

Summary
Background
We already demonstrated that adoptive transfer of alveolar macrophages (AMs) from non-allergic rats into AM-depleted allergic rats prevents airway hyperresponsiveness (AHR). We also showed that AMs from non-sensitized, but not from sensitized, allergy-prone rats can prevent AHR following allergen challenge in sensitized allergic animals, establishing the importance of rat immunological status on the modulation of AM functions and suggesting that an allergic lung environment alters AM functions.
Objective
We investigated how the activation of allergic AMs can be modulated to reinstitute them with their capacity to reduce AHR.
Methods
AMs from sensitized Brown Norway rats were cultured ex vivo for up to 18 h in culture media to deprogram them from the influence of the allergic lung before being reintroduced into the lung of AM-depleted sensitized recipient. AHR and cytokines in bronchoalveolar lavage (BAL) were measured following allergen challenge. AMs stimulated ex vivo with Bacillus Calmette-Guerin(BCG) were used as positive controls as BCG induces a T-helper type 1 activation in AMs.
Results
AMs ex vivo cultured for 4–18 h reduced AHR to normal level. Interestingly, pro-allergic functions of AMs were dampened by 18 h culture and they reduced AHR even after spending 48 h in an allergic lung microenvironment. Furthermore, transfer of cultured AMs caused an increase in the levels of IFN-γ and IL-12 in BAL when compared with their ovalbumin control. After 18 h of ex vivo culture, AMs expressed reduced levels of TNF, IL-1α, IL-6, and Arginase-2 mRNAs compared with freshly isolated AMs, suggesting that ex vivo culture exempted AMs from lung stimuli that affected their functions.
Conclusions
There is a significant crosstalk between lung microenvironment and AMs, affecting their functions. It is also the first report showing that sensitized AMs can be modulated ex vivo to reduce lung pro-allergic environment, opening the way to therapies targetting AMs.
Keywords: alveolar macrophages and asthma, bronchoalveolar lavage, lung microenvironment, Th1 type cytokines
Introduction
Asthma incidence has increased over the last two decades. Numerous studies have shown the pivotal role of T-helper type 2 (Th2) cytokines [1, 2]; therefore, asthma is often referred as a Th2-driven disease. Although the implication of different cell types such as mast cells, lymphocytes, and eosinophils in asthma is well known [3], the role of alveolar macrophages (AMs) is not well defined [4, 5]. AMs are found throughout the respiratory tract from the alveolus to the larynx and play a key role in the maintenance of lung immunological homeostasis [6]. AMs can produce both Th1 and Th2-type cytokines [7], giving them the opportunity to influence the chronic Th2 inflammation observed in asthma and to modulate the issue of the disease. Interestingly, depletion of AMs in rats with immunological memory results in a profound state of airway hyperresponsiveness (AHR), an increased IgE response, and an influx of activated T and B cells into the lung, showing the importance of these cells in asthma pathogenesis [8–10].
The Brown Norway rat sensitized to ovalbumin (OVA) is a widely used model of allergic asthma. It mimics human asthma with early and late-phase reactions [11], increased serum-specific IgE level [12], and increased bronchial responsiveness to methacholine [13]. We demonstrated that Brown Norway rats recruit more eosinophils and AMs to the lung after allergen challenge compared with Sprague–Dawley rats, an allergy-resistant strain [14], and that these AMs produce more Th2-type cytokines (IL-10 and IL-13) than their allergy-resistant counterpart [7]. Recently, we showed that transfer of AMs from sensitized Sprague–Dawley rats into AM-depleted sensitized Brown Norway rats 24 h before allergen challenge normalizes airway responsiveness to methacholine. In stark contrast, in the same experiment sets, transfer of AMs from a sensitized Brown Norway rats to another Brown Norway rat did not modulate AHR [8]. Most interestingly, transfer of AMs from unsensitized Brown Norway rats into AM-depleted sensitized Brown Norway rats reduced AHR [15], suggesting that AHR reduction is a common function of AMs from both allergy-prone or allergy-resistant strains and that allergic sensitization eliminates this function only in allergy-prone strains.
In regard to these previous results showing that AM ability to prevent AHR is suppressed by allergen sensitization [15], we hypothesized that lung microenvironment in sensitized animals constantly restrains this AM function in allergic lung and prevents them from reducing AHR. Therefore, we investigated the possibility that AM-modulated reduction of AHR can be restored by releasing AMs from the lung microenvironment by ex vivo culture in a neutral milieu before their reinstillation in AM-depleted sensitized rat. Bacillus Calmette-Guerin (BCG) stimulation was also used as a positive control for its renowned induction of a Th1 response that is antagonist to the allergic lung Th2 response.
Thus, AMs isolated from sensitized Brown Norway rats were kept in culture for 0, 1, 4, or 18 h before their reintroduction into AM-depleted sensitized rats (see Fig. 1 for the experimental setting). Ex vivo culture of AMs resulted in AHR reduction in a culture time-dependent manner, with a maximum effect between 4 and 18 h. Reinstillation of AMs ex vivo cultured resulted in an increase of Th1-associated cytokines (IFN-γ and IL-12) in bronchoalveolar lavage (BAL) without affecting IL-13 level.
Fig. 1.
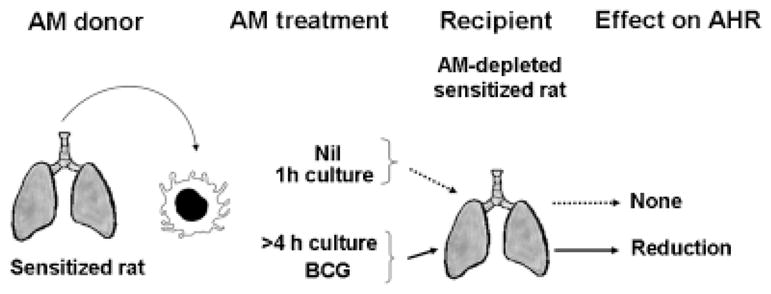
Hypothesis and experimental design. Transfer of freshly isolated AMs from sensitized rats, or AMs kept in culture for 1 h, does not reduce AHR. Sensitized AMs kept in culture for more than 4 h or stimulated with BCG reduce AHR. AM, alveolar macrophage; AHR, airway hyperresponsiveness; BCG, Bacillus Calmette-Guerin.
Materials and methods
Animals
Brown Norway rats (BN/Ssn) aged 72–80 days were obtained from Harlan–Sprague–Dawley (Indianapolis, IN, USA). They were maintained in filter top cages in virus/ pathogen-free conditions at Laval Hospital animal facility on a 12 h light/dark cycle. Food and water were given ad libitum. The protocol was approved by Laval University Animal Care Committee in accordance with the guidelines of the Canadian Council on Animal Care.
In vivo ovalbumin sensitization and challenge
Sensitization and challenge were performed as previously described [14] with 1 mg/mL OVA grade V (Sigma-Aldrich Canada Ltd, Oakville, ON, Canada) and 100 mg/mL Al(OH3) (EMD Chemicals Inc., Gibbstown, NJ, USA). Challenges were performed using 5% aerosolized OVA and 0.9% saline solution for control rats.
Alveolar macrophages depletion
AMs were depleted from recipient animals using a suspension of multilamellar liposomes encapsulating dichloromethylene-diphosphate (Cl2MDP) (Roche Diagnostics Canada, Laval, QC, Canada) prepared according to the Van Rooijen method [16]. The liposomes were instilled in recipient rats 3 days before adoptive transfer, the time when the depletion was optimal (depletion of 85% AMs).
Alveolar macrophages isolation and treatment
AMs were retrieved by BAL as previously described [14]. Cells from BAL were centrifuged and resuspended in RPMI 1640 medium. The purity of AMs was >98% according to Diff-Quik staining. Culture and BCG stimulation (5 ×106 CFU/mL, Calbiochem, San Diego, CA, USA) were performed in RPMI 1640 medium in non-adherent tubes. Cells were washed twice with phosphate-buffered saline (PBS) before being resuspended in saline and instilled in AM-depleted recipient as described previously [8]. The optimal number of AMs (2 ×106 AMs/animal) was instilled in the recipient at day 20 after sensitization, the recipient being depleted of AMs at day 17 [8]. The chronology of events is described in Fig. 2.
Fig. 2.

Chronology of the protocol for 18 h experiments. Animals were sensitized on day 0. Recipient rats were depleted of their AMs on day 17. BAL was performed in donor rat on day 19 to retrieve AMs. These cells were cultured for 18 h, before being transferred into AM-depleted rats. Antigen challenge was performed the next day (day 21) and AHR was measured or BAL performed 24 h later. BAL, bronchoalveolar lavage; AM, alveolar macrophage; AHR, airway hyperresponsiveness; OVA, ovalbumin.
Airway hyperresponsiveness measurement
AHR was measured 24 h after allergen challenge, as described previously [14]. Briefly, rats were anaesthetized intraperitoneally with 1.5 g/kg urethane (Sigma-Aldrich Canada Ltd.). The trachea of the rat was cannulated and connected to the Quadra-T CombiChamber (Scireq, Montreal, PQ, Canada), which calculates pulmonary resistance (RL). Animals were exposed to aerosolized doubling concentrations (1–128 mg/mL) of methacholine for 30 s and lung resistance (RL) measurements were taken every minute for 5 min. Peak values from each concentration were used to plot a concentration–response curve. The concentration required to induce 200% increase in resistance, EC200RL, was calculated and noted as representative of AHR.
Analysis of cytokines in bronchoalveolar lavage
BAL was performed 24 h after OVA or saline challenge and cytokine levels were measured in the first 5 mL BAL fluid after centrifugation to eliminate cells. ELISA was performed with OptEIA kits from Biosciences (Mississauga, ON, Canada) for IL-10, IFN-γ, TNF, and IL-4. IL-12 p40, IL-12p70, and IL-13 levels were measured with kits from Biosource International (Camarillo, CA, USA). The sensitivity of the assays was 10 pg/mL for IL-4, IL-10, IL-12 (p40 and p70), IL-13, and IFN-γ, and 30 pg/mL for TNF.
Mediator production
Total RNA from freshly isolated AMs or 18 h cultured AMs was extracted using TRIzol reagent (Invitrogen, Burlington, ON, Canada) and quantified using RiboGreenTM quantification reagent (Molecular Probes Inc., Eugene, OR, USA). Readings were performed on a Fluoroskan Ascent FL (Labsystems, Franklin, MA, USA). For cDNA synthesis, 1 μg of total RNA was reverse transcribed by Moloney murine leukemia virus reverse transcriptase enzyme (Invitrogen) using a MinicyclerTM (MJ Research, Waltham, MA, USA). Multiplex PCR (Biosource), designed to detect simultaneously the expression of rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IL-1α, IL-1β, IL-6, TNF, and IL-1Ra genes, was performed according to the manufacturer’s protocol. The expression of Arginase-2 was investigated using sense primer 5′-GAG AAG CTG GCT TGC TGA AG-3′ and antisense primer 5′-GAG CGA GGA TAC ACG ACC AG-3′. The ratio of the band density of each cytokine over the housekeeping gene, GAPDH, was calculated.
Statistical analysis
The log of EC200RL was used for statistical analysis. Both AHR and cytokine levels were analyzed for statistical significance by ANOVA, combined with Fisher’s protected least-significant difference test using StatView 5.0 (SAS Institute Inc., Cary, NC, USA) with a level of significance of 5%.
Results
Alveolar macrophage depletion
AMs were depleted using intra-tracheal injection of liposomes containing clodronate as previously demonstrated [8]. The number and type of cells in BAL were determined 3 days after liposome instillation, corresponding to the optimal depletion of AMs. The instillation of liposomes dramatically reduced AM number by about 85%, while the number of lymphocytes, eosinophils, and neutrophils was not significantly affected by the treatment (Fig. 3).
Fig. 3.

Bronchoalveolar lavage (BAL) was performed in rats that received PBS- or clodronate-liposomes 3 days earlier. Alveolar macrophages (AM) were significantly depleted (‡P<0.001) without affecting other cell types. Means ±SEM of three animals in each group. PBS, phosphate-buffered saline.
Ex vivo culture
AMs were cultured in non-adherent tissue culture tubes to avoid cell adherence and facilitate cell retrieval for subsequent transfer. Culture was performed for 1, 4, or 18 h and cytokines were measured in supernatant of 18 h culture. As the allergic lung microenvironment is of Th2 nature and we suspected this Th2 pressure to suppress AM capacity to reduce AHR, we used BCG as a positive control to create a Th1-promoting environment that would counteract the Th2 influence that AMs had in the donor sensitized lung. AMs were therefore also cultured in the presence of 5 ×106 CFU/mL of BCG, an optimal dose to elicit AM IL-12 production in vitro according to preliminary results (data not shown). BCG-stimulated AMs released, in the culture media after 18 h, significantly more IFN-γ (206.6 ± 85.8 pg/106 AMs), IL-12 p40 (386.2 ± 149.4 pg/106 AMs), IL-10 (185.9 ± 58.7 pg/106 AMs), and IL-13 (20.8 ± 8.5 pg/106 AMs) compared with unstimulated AMs that produce very small amounts of these cytokines (Fig. 4). These data demonstrate that although BCG stimulates AMs to release both Th1- and Th2-type cytokines, Th1-type cytokines were predominantly produced, confirming the effectiveness of this Th1 stimulation and hence the relief of these AMs from their previous Th2 influence in the allergic lung.
Fig. 4.

In vitro production of cytokines by AMs stimulated or not with BCG (5 ×106 CFU/106 AMs) in non-adherent tissue culture tubes. BCG significantly (*P≤0.05) increased the production of IFN-γ, IL-12p40, IL-10, and IL-13. Means ± SEM of seven experiments. AMs, alveolar macrophages; BCG, Bacillus Calmette-Guerin.
Airway hyperresponsiveness
Rats were sensitized with OVA on day 0 and challenged on day 21 to either aerosolized saline (control) or OVA and AHR was measured 24 h later (day 22). In order to investigate the role of local allergic lung milieu of sensitized allergy-prone rats on AMs, recipient rats were depleted of their AMs on day 17, and received or not (depletion controls) AMs (from sensitized rats) that were ex vivo cultured in the absence (neutral medium) or presence of BCG. AM transfer was performed on day 20 (Fig. 2), corresponding to the optimal AM depletion as shown previously [8].
Respiratory distress corresponding to the early-phase reaction was visually observed only in OVA-challenged animals without AM transfer (data not shown). AHR was observed in OVA-challenged rats (Fig. 5a), as reflected by a lower methacholine concentration needed to double lung resistance (RL) (EC200RL of 75.8 ± 11.0 mg/mL) compared with saline-challenged rats with an EC200RL of 112.2 ± 6.7 mg/mL. AM-depleted rats challenged with saline did not show significant AHR (97.7 ± 6.7 mg/mL), whereas OVA challenge significantly increased AHR (59.8 ± 7.1 mg/mL) in AM-depleted rats (Fig. 5a), confirming our previous results [8, 15].
Fig. 5.

AHR of control rats, AM-depleted rats, and repleted with ex vivo cultured AM (Sham or BCG treated). AHR was measured following methacholine challenge and EC200 values were calculated. These values were transformed to log to best fit the natural exponential phenomenon. Hatched and closed bars represent saline and OVA challenge, respectively. (a) AMs were treated for 18 h before being instillated into AM-depleted rats. As expected, OVA challenge significantly increased AHR (*P<0.05) compared with saline in the control group and AM-depleted rats, whereas Sham- and BCG-treated AM abrogated AHR. Means ± SEM of 10 experiments. (b) AMs were cultured for 0, 1, or 4 h before being instillated into AM-depleted rats. Allergen challenge (OVA) significantly increased AHR, whereas transfer of 4 h cultured AMs abrogated AHR. Shorter treatment periods showed variable data (a and b are different, P≤0.05). Means ± SEM of three to four experiments. OVA, ovalbumin; AM, alveolar macrophage; AHR, airway hyperresponsiveness; BCG, Bacillus Calmette-Guerin.
Ex vivo culture (18 h) of AMs allowed the normalization of methacholine airway responsiveness (101.7 ± 9.7 mg/ mL) (Fig. 5a). In comparison, BCG (115.1 ± 6.0 mg/mL) stimulation for 18 h, used as a control for induction of Th1 response in AMs, also optimally reduced AHR (Fig. 5a). Very interestingly, this modulation of AHR was dependent on time of culture, showing an optimal effect between 4 and 18 h of ex vivo culture (Fig. 5b). Transfer of freshly isolated AMs (0 h) did not modulate AHR, suggesting that the effect of Th2 pressure from the donor lung environment needs time to vanish from AMs originating from allergic rats. These results strongly suggest that allergic lung milieu influences AM response and affects their capacity to reduce AHR.
Cytokine production
As we noted that AMs were modifying lung physiological response to metacholine (AHR), we were interested in identifying the modifications of local inflammation induced by AM. Therefore, we investigated cytokine levels in the first 5 mL of lung BAL fluid performed 24 h after challenge. OVA challenge significantly increased pro-inflammatory TNF release compared with saline challenge (32.3 ± 12.9 and 7.5 ± 2.5 pg/mL, respectively; data not shown). However, ex vivo treatment of AMs with BCG or medium alone did not significantly modulate TNF production when compared with OVA control (33.8 ± 15.8 and 35.7 ± 10.9, respectively).
Th1-type cytokines were measured in BAL fluid. IFN-γ release was not increased in OVA-challenged rats compared with control (Fig. 6a). However, IFN-γ levels in the BAL of rats that received cultured and BCG-stimulated AMs were significantly increased. Similar results were observed with IL-12p40 levels (Fig. 6b), while IL-12p70 levels remained below the detection limit. It was impossible in this context to identify the producing cells, but globally it is clear that AMs were able to modulate lung cytokine environment either by their direct production or by inducing cytokine production of other cell types.
Fig. 6.

Dosage of Th1 type cytokines in the bronchoalveolar lavage (BAL) fluid of control rats and rats repleted with AMs treated or not with BCG. Cytokine levels were measured in the first 5 mL of BAL from investigated rats using ELISA. Hatched and closed bars represent saline and ovalbumin (OVA) challenge, respectively. In the controls, OVA challenge did not increase IFN-γ (a) and IL-12p40 (b) production. Transfer of cultured and BCG-stimulated AMs significantly (*P<0.05) increased the levels of IFN-γ and IL-12p40 in BAL compared with saline and OVA groups. Means ± SEM of eight to 10 experiments. AMs, alveolar macrophage; Th1, T-helper type 1; BCG, Bacillus Calmette-Guerin.
Levels of Th2-type cytokines, IL-10, IL-13, and IL-4 were also investigated in BAL fluid. Ovalbumin challenge significantly increased IL-10 release in BAL (234.9 ± 116.2 pg/mL) compared with saline control (15.3 ± 3.5 pg/mL), without modulating IL-13 release (Figs 7a and b), whereas IL-4 levels remained undetectable in all groups (data not shown). Transfer of cultured and BCG-stimulated AMs did not significantly modulate IL-10 and IL-13 levels. Overall, it is clear that while the Th2 response was not extensively affected, the Th1 response was increased by ex vivo treatments. This increase in Th1 (IFN-γ, IL-12) cytokines did not result in the exacerbation of AHR, but in its reduction, suggesting a modulation of cytokine production that would be regulatory (homeostatic) to reduce Th2-type response more than a pro-inflammatory Th1 response. This is also supported by the fact that TNF was not modulated.
Fig. 7.

Dosage of Th2 type cytokines in the bronchoalveolar lavage fluid of control rats and rats repleted with AMs treated or not with BCG. Hatched and closed bars represent saline and ovalbumin (OVA) challenge, respectively. OVA challenge significantly (*P<0.05) increased IL-10 production compared with saline (a), but not IL-13 (b). Means ± SEM of eight to 10 experiments. Th2, T-helper type 2; AMs, alveolar macrophage; BCG, Bacillus Calmette-Guerin.
Differences between freshly isolated alveolar macrophages and 18 h cultured alveolar macrophages
Given the difference in the modulation of AHR and the difference in the modulation of lung cytokine milieu by freshly isolated AMs and 18 h cultured AMs, we were interested to investigate the production of cytokines in AMs cultured for 18 h (Fig. 8a). Freshly isolated AMs produced significantly more IL-6, IL-1α, and TNF compared with 18 h cultured AMs (Fig. 8b), suggesting that culture period can down-regulate the production of pro-inflammatory cytokines and therefore reduce the support to local inflammation upon reinstallation to the lung. Furthermore, given that IL-4 and IL-13, two important Th2 cytokines found in allergic lung, can drive AMs towards the alternative activation pathway [17], we investigated the mRNA expression of Arginase-2 and FIZZ-1, which are associated with alternatively activated macrophages [18, 19]. Arginase-2 expression was significantly reduced by the ex vivo culture of 18 h (Fig. 8c), as was FIZZ-1, but the latter did not reach significance at this time-point (data not shown). This reinforces our conclusion that ex vivo culture allows AMs to deprogram themselves from the Th2 pressure that they are submitted to in the allergic lung.
Fig. 8.

Cytokine expression in freshly isolated AMs and 18 h cultured AMs. RNA was extracted in freshly isolated (open bars) and 18 h cultured AMs (closed bars) to measure cytokine expression using Multiplex PCR (a). The ratio of the band density of each cytokine over the housekeeping gene, GAPDH, was calculated (b). Cultured AMs expressed significantly (*P<0.05) less IL-6, IL-1α, and TNF compared with freshly isolated AMs. Mean ±3 to 5 experiments. Cultured AMs express significantly (**P<0.01) less Arginase-2 than freshly isolated AMs (c). Mean ± 7 experiments. AMs, alveolar macrophage; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
We showed some years ago that AMs from allergy-resistant rats, sensitized or not, can prevent AHR upon transfer [8]. We have also previously demonstrated that AMs from unsensitized allergy-prone rats can abrogate AHR when transferred into AM-depleted allergic rats, while AMs from sensitized donors did not [15]. This clearly stated that AMs have the intrinsic capacity to reduce AHR, and this capacity is suppressed only in allergy-prone rats upon allergic sensitization. Hence, this study highlighted the importance of lung environment modifications by allergen sensitization. In the present study, we demonstrated that AMs from sensitized allergic rats can be modulated ex vivo to abrogate AHR when reintroduced into allergic rats, clearly establishing a preponderate role for the environment in modulating AM functions and the reciprocal action of AMs on the lung microenvironment.
We already demonstrated that the optimal AM depletion by clodronate liposomes occurs at 3 days [8], and here we showed that this depletion does not affect lymphocytes, neutrophils or eosinophils. AMs are the main phagocyte engulfing particles, whereas dendritic cells (DCs) are not substantially performing it in the lung [20]. Furthermore, a study demonstrated that AMs and not DCs are selectively depleted due to the size of liposomes [21]. Interestingly enough, the Dr Lambrecht team reported that DC depletion during allergen challenge improves the status of asthma [22], contrasting strikingly with what we observed with AM depletion. Dr Lambrecht’s study strongly suggests that DCs are not depleted in our present study, as in our control where AMs are depleted, AHR is still observed (Fig. 5a), which would not happen if DCs were depleted, as in his model. We also make the same observation in Fig. 5b, where the AM-depleted rat receiving AMs that are not ex vivo cultured exhibits AHR of the asthmatic rat. As DCs are needed to induce asthma upon allergen challenge, it is improbable that DCs are noticeably depleted from the lungs in our model.
To verify the effect of ex vivo culture or BCG stimulation on AMs, we first assessed the production of cytokines from AMs kept in culture or stimulated with BCG ex vivo for 18 h, and showed a very small induction of cytokine production in non-stimulated cells but a strong induction of IFN-γ, IL-12 and IL-10 in the supernatant of BCG-stimulated AMs. These data are in agreement with a Th1 response from AMs and therefore established that BCG-stimulated AMs were engaged in a Th1-oriented response at the moment of instillation. It was impossible in our context to assess directly at the AM level their Th1/2 activation status after transfer, but one could speculate that upon re-introduction in a Th2-prone environment, AM would readapt their reaction after a given period of time. However, our results suggest the opposite as cultured AM can install a Th1 microenvironment in the lung after introduction, and therefore prevent their return to pro-allergic functions.
We then proceeded to assess AHR in all rat groups. The saline-challenged group shows minimal AHR (needs a high concentration of metacholine to double lung resistance), while the OVA-challenged control group shows important AHR (needs a lower concentration of metacholine). As observed previously [8], depletion of AMs and subsequent saline challenge did not result in increased AHR, establishing the innocuousness of the technique on AHR measurement. Furthermore, AHR was slightly increased in OVA-challenged, AM-depleted rats, in comparison with OVA-challenged rats, which is in accordance with our previous results [8] and highlights the importance of AMs in containing AHR after allergen challenge. It is interesting to note that AM modulatory capacity was modified with only 4 h of ex vivo culture, but that once reintroduced in the lung, the effect of this ex vivo treatment was maintained even after remaining 48 h in the lung. Added to the fact that AMs from both allergy-prone and allergy-resistant strains can reduce AHR, this could mean that AMs have the intrinsic capacity to reduce AHR and hence are more easily directed towards this natural function than towards functions supporting allergic inflammation. However, this suggestion will require more investigation.
As AMs from allergic donors are extracted from a Th2 lung environment, we wanted to investigate the effect of a Th1 stimulation of AMs as this is known to greatly modify macrophage capacities and antagonize the previous Th2 pressure. As it is well established that BCG is a potent Th1 stimulus, we used it to stimulate AMs ex vivo. Before transfer, AMs were carefully washed to remove any BCG from suspension in order to investigate the effect of AM stimulation and not the effect of BCG instillation in the lungs, which has already been shown to suppress several pathologic features in a mouse model of asthma when given intranasally [23]. To our knowledge, there is no report on ex vivo stimulation of AMs with BCG and the modulation of AHR and cytokine levels after reintroduction of the cells into the animal. Interestingly, the effect on AHR was similar at 18 h between non-stimulated and BCG-stimulated AMs (Fig. 5), suggesting that a Th1 response is not necessary but that an AM response may be modified by alteration of its environmental pressure.
We can exclude the possibility that mediators from the culture media or BCG influenced the lung as AMs were carefully washed twice in PBS before being resuspended in saline and re-instilled in the lung. The only mediators that will affect the lung are the ones produced by AMs upon engagement of signaling pathways triggered ex vivo. Our study clearly demonstrates the pivotal role of AMs as a regulatory cell in asthma as suggested by Smit [24]. Of uttermost importance, we show that AMs from asthmatics are not ‘committed’ to an asthmatic phenotype, but are maintained in such a state by a constant influence of their residing milieu. Therefore, identification and removal of such stimuli maintaining AMs in an asthmatic phenotype could be envisaged as a method to improve AHR in asthmatic subjects if the observation is confirmed in humans.
Given the importance of Th1 and Th2 cytokine levels in allergic asthma, we measured their levels in BAL fluid 24 h after allergen challenge to understand the mechanisms of AM protection. As expected, Th1 cytokine levels, IFN-γ and IL-12, were not affected by OVA challenge (Fig. 6). However, the transfer of cultured or BCG-stimulated AMs significantly increased the IFN-γ and IL-12p40 levels in BAL. IFN-γ is well known to down-regulate Th2-type cytokines [25]. Furthermore, IFN-γ has been shown to inhibit allergen-induced eosinophilic inflammation [26] and IgE-stimulated mast cells [27]. Thus, an increased level of IFN-γ in the BAL of rats that received cultured AMs may explain, at least in part, the reduction of the early-phase reaction and AHR.
IL-12 has been shown to be important during the sensitization process to reduce Th2 response [28], but it can enhance this response in certain circumstances [29]. The timing and the concentration seem important for IL-12 modulation of Th2 response [30]. Transfer of cultured AMs increased IL-12p40 levels in BAL fluid. Although IL-12p40 has been suggested to act as a competitive antagonist of IL-12 [31], the homodimer, IL-12p80, has been suggested to function as a proinflammatory protein that enhances leukocyte accumulation and provides protective immunity toward mycobacterial infection [32, 33]. Furthermore, the increase of the p40 subunit may correspond to an increase of IL-23, which is composed of p19 and p40 subunits [34]. IL-23 has been shown to share some of the IL-12 biological effects, suggesting a protective role in asthma [35]. The increase of IL-12p40 observed in our study may then participate in the reduction of AHR following allergen challenge.
Although the transfer of cultured AMs increased Th1 cytokine levels in BAL, it did not modulate Th2 cytokine levels. The implication of IL-10 as a Th2 cytokine has been controversial [36, 37], but in our model OVA challenge significantly increased the release of IL-10 in BAL. An increase of IL-10 production by cells in the sputum was also observed in humans following in vivo allergen challenge [38]. However, given the anti-inflammatory properties of IL-10, it is possible that the observed increase is the result of the organism trying to control the allergic inflammatory response.
In order to understand changes happening in AMs, we investigated the modulation in gene expression of cytokines (IL-6, IL-1α, TNF) and alternative activation markers (Arginase-2, and FIZZ-1) in cultured AMs. These three cytokines play an important role in inflammation and their levels are significantly higher in patients with asthma [39]. IL-6 and IL-1α have been shown to induce Th differentiation toward Th2 cells [40, 41] and IL-6 to suppress T regulatory cells [40]. Furthermore, study with IL-1 receptor-deficient mice revealed the critical role of IL-1α in promoting eosinophil recruitment [42] and asthmatic response [43]. Thus, a reduction in IL-6 and IL-1α production may participate in the inhibition of AHR observed in animals that received cultured AMs. Interestingly, the alternative activation status of AM was reduced by ex vivo cultures, indicating that the general activation of AMs was modulated by this culture. Our data suggest that the pro-allergic phenotype of AMs might be linked to their alternative activation and, most important, that it is reversible. Taken together, our data suggest that modifications in the AM milieu are quickly sensed by AMs, which can rapidly react by down-regulating their production of inflammatory cytokines and modification of their functions leading to a reduction of AHR.
TNF has been shown to enhance airway responsiveness in experimental animals and in human subjects [44, 45]. Transfer of AMs from unsensitized rat in sensitized rat reduced the TNF level in BAL and AHR [15]. Although TNF production was inhibited in cultured AMs, TNF level in BAL was not reduced by the transfer of cultured AMs, suggesting that an other mechanism may be involved in the protection against AHR. TNF production by other cell types, such as epithelial cells and mast cells, may mask the reduction of AM TNF production.
In summary, allergen-sensitized AMs can be deprogrammed ex vivo to protect against AHR development. The potency of this inhibition may be the result of multiple mechanisms. Transfer of cultured AMs reduced the early-phase reaction probably by down-regulating mast cell activation and caused an increase in IFN-γ and IL-12 in BAL. Furthermore, cultured AMs produce less IL-6, IL-1α, and TNF, and express less alternative activation markers, suggesting that AMs may reduce the inflammatory response following allergen challenge. Although more work will have to be performed to understand the precise mechanisms, our work clearly established that AMs have the intrinsic capacity to reduce AHR, a capacity that is suppressed in allergen-sensitized allergy-prone rats only. This suppression of function is not long-lasting and requires the constant pressure of Th2 response in the lung as a culture of 4 h in neutral media deprogrammed AMs and reinstituted them with their capacity to regulate AHR. We are convinced that this might be an important therapeutic strategy, as affecting only AMs is sufficient to change the homeostasis of the lung and as liposomes represent a specific method of delivering a drug to AMs. In order to better understand the mechanisms of such a modulation, further work is actually ongoing in our laboratory.
Acknowledgments
This study was supported by the Canadian Institutes of Health Research and the Foundation Alphonse L’Espérance (bourse Claude-Marie Gagnon). P. P. had a studentship from the Réseau en Santé Respiratoire du FRSQ and E. C. had a studentship from FRSQ.
References
- 1.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol. 1999;17:255–81. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 2.Chung KF, Barnes PJ. Cytokines in asthma. Thorax. 1999;54:825–57. doi: 10.1136/thx.54.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–45. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 4.Magnan A, van Pee D, Bongrand P, Vervloet D. Alveolar macrophage interleukin (IL)-10 and IL-12 production in atopic asthma. Allergy. 1998;53:1092–5. doi: 10.1111/j.1398-9995.1998.tb03821.x. [DOI] [PubMed] [Google Scholar]
- 5.Tang C, Rolland JM, Ward C, et al. Differential regulation of allergen-specific T(H2)- but not T(H1)-type responses by alveolar macrophages in atopic asthma. J Allergy Clin Immunol. 1998;102:368–75. doi: 10.1016/s0091-6749(98)70122-8. [DOI] [PubMed] [Google Scholar]
- 6.Kraal G, Broug E, Thepen T, Van Iwaarden JF, Persoons JHA. The role of alveolar macrophages in pulmonary immune function. In: Lipscomb MF, Russell SW, editors. Lung macrophages and dendritic cells in health and disease. New York: Marcel Dekker Inc; 1997. pp. 203–20. [Google Scholar]
- 7.Sirois J, Bissonnette EY. Alveolar macrophages of allergic resistant and susceptible strains of rats show distinct cytokine profiles. Clin Exp Immunol. 2001;126:9–15. doi: 10.1046/j.1365-2249.2001.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Careau E, Bissonnette EY. Adoptive transfer of alveolar macrophages abrogates bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2004;31:22–7. doi: 10.1165/rcmb.2003-0229OC. [DOI] [PubMed] [Google Scholar]
- 9.Thepen T, Hoeben K, Breve J, Kraal G. Alveolar macrophages down-regulate local pulmonary immune responses against intratracheally administered T-cell-dependent, but not T-cell-independent antigens. Immunology. 1992;76:60–4. [PMC free article] [PubMed] [Google Scholar]
- 10.Strickland DH, Thepen T, Kees UR, Kraal G, Holt PG. Regulation of T-cell function in lung tissue by pulmonary alveolar macrophages. Immunology. 1993;80:266–72. [PMC free article] [PubMed] [Google Scholar]
- 11.Eidelman DH, Bellofiore S, Martin JG. Late airway responses to antigen challenge in sensitized inbred rats. Am Rev Respir Dis. 1988;137:1033–7. doi: 10.1164/ajrccm/137.5.1033. [DOI] [PubMed] [Google Scholar]
- 12.Waserman S, Olivenstein R, Renzi P, Xu LJ, Martin JG. The relationship between late asthmatic responses and antigen-specific immunoglobulin. J Allergy Clin Immunol. 1992;90:661–9. doi: 10.1016/0091-6749(92)90140-w. [DOI] [PubMed] [Google Scholar]
- 13.Bellofiore S, Martin JG. Antigen challenge of sensitized rats increases airway responsiveness to methacholine. J Appl Physiol. 1988;65:1642–6. doi: 10.1152/jappl.1988.65.4.1642. [DOI] [PubMed] [Google Scholar]
- 14.Careau E, Sirois J, Bissonnette EY. Characterization of lung hyperresponsiveness, inflammation, and alveolar macrophage mediator production in allergy resistant and susceptible rats. Am J Respir Cell Mol Biol. 2002;26:579–86. doi: 10.1165/ajrcmb.26.5.4737. [DOI] [PubMed] [Google Scholar]
- 15.Careau E, Proulx LI, Pouliot P, Spahr A, Turmel V, Bissonnette EY. Antigen sensitization modulates alveolar macrophage functions in an asthma model. Am J Physiol Lung Cell Mol Physiol. 2006;290:L871–9. doi: 10.1152/ajplung.00219.2005. [DOI] [PubMed] [Google Scholar]
- 16.Van Rooijen N. The liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods. 1989;124:1–6. doi: 10.1016/0022-1759(89)90178-6. [DOI] [PubMed] [Google Scholar]
- 17.Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, et al. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211:487–501. doi: 10.1016/j.imbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Gotoh T, Mori M. Arginase II downregulates nitric oxide (NO) production and prevents NO-mediated apoptosis in murine macrophage-derived RAW 264. 7 cells. J Cell Biol. 1999;144:427–34. doi: 10.1083/jcb.144.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiman RM, Thompson RW, Feng CG, et al. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006;74:1471–9. doi: 10.1128/IAI.74.3.1471-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. Modulation of dendritic cell trafficking to and from the airways. J Immunol. 2006;176:3578–84. doi: 10.4049/jimmunol.176.6.3578. [DOI] [PubMed] [Google Scholar]
- 21.Korf J, Stoltz A, Verschoor J, De Baetselier P, Grooten J. The mycobacterium tuberculosis cell wall component mycolic acid elicits pathogen-associated host innate immune responses. Eur J Immunol. 2005;35:890–900. doi: 10.1002/eji.200425332. [DOI] [PubMed] [Google Scholar]
- 22.van Rijt LS, Jung S, Kleinjan A, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. 2005;201:981–91. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erb KJ, Holloway JW, Sobeck A, Moll H, Le Gros G. Infection of mice with Mycobacterium bovis-Bacillus Calmette-Guerin (BCG) suppresses allergen-induced airway eosinophilia. J Exp Med. 1998;187:561–9. doi: 10.1084/jem.187.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smit JJ, Folkerts G, Nijkamp FP. Ramping up allergies: Nramp1 (Slc11a1), macrophages and the hygiene hypothesis. Trends Immunol. 2004;25:342–7. doi: 10.1016/j.it.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 25.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–95. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lack G, Bradley KL, Hamelmann E, et al. Nebulized IFN-gamma inhibits the development of secondary allergic responses in mice. J Immunol. 1996;157:1432–9. [PubMed] [Google Scholar]
- 27.Swieter M, Ghali WA, Rimmer C, Befus D. Interferon-alpha/beta inhibits IgE-dependent histamine release from rat mast cells. Immunology. 1989;66:606–10. [PMC free article] [PubMed] [Google Scholar]
- 28.Kips JC, Brusselle GJ, Joos GF, et al. Interleukin-12 inhibits antigen-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 1996;153:535–9. doi: 10.1164/ajrccm.153.2.8564093. [DOI] [PubMed] [Google Scholar]
- 29.Germann T, Guckes S, Bongartz M, et al. Administration of IL-12 during ongoing immune responses fails to permanently suppress and can even enhance the synthesis of antigen-specific IgE. Int Immunol. 1995;7:1649–57. doi: 10.1093/intimm/7.10.1649. [DOI] [PubMed] [Google Scholar]
- 30.Sur S, Lam J, Bouchard P, Sigounas A, Holbert D, Metzger WJ. Immunomodulatory effects of IL-12 on allergic lung inflammation depend on timing of doses. J Immunol. 1996;157:4173–80. [PubMed] [Google Scholar]
- 31.Gillessen S, Carvajal D, Ling P, et al. Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur J Immunol. 1995;25:200–6. doi: 10.1002/eji.1830250133. [DOI] [PubMed] [Google Scholar]
- 32.Kopp T, Kieffer JD, Rot A, Strommer S, Stingl G, Kupper TS. Inflammatory skin disease in K14/p40 transgenic mice: evidence for interleukin-12-like activities of p40. J Invest Dermatol. 2001;117:618–26. doi: 10.1046/j.1523-1747.2001.01441.x. [DOI] [PubMed] [Google Scholar]
- 33.Holscher C, Atkinson RA, Arendse B, et al. A protective and agonistic function of IL-12p40 in mycobacterial infection. J Immunol. 2001;167:6957–66. doi: 10.4049/jimmunol.167.12.6957. [DOI] [PubMed] [Google Scholar]
- 34.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 35.Wohlleben G, Erb KJ. Immune stimulatory strategies for the prevention and treatment of asthma. Curr Pharm Des. 2006;12:3281–92. doi: 10.2174/138161206778194114. [DOI] [PubMed] [Google Scholar]
- 36.Adachi M, Oda N, Kokubu F, Minoguchi K. IL-10 induces a Th2 cell tolerance in allergic asthma. Int Arch Allergy Immunol. 1999;118:391–4. doi: 10.1159/000024145. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Wang S, Fan Y, Han X. IL-10 deficiency prevents IL-5 overproduction and eosinophilic inflammation in a murine model of asthma-like reaction. Eur J Immunol. 2000;30:382–91. doi: 10.1002/1521-4141(200002)30:2<382::AID-IMMU382>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 38.Bettiol J, Sele J, Henket M, et al. Cytokine production from sputum cells after allergenic challenge in IgE-mediated asthma. Allergy. 2002;57:1145–50. doi: 10.1034/j.1398-9995.2002.23586.x. [DOI] [PubMed] [Google Scholar]
- 39.Tillie-Leblond I, Pugin J, Marquette CH, et al. Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. Am J Respir Crit Care Med. 1999;159:487–94. doi: 10.1164/ajrccm.159.2.9805115. [DOI] [PubMed] [Google Scholar]
- 40.Doganci A, Eigenbrod T, Krug N, et al. The IL-6R alpha chain controls lung CD4+CD25+Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–25. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huber M, Beuscher HU, Rohwer P, Kurrle R, Rollinghoff M, Lohoff M. Costimulation via TCR and IL-1 receptor reveals a novel IL-1alpha-mediated autocrine pathway of Th2 cell proliferation. J Immunol. 1998;160:4242–7. [PubMed] [Google Scholar]
- 42.Broide DH, Campbell K, Gifford T, Sriramarao P. Inhibition of eosinophilic inflammation in allergen-challenged, IL-1 receptor type 1-deficient mice is associated with reduced eosinophil rolling and adhesion on vascular endothelium. Blood. 2000;95:263–9. [PubMed] [Google Scholar]
- 43.Schmitz N, Kurrer M, Kopf M. The IL-1 receptor 1 is critical for Th2 cell type airway immune responses in a mild but not in a more severe asthma model. Eur J Immunol. 2003;33:991–1000. doi: 10.1002/eji.200323801. [DOI] [PubMed] [Google Scholar]
- 44.Kips JC, Tavernier J, Pauwels RA. Tumor necrosis factor causes bronchial hyperresponsiveness in rats. Am Rev Respir Dis. 1992;145:332–6. doi: 10.1164/ajrccm/145.2_Pt_1.332. [DOI] [PubMed] [Google Scholar]
- 45.Thomas PS, Yates DH, Barnes PJ. Tumor necrosis factor-alpha increases airway responsiveness and sputum neutrophilia in normal human subjects. Am J Respir Crit Care Med. 1995;152:76–80. doi: 10.1164/ajrccm.152.1.7599866. [DOI] [PubMed] [Google Scholar]