4 clustering aptamers based upon sequence4,10,11 or structural similarity,4 identifying sequence/structure motifs,9,12,13,14,15,16 and using fold enrichment4,17,18 as a guide to identify candidate aptamers. New strategies to analyze aptamer HTS data are constantly being developed, updated, and validated.
All of these tools, like using Galaxy, include advantages and disadvantages. The Galaxy Project's free public webserver is an ideal platform for aptamer HTS bioinformatics with several key advantages. These include immediate implementation, reliable updating due to multiinstitutional and National Institutes of Health support, an extensive community support through the Galaxy wiki, flexible and powerful tools, and ease in sharing data and data analysis. Indeed, we previously used Galaxy Webserver tools for analysis of aptamer HTS data,8 yet this required a significant degree of expertise in Galaxy tool execution for proper data manipulation. Moreover, for the more complex aptamer HTS data analysis, the proper execution of each individual Galaxy tool can become very confusing and time-consuming. To address these issues, we have now created Galaxy Workflows for semiautomated aptamer HTS data preprocessing, generation of an aptamer NrD, and subsequent analysis with filtering (Figure 1). A key advantage of this approach is that users can run the Galaxy Workflows quickly and efficiently without a sophisticated understanding of how to use the individual Galaxy Webserver tools.
We demonstrate that these Workflows can be used to analyze aptamer abundance and persistence, two properties exhibited by selected aptamers during the SELEX process. Selected aptamers, as compared to nonselected aptamers, will both accumulate and persist throughout the SELEX process. We show that analyzing the abundance and persistence of aptamers using Galaxy Workflows can rapidly and efficiently isolate selected aptamers and eliminate nonselected aptamers. Since abundance and persistence analyses are highly dependent on the size of the dataset, the appropriate cutoff values should be determined empirically for each dataset.
We envision aptamer researchers using Galaxy and Galaxy Workflows to analyze aptamer HTS data as a first pass analysis to identify candidate aptamer sequence for further experimental validation and for more in-depth aptamer bioinformatics analysis, such as sequence/structure clustering and motif identification. Filtering aptamer HTS data of nonselected aptamers, by analyzing aptamer abundance and persistence, will likely improve the results of subsequent, more in-depth analyses. The availability and accessibility of Galaxy will broaden the number of aptamer researchers that can analyze their own data. These Galaxy Workflows can easily be modified to suit any aptamer library and then be shared with other Galaxy users. Moreover, analysis of larger datasets, with >50 million reads, demonstrates the scalability of this approach with reasonable runtimes. Using these Galaxy Workflows, an aptamer researcher could process, compile, analyze, and filter an aptamer selection contained within a barcoded FASTQ dataset of >100 million reads within a single day. As Galaxy becomes more widely adopted to analyze aptamer HTS data, the availability of additional sophisticated tools, including for in-depth analyses, will also grow.
Materials and methods
All of the Galaxy Workflows were built using the Workflow Editor and tools available within the free public Galaxy webserver. The Supplementary Material file contains detailed descriptions of Galaxy access and usage (see Supplementary Materials: Section 2) and Galaxy Workflow methodology and usage (see Supplementary Materials: Section 3) as well as screenshots of actual Galaxy Workflow runs using an example database (see Supplementary Materials: Supplementary Figures S1–11).
HTS Aptamer Dataset and Galaxy Workflows. The example HTS Aptamer Dataset, “Analyzing HT-SELEX data with the Galaxy Project tools,” was attained from Galaxy.8 The example HTS Aptamer Dataset is a multiplexed FASTA file containing the starting nonselected aptamer library (Round 0) along with six rounds of aptamer selection data (Rounds 1, 3, 5, 6, 7, 8). A barcode text file was created using the barcodes provided within the annotation/notes field of the HTS Aptamer Dataset. All Galaxy Workflows were obtained from Galaxy under “Shared Data” and “Published Workflows” by searching for “thielwh” and “aptamer” (see Supplementary Materials: Section 2 and Supplementary Figure S2). Test Datasets 01/02 and 03/04 were sequenced within the same Illumina HTS runs. Test Dataset 05 is a simulated dataset created by combining Test Datasets 03 and 04. Table 1 summarizes the number of barcodes, reads, and selection rounds within each Test Dataset.
Preprocessing. The first two Workflows, Preprocess Workflow 1 and Preprocess Workflow 2, were used sequentially to process the example HTS Aptamer Dataset FASTA file and output multiple FASTA formatted files that contained the variable region sequence information with read counts (see Supplementary Materials: Section 3.1 and Supplementary Figure S3–S6). Running the Preprocess Workflow 1 resulted in a table with links to FASTA files of each selection round, which were uploaded to Galaxy and renamed to indicate the round number (R00, R01, R02, R03, R05, R06, R07, and R08). The Preprocess Workflow 2 was run separately for each selection round with “Step 5: Filter sequence by length” set to ±10% nucleotides of a 20 nt Sel2 variable region sequence length (18, 22 nt). The Preprocess Workflow 2 yielded FASTA formatted files labeled with a “-PP” (e.g., R00-PP) to indicate “preprocessed.”
Nonredundant database. The third Workflow, NrD Workflow, was used to build an NrD of all unique variable region sequences using the output FASTA formatted files from Preprocess Workflow 2 (see Supplementary Materials: Section 3.2 and Supplementary Figures S7–S8). The NrD database contained all unique variable region sequences across all selection rounds with associated read counts (“Abundance”) and round representation (“Persistence”). The NrD Workflow: 7 round was used to compile an NrD of the R00-PP through R08-PP FASTA formatted files. The simple calculation tool within Galaxy, Compute, was used to sum all the reads from the rounds.8 Next, the sorting tool within Galaxy, Sort, was used to sort the aptamer sequences based upon the calculated total read count.8
Abundance and persistence analysis and filtering. The fourth and fifth Workflows, Abundance NrD Analysis Workflow and Persistence NrD Analysis Workflow, were used to analyze and compare the selection rounds within the NrD database for aptamer abundance and aptamer persistence, respectively (see Supplementary Materials: Section 3.3 and Supplementary Figures S9–S10). The Abundance NrD Analysis Workflow: Compare 5 rounds and the Persistence NrD Analysis Workflow: Compare 5 rounds were run using the Aptamer HTS Dataset NrD and selecting the columns for round 0, 1, 3, 5, and 8. The sixth and seventh Workflows, Abundance NrD Filter Workflow and Persistence NrD Filter Workflow, enabled filtering of the NrD based upon the aptamer abundance and persistence analyses (see Supplementary Materials: Section 3.4 and Supplementary Figure S11). The Abundance NrD Filter Workflow was run with the expression “c4>2 or c5>2 or c6>2 or c7>2 or c8>2 or c9>2” and the Persistence NrD Filter Workflow was run with the expression “c2>=2.” The “c” refers to the column designation and is used by Galaxy to denote columns within the NrD (e.g., column 4 = “c4”).
Calculation of Workflow runtimes. Runtimes for each Galaxy Workflow were determined as the duration between the start time of a Workflow and the stop time of the Workflow. The start time was determined by the “Created:” time under “View Details” after the execution of a Workflow. The stop time was determined by selecting “Yes” for an email notification with the completion of the Workflow and noting the time stamp of the email.
SUPPLEMENTARY MATERIAL Figure S1. Schematic of SELEX process and the pipeline using Galaxy Workflows to analyze aptamer HTS data. Figure S2. Galaxy website interface. Figure S3. Pre-Process Workflow 1. Figure S4. Pre-Process Workflow 1 input and output. Figure S5. Pre-Process Workflow 2. Figure S6. Identifying full or partial 3' constant region sequence. Figure S7. The example Aptamer HTS Dataset non-redundant database (NrD). Figure S8. NrD Workflow. Figure S9. Abundance NrD Analysis Workflow. Figure S10. Persistence NrD Analysis Workflow. Figure S11. Abundance NrD Filter Workflow and Persistence NrD Filter Workflow. Figure S12. Relationship between number of reads and Galaxy Workflow runtimes. Supplementary Material
Acknowledgments
This work was supported in part by an American Heart Association Scientist Development Grant (14SDG18850071 to W.H.T.). The funders had no role in the design of the study, collection and analysis of the data, or the decision to publish. We thank Paloma H. Giangrande, George J. Weiner, Suresh Veeramani, Sue E. Blackwell, David D. Dickey, and Sven Kruspe and Kevin T. Urak for their helpful discussions and providing aptamer HTS data for testing. We thank Kristina W. Thiel for her helpful discussions and preparation of this manuscript.
Supplementary Material
References
- Tuerk, C and Gold, L (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505–510. [DOI] [PubMed] [Google Scholar]
- Ellington, AD and Szostak, JW (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822. [DOI] [PubMed] [Google Scholar]
- Zimmermann, B, Gesell, T, Chen, D, Lorenz, C and Schroeder, R (2010). Monitoring genomic sequences during SELEX using high-throughput sequencing: neutral SELEX. PLoS One 5: e9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel, WH, Bair, T, Peek, AS, Liu, X, Dassie, J, Stockdale, KR et al. (2012). Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS One 7: e43836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks, J, Nekrutenko, A and Taylor, J; Galaxy Team (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg, D, Von Kuster, G, Coraor, N, Ananda, G, Lazarus, R, Mangan, M et al. (2010). Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol Chapter 19: Unit 19.10.1–Unit 19.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine, B, Riemer, C, Hardison, RC, Burhans, R, Elnitski, L, Shah, P et al. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res 15: 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel, WH and Giangrande, PH (2016). Analyzing HT-SELEX data with the Galaxy Project tools—a web based bioinformatics platform for biomedical research. Methods 97: 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blind, M, and Blank, M (2015). Aptamer selection technology and recent advances. Mol Ther Nucleic Acids 4: e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoinka, J, Berezhnoy, A, Sauna, ZE, Gilboa, E, and Przytycka, TM (2014). AptaCluster—a method to cluster HT-SELEX aptamer pools and lessons from its application. In: Sharan, R (ed). Research in Computational Molecular Biology, vol. 8394. Springer International Publishing: Pittsburgh, PA. pp 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam, KK, Chang, JL and Burke, DH (2015). FASTAptamer: a bioinformatic toolkit for high-throughput sequence analysis of combinatorial selections. Mol Ther Nucleic Acids 4: e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoinka, J, Zotenko, E, Friedman, A, Sauna, ZE and Przytycka, TM (2012). Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 28: i215–i223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, P, Meyer, S, Hou, Z, Propson, NE, Soh, HT, Thomson, JA et al. (2014). MPBind: a Meta-motif-based statistical framework and pipeline to Predict Binding potential of SELEX-derived aptamers. Bioinformatics 30: 2665–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroli, J, Taccioli, C, De La Fuente, A, Serafini, P and Bicciato, S (2016). APTANI: a computational tool to select aptamers through sequence-structure motif analysis of HT-SELEX data. Bioinformatics 32: 161–164. [DOI] [PubMed] [Google Scholar]
- Thiel, KW, Hernandez, LI, Dassie, JP, Thiel, WH, Liu, X, Stockdale, KR et al. (2012). Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res 40: 6319–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoinka, J, Berezhnoy, A, Dao, P, Sauna, ZE, Gilboa, E and Przytycka, TM (2015). Large scale analysis of the mutational landscape in HT-SELEX improves aptamer discovery. Nucleic Acids Res 43: 5699–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, M, Xiao, Y, Nie, J, Stewart, R, Csordas, AT, Oh, SS et al. (2010). Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc Natl Acad Sci USA 107: 15373–15378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütze, T, Wilhelm, B, Greiner, N, Braun, H, Peter, F, Mörl, M et al. (2011). Probing the SELEX process with next-generation sequencing. PLoS One 6: e29604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.