

Abstract
The family of pediatric fibroblastic and myofibroblastic proliferations encompasses a wide spectrum of pathologic entities with overlapping morphologies and ill-defined genetic abnormalities. Among the superficial lesions, lipofibromatosis (LPF), composed of an admixture of adipose tissue and fibroblastic elements, in the past has been variously classified as infantile fibromatosis or fibrous hamartoma of infancy. In this regard, we have recently encountered a group of superficial soft tissue tumors occurring in children and young adults, with a notably infiltrative growth pattern reminiscent of LPF, variable cytologic atypia and a distinct immunoprofile of S100 protein and CD34 reactivity, suggestive of neural differentiation. SOX10 and melanocytic markers were negative in all cases tested. In contrast, a control group of classic LPF displayed bland, monomorphic histology and lacked S100 protein immunoreactivity. In order to define the pathogenetic abnormalities in these seemingly distinctive groups, we performed RNA sequencing for fusion gene discovery in 2 cases each, followed by screening for any novel alterations identified in a larger cohort representing both entities. The 2 index LPF-like neural tumors (LPF-NT) showed TPR-NTRK1 and TPM3-NTRK1 gene fusions, which were further validated by FISH and RT-PCR. Subsequent FISH screening of 14 LPF-NT identified recurrent NTRK1 gene rearrangements in 10 (71%) cases. Of the NTRK1 negative LPF-NT cases, one case each showed ROS1 and ALK gene rearrangements. In contrast none of the 25 classic LPF showed NTRK1 gene rearrangements, although regional abnormalities were noted in the 1q21–22 region by FISH in a majority of cases. Furthermore, NTRK1 immunostaining was positive only in NTRK1-rearranged S100 positive LPF-NT, but negative in classic LPF. These results suggest that NTRK1 oncogenic activation through gene fusion defines a novel and distinct subset of soft tissue tumors resembling LPF, but displaying cytologic atypia and a neural immunophenotype, provisionally named LPF-like neural tumors.
Keywords: lipofibromatosis, neural, NTRK1, TPR, TPM3, LMNA
INTRODUCTION
Pediatric fibroblastic / myofibroblastic mesenchymal neoplasms are a morphologically diverse group of often locally aggressive soft tissue tumors that encompass a wide spectrum of pathologic entities. Classification of these tumors has been controversial, due to their rare incidence, morphologic similarities, as well as similar clinical presentation overlapping with true sarcomatous processes, such as infantile fibrosarcoma and spindle cell rhabdomyosarcoma. Aside from the more common and well characterized desmoid-type fibromatosis, other pediatric lesions in this spectrum, including lipofibromatosis (LPF), calcifying aponeurotic fibroma and myofibromatosis, lack well-defined genetic abnormalities and are often diagnostically challenging.1 Apart from a single case report of LPF harboring a t(4;9;6) translocation,2 there are no other studies to date investigating their molecular alterations. Most LPFs affect young children and occur in the soft tissues of the trunk and distal extremities (hands and feet) and are characterized by a highly infiltrative growth pattern in the surrounding tissues.3 The extent of disease is often underestimated by pre-operative imaging, leading in most instances to incomplete excision and subsequent local recurrence. Immunohistochemically, they often show variable but inconsistent staining for CD34 and SMA, and are negative for other diagnostic markers. We have encountered in our consultation practice a subset of lesions that resemble the growth pattern of LPF, but show variable cytologic atypia and an immunoprofile more closely related to neural tumors, with positivity for S100 and CD34.
In an attempt to further our understanding of the genetic alterations in these lesions, we sought to investigate tumors in the LPF morphologic spectrum by various methodologies, including whole transcriptome sequencing and FISH.
METHODS
The Department of Pathology files at Memorial Sloan Kettering Cancer Center and the personal consultation files of the senior authors (CRA, CDMF) were searched for lipofibromatosis (LPF) and lipofibromatosis-like tumors with available tissue for molecular analysis, diagnosed between 1993–2015. Hematoxylin and eosin sections and immunohistochemical stains performed at the time of diagnosis were reviewed. The gross and microscopic findings, including tumor size, anatomic location and tumor morphology were recorded. Clinical and follow-up data were obtained from the referring pathologists (see Acknowledgements) and clinical database. The study was approved by the Institutional Review Boards at all participating institutions.
RNA Sequencing
Four cases were analyzed by RNA sequencing. Total RNA was prepared for RNA sequencing in accordance with the standard Illumina mRNA sample preparation protocol (Illumina). Briefly, mRNA was isolated with oligo(dT) magnetic beads from total RNA (2μg). The mRNA was fragmented by incubation at 94°C for 2.5 min in fragmentation buffer (Illumina). To reduce the inclusion of artifact chimeric transcripts into the sequencing library, an additional gel size-selection step was introduced prior to the adapter ligation step.4 The adaptor-ligated library was then enriched by PCR for 15 cycles and purified. The library was sized and quantified using DNA1000 kit (Agilent) on an Agilent 2100 Bioanalyzer according to the manufacturer’s instructions. Paired-end RNA sequencing at read lengths of 50 or 51 bp was performed with the HiSeq 2000 (Illumina). A total of about 60.9 million paired-end reads were generated, corresponding to about 3.1 billion bases.
Analysis of RNA Sequencing Results with FusionSeq
All reads were independently aligned with STAR (ver 2.3)5 and TopHat2 (ver 2.0.14)6 against the human reference genome (hg19), followed by FusionSeq7 and TopHat-Fusion (ver 0.1.0)8 respectively to identify fusion candidates. In the first analysis mapped reads were converted into Mapped Read Format 9 and analyzed with FusionSeq to identify potential fusion transcripts. We then applied TopHat-Fusion analysis to detect potential gene fusion events with spanning reads > 20 bp. In contrast to FusionSeq, TopHat-Fusion aligns reads without relying on existing annotation, and directly detects individual reads that span the fusion event. This tool proved to be more sensitive in detecting small intra-chromosomal inversions, which most likely were filtered out by the Fusion Seq algorithm. In addition, RNA sequencing data was analyzed for gene mutation calls. BAM files were generated by STAR alignment, followed by PicardTools (ver 1.130) standard preprocessing, which include marking of duplicate reads, recalibration of base quality scores and local realignment. MuTect (var 1.15)10 and VarScan (var 2.3.8)11 variant callers were both applied for mutation detection, followed by vcf2maf (https://github.com/ckandoth/vcf2maf) for converting VCF into MAF files, with the annotation added by Variant Effect Predictor tool provided by Ensembl. (http://useast.ensembl.org/info/docs/tools/vep/index.html). Variants with missense and frame shift mutation in the 340 genes from the IMPACT panel were considered, and potential mutation locations were compared to NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp), cBioPortal (http://www.cbioportal.org/), and COSMIC (http://cancer.sanger.ac.uk/cosmic). Sanger PCR validation was performed subsequently.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
An aliquot of the RNA extracted above from frozen tissue (Trizol Reagent; Invitrogen; Grand Island, NY) was used to confirm the novel fusion transcript identified by FusionSeq. RNA quality was determined by Eukaryote Total RNA Nano Assay and cDNA quality was tested for PGK housekeeping gene (247 bp amplified product). Three micrograms of total RNA was used for cDNA synthesis by SuperScript ® III First-Strand Synthesis Kit (Invitrogen, Carlsbad, CA). RT-PCR was performed using the Advantage-2 PCR kit (Clontech, Mountain View, CA) for 30 cycles at a 64.5°C annealing temperature. Primers used are listed in Supplementary Table 1. Amplified products were purified and sequenced by Sanger method.
Fluorescence In Situ Hybridization (FISH)
FISH on interphase nuclei from paraffin-embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking genes NTRK1, TPM3, TPR, LMNA that were identified as potential fusion partners in the RNA-seq experiments or in the literature. BAC clones were chosen according to UCSC genome browser (http://genome.ucsc.edu), see Supplementary Table 2. The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with an incomplete set of signals were omitted from the score.
Immunohistochemistry (IHC)
Immunohistochemistry was performed on formalin-fixed paraffin-embedded tissue using 4 μm sections cut from paraffin blocks. Staining was performed using a fully automated system (Benchmark ULTRA, Ventana Medical Systems, Tuscon, AZ, USA) and the following antibodies were used: NTRK1 (Ab76291, 1:1,500, ABCAM), SOX10 (383A-75,1:50, CELL MARQUE), S100 (Z0311, 1:8,000, DAKO), H3 (9733, 1:100, CELL SIGNALING), CD34 (790-2927, VENTANA), SMA (VPS281, 1:50, VECTOR), Desmin (760-2513, VENTANA), Melan A (790-2990, VENTANA), STAT6 (SC-621, 1:2,500, SANTA CRUZ) and HMB45 (M0634, 1:100, DAKO).
RESULTS
Novel TPR-NTRK1 and TPM3-NTRK1 gene fusions in the S100 positive LPF-NT index cases
The FusionSeq bioinformatic algorithm detected an intra-chromosomal TPR-NTRK1 candidate gene fusion in the first index case (case 1, 7 year-old female, hand). The TPR (Translocated Promoter Region) gene on 1q25 and NTRK1 (Neurotrophic Tyrosine Kinase Receptor 1) gene on 1q21–22 are located 29 Mb apart with opposite directions of transcription. For a functional fusion transcript to occur, the translocation requires a break and inversion of the NTRK1 gene, with subsequent fusion to the 5’ portion of the TPR gene. The RT-PCR confirmed the candidate gene fusion showing exon 10 of TPR being fused to NTRK1 exon 9 (Fig 1). FISH break-apart test also confirmed the TPR and NTRK1 (Fig 1) gene rearrangements, while the FISH fusion assay confirmed the gene fusion.
Figure 1. TPR-NTRK1 gene fusion in LPF-like NT (case 1).

(A) Schematic representation of the intra-chromosomal fusion of TPR locus located on 1q25 with NTRK1 on 1q21–22 (29 Mb apart). As the 2 genes have opposite directions of transcription, the fusion requires a break and inversion of the NTRK1 gene. (B) Fusion candidates were validated by RT-PCR showing TPR exon 10 fused to NTRK1 exon 9. (C) FISH using NTRK1 break-apart probe shows a constant split between the red (centromeric) and green (telomeric) signals, in keeping with an intra-chromosomal inversion. (D) Exon expression of NTRK1 showing the increased expression following the break-point on exon 9, when compared to other fusion negative neoplasms.
Morphologically, the tumor showed relatively uniform spindle cells arranged in streaming fascicles, infiltrating the subcutaneous adipose tissue (LPF-like) (Fig. 3). The tumor cells showed mild cytologic atypia. Immunohistochemically, the tumor cells were multifocally positive for S100, positive for SMA and CD34 and negative for desmin, HMB45, Melan A and SOX10.
Figure 3. Morphologic features of TPR-NTRK1 and TPM3-NTRK1 fusion positive LPF-like NT.
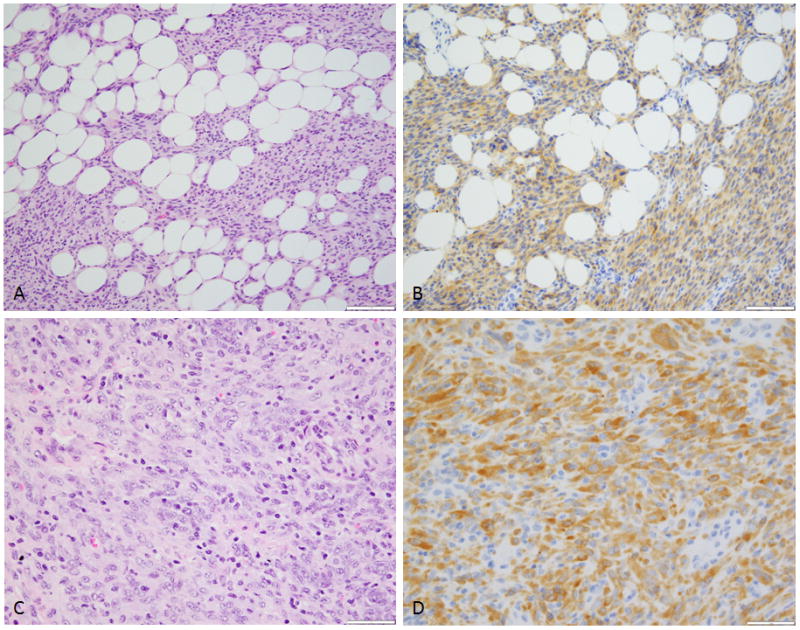
(A) Low power view reveals distinctive lipofibromatosis-like pattern of infiltration into fat, a hallmark feature of these lesions and (B) showing diffuse NTRK1 immunoreactivity (Case 1). (C) Higher power showing a densely cellular spindle cell neoplasm with mild nuclear atypia and (D) strong and diffuse NTRK1 immunostaining (Case 2).
The second index case (case 2, 10 year-old female, antecubital fossa) showed a TPM3-NTRK1 gene fusion by RNA sequencing. Similar to case 1, TPM3 and NTRK1 genes are also located on the long arm of chromosome 1, but at a closer distance of 2.7 Mb (1q22–23) from each other, and require a NTRK1 break/inversion event due to opposite directions of transcription. The RT-PCR validated the candidate fusion, with a TPM3 exon 6 being fused to NTRK1 exon 9 (Fig 2). Further, FISH assay confirmed the NTRK1 gene rearrangement.
Figure 2. TPM3-NTRK1 gene fusion in LPF-like NT (case 2).

(A) Schematic representation of the intra-chromosomal fusion of TPM3 locus located on 1q21.2 with NTRK1 on 1q21–22 (2.7Mb apart). The fusion involves a break and inversion of the TPM3 gene. (B) Fusion candidates were validated by RT-PCR showing TPM3 exon 6 fused to NTRK1 exon 9. (C) NTRK1 shows increased expression following the break-point on exon 9, when compared to other fusion negative neoplasms.
Morphologically, case 2 showed a cellular neoplasm composed of plump spindle cells with mild cytologic atypia. The tumor showed a fascicular pattern with focal infiltration within subcutaneous tissue (LPF-like) (Fig. 3). Immunohistochemically, the tumor cells were diffusely positive for both S100 protein and CD34, while being negative for SMA, desmin, STAT6, HMB45, Melan A and SOX10.
Clinicopathologic features of LPF-like NT cases
Based on the findings identified in the 2 index cases, we searched our consultation files for similar cases showing an infiltrative growth pattern reminiscent of LPF, but harboring an immunoprofile closer to neural tumors, with S100 protein reactivity. Originally rendered diagnoses included atypical LPF, low grade malignant peripheral nerve sheath tumor, and unusual benign peripheral nerve sheath tumor with infiltrative growth.
Together with the index cases there were 14 cases that fit the above morphologic and immunohistochemical description (Table 1). There were 8 females and 6 males, with an age range from 4 years to 38 years (median 13.5 years). Tumors were mostly subcutaneous and had a wide anatomic distribution, including upper extremity (6, arm, forearm, antecubital fossa and hand), lower extremity (5, buttock, thigh, leg and foot), head and neck (2, scalp, neck), and flank (1). Maximum dimension ranged from 1.3 to 5.4 cm.
TABLE 1.
Clinicopathologic features, genetic abnormalities and follow-up of LPF-NT cases.
| Case # | Age/Sex | Location | Mitoses /10HPF | Immunohistochemistry | Genetic abnormality | Follow-up | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S100 | CD34 | NTRK1 | LR | Duration | status | |||||
| 1* | 7 / F | hand | 1 | pos | pos | pos | TPR-NTRK1 | x 1 | 85 m | NED |
| 2* | 10 / F | antecubital | 0–1 | pos | pos | pos | TPM3 - NTRK1 | 10 m | NED | |
| 3 | 4 / F | thigh | 2 | pos | pos | pos | LMNA-NTRK1 | 8 m | NED | |
| 4 | 28 / F | flank | 0–1 | pos | n/a | neg | LMNA-NTRK1 | 2 m | NED | |
| 5 | 15 / M | forearm | 1 | pos | n/a | rare focal | LMNA-NTRK1 | n/a | n/a | |
| 6 | 12 / M | arm | 0–1 | pos | pos | pos | LMNA-NTRK1 | 1 m | NED | |
| 7 | 27 / F | forearm | 0–1 | pos | n/a | pos | NTRK1 | 54 m | NED | |
| 8 | 25 / M | foot | 0–1 | pos | focal | focal | NTRK1 | x1 | 60 m | NED |
| 9 | 38 / F | scalp | 1 | pos | pos | focal | NTRK1 | n/a | n/a | |
| 10 | 10 / M | leg | 0–1 | pos | pos | pos | NTRK1 | x 2 | 48 m | NED |
| 11 | 12 / M | thigh | 8 | pos | pos | n/a | ROS1 | x 1 | 9 m | NED |
| 12 | 14 / F | buttock | 0–1 | pos | neg | n/a | ALK | 36 m | NED | |
| 13 | 36 / M | arm | 0–1 | pos | pos | n/a | none | 36 m | NED | |
| 14 | 19 / F | neck/face | 0–1 | pos | pos | n/a | none | x 3 | 60 m | NED |
subjected to RNA sequencing; F, female; M, male; R, right; L, left; n/a, not available; pos, positive; neg, negative; LR, local recurrence; m, months; NED, no evidence of disease.
All cases showed distinct morphology of a cellular spindle cell neoplasm arranged in streaming fascicles and an invariably infiltrating pattern into surrounding subcutaneous adipose tissue and, in three cases, into skeletal muscle (Fig 4). The lesional cells showed palely eosinophilic cytoplasm, with indistinct cell borders, arranged in a sheet-like growth pattern. The cells showed fusiform nuclei with mild nuclear atypia and hyperchromasia, inconspicuous nucleoli, with some cases showing scattered pleomorphic cells (Fig 4).
Figure 4. Morphologic spectrum of NTRK1 positive LPF-like NT.
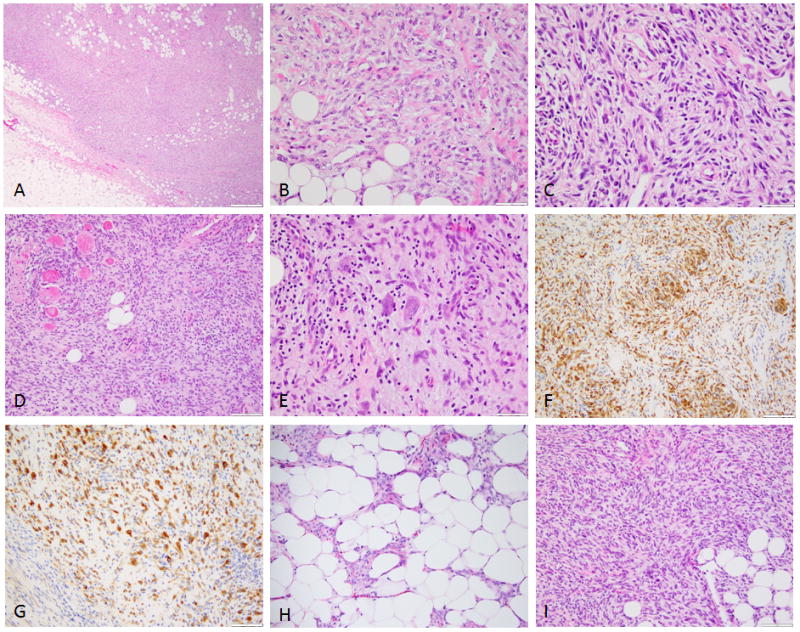
(A) Low power showing an infiltrative pattern within subcutaneous tissue and (B) at high power showing uniform spindle cells with open chromatin, separated by thick collagen bundles in a haphazard arrangement (Case 3, LMNA-NTRK1); (C) short fascicles of spindle cells with moderate nuclear atypia and hyperchromasia (Case 4, LMNA-NTRK1); (D) cellular LPF-like NT infiltrating fat and skeletal muscle, which at higher power (E) showed scattered pleomorphic nuclei and was strongly and diffusely positive for both (F) S100 and (G) NTRK1 (Case 6, LMNA-NTRK1); (H) Spindle cells intimately associated with adipose tissue (LPF-like, case 14); (I) densely cellular lesion with streaming fascicles infiltrating fat (Case 9, NTRK1 rearrangement positive).
Despite the increased cellularity and cytologic atypia, the mitotic activity was low, with 13 of the 14 cases showing less than 2 mitoses per 10 high power fields. Only one tumor (case 11) showed mitotic activity of up to 8 per 10 high power fields. None of the tumors showed areas of necrosis.
Immunohistochemically, all cases showed multifocal and quite extensive positivity for S100 protein (Fig 4). CD34 showed positivity ranging from focal to multifocal diffuse staining in 10 of the 11 cases tested. Focal SMA positivity was seen in 3 of 8 cases tested. None of the cases showed positivity for desmin, GFAP or melanocytic markers (SOX10, HMB45, Melan A). H3K27me3 expression was retained in all 6 cases analyzed. In one case initially diagnosed as a solitary fibrous tumor (case 2) due to diffuse CD34 reactivity, STAT6 was performed and was negative.
Most LPF-like NT show NTRK1 gene rearrangements, with common LMNA-NTRK1 gene fusions
Ten of the 14 (71 %) LPF-NT showed NTRK1 gene rearrangements by FISH analysis. In addition to TPR and TPM3 gene partners identified in the index cases, we additionally tested for LMNA gene abnormalities, as it has been previously described as a common NTRK1 fusion partner 12. Indeed, 4 NTRK1-positive LPF-NT showed LMNA gene rearrangement. LMNA is also located in close proximity to NTRK1 on 1q22 (0.7 Mb apart, centromeric). Both genes have similar directions of transcription and based on the FISH pattern noted in all 4 cases, the LMNA-NTRK1 fusion results from a simple interstitial deletion (Supplementary Fig 1). In the remaining 4 NTRK1-positive LPF-NT cases, FISH showed an NTRK1 inversion pattern, but no gene fusion partner was identified.
Four cases showed no NTRK1 gene rearrangements by FISH. Interestingly, one of these cases (case 11) showed ROS1 gene rearrangement and another (case 12) showed an ALK gene rearrangement (Supplementary Fig 2). Immunohistochemical stains for ROS1 and ALK were negative in the above 2 cases, respectively. No gene abnormalities were identified in the remaining 2 cases.
Clinical follow-up was available in 12 patients ranging from 1 month to 85 months (median 36 months) in duration. Local recurrence was seen in five (42%) cases (cases 1, 8, 10, 11 and 14), but none metastasized. All of the local recurrences occurred in patients’ in whom the initial excision had positive margins. The patients with negative surgical margins or subsequent re-excision with negative margins showed no evidence of relapse. All 12 patients with follow-up available were alive with no evidence of disease at the last follow-up.
NTRK1 immunohistochemistry is a sensitive marker for NTRK1-rearranged LPF-NT
NTRK1 cytoplasmic immunopositivity was noted in 9 of 10 cases with NTRK1 associated genetic abnormalities, with 6 of the 9 positive cases showing relatively diffuse staining and the remaining 3 showing focal positivity (Fig. 3, 4).
No NTRK1 gene rearrangements were detected in classic LPF
RNA sequencing performed on 2 classic LPFs showed no gene fusion candidates or other mutations. All tumors tested were negative for S100 protein by immunohistochemistry. Additional FISH performed on a control group of 25 classic LPFs showed no NTRK1 gene break-apart using gene specific probes. However, regional but variable abnormalities were detected in the 1q21–22 locus, which encompassed the NTRK1 gene in 19 of the 25 cases. Furthermore, NTRK1 immunohistochemistry performed in 12 LPF cases showed no positivity. Morphologically, all of the LPF cases showed a spindle cell component intricately admixed with mature adipose tissue. The spindle cells showed minimal to no cytologic atypia and were arranged in fascicles that infiltrated the adipose tissue. At the interface of the lesional cells with adipose tissue, some of the adipocytes were small in size, giving the false appearance of uni-vacuolated lipoblasts (pseudolipoblasts). Mitotic activity was low (0–1 per 10 high power fields) and no necrosis was identified.
DISCUSSION
We herein report a group of previously uncharacterized soft tissue tumors that have a distinct morphology, an immunoprofile characterized by S100 and CD34 positivity and recurrent gene fusions involving the NTRK1 gene. However, based on the immunoprofile, the line of differentiation of these tumors remains intriguing. CD34 positivity alone raises the possibility of fibroblastic differentiation, but the additional multifocal positivity for S100 protein suggests a neural or neuroectodermal lineage. Indeed, in many of these cases the cytologic atypia and the infiltrative growth pattern within adipose tissue were concerning for a low grade malignant peripheral nerve sheath tumor (MPNST). None of the patients with clinical data available had a history of type 1 neurofibromatosis (NF1). Further SOX10 immunostaining showed no reactivity in all 10 cases tested, while H3K27me3 expression, a marker recently reported to be lost in the majority of high grade MPNST, was retained in all 6 cases analyzed. 13,14
Mesenchymal neoplasms associated with recurrent gene fusions often lack a defined histogenesis or a predictable immunoprofile, with most tumors in this category having an ambiguous line of differentiation. One such example is ossifying fibromyxoid tumor, a translocation-positive neoplasm of unknown lineage, with consistent S100 protein reactivity. Another example is the biphenotypic sinonasal sarcoma, a recently described tumor with dual neural and myogenic differentiation, harboring a PAX3-MAML3 gene fusion. 15 In the latter example, the so-called ‘neural phenotype’ is defined on the basis of its S100 protein expression, in the absence of SOX10 staining. The results of our study provide yet another example with an NTRK1-fusion positive mesenchymal tumor showing possible neural differentiation based on its S100 protein reactivity.
The Neurotrophic Tyrosine Kinase Receptor 1 (NTRK1) gene encodes the TRKA receptor tyrosine kinase, the high affinity receptor for the Nerve Growth Factor (NGF). TRKA is a member of the neurotrophin receptor family of receptor tyrosine kinases that also includes TRKB and TRKC (encoded by the NTRK2 and NTRK3 genes, respectively).16–19 During embryogenesis, TRKA expression is critical for development and maturation of the central and peripheral nervous systems. In adults it is expressed in basal forebrain where it plays a role in memory processes, and in sympathetic and sensory neurons, where it is implicated in pain and temperature sensing.20–22 In normal tissues TRKA undergoes physiological activation through NGF-mediated dimerization, which induces autophosphorylation of specific tyrosine residues and transphosphorylation of a series of substrates, leading to activation of the PI3K/AKT, Ras/MAPK and PLC-g pathways.23
Genetic alterations of the NTRK1 gene, including translocations, amplifications, deletions and point mutations, have been observed in different tumor types, suggesting a potential role for TRKA in oncogenesis. NTRK1-related chromosomal rearrangements were first described in human papillary thyroid carcinoma (PTC), where it can be fused with either TPM3 or TPR genes on chromosome 1q25, or with TFG (TRK Fused Gene) on chromosome 3q11–12. 24,25 In a recent study, sequencing of 36 ‘pan-negative’ (i.e. wild-type for EGFR, KRAS, ALK, and ROS1) lung adenocarcinomas led to the identification of the novel MPRIP-NTRK1 and CD74-NTRK1 gene fusions in non-small cell lung cancer.26 More recently, NTRK1 chromosomal rearrangements have been identified in additional tumor types, including Spitzoid melanocytic neoplasms, intrahepatic cholangiocarcinoma, glioblastoma and pediatric high grade glioma 27–30, suggesting that oncogenic activation through gene fusions might occur at low frequency across many cancer types. Interestingly, a significant number of NTRK1-associated gene fusions reported 12 are the result of intra-chromosomal gene fusions, including RABGAP1L, TPM3, TPR, CHTOP, ARHGEF2, NFASC, BCAN and LMNA on chromosome 1. Depending on the directions of transcription of the NTRK1 gene and its fusion partner, these intra-chromosomal fusions can occur either through a simple interstitial deletion (e.g. LMNA-NTRK1) or through a more complex break/inversion mechanism (e.g. TPM3-NTRK1 or TPR-NTRK1) if the 2 genes are transcribed in opposite directions. In the latter scenario the low resolution of FISH might be suboptimal to detect these abnormalities in most cases, being closely dependent upon the level of proximity between the two genes.
Wiesner et al. 28 reported kinase fusions in Spitzoid melanocytic neoplasms including Spitz naevi, atypical Spitz tumors and spitzoid melanomas. Kinase fusions involved ROS1, ALK, NTRK1, BRAF and RET genes. NTRK1 gene fusions identified in these cases (16%) included LMNA-NTRK1 and TP53-NTRK1. Our study cohort of LPF-NT cases, in spite of the S100 protein positivity, is different from the Spitzoid neoplasms, lacking dermal or cutaneous involvement. Additionally, all of the melanocytic markers performed using immuno-histochemistry (SOX10 and HMB45) were negative, excluding melanocytic differentiation.
In the setting of soft tissue tumors, Doebele et al.31 identified 5 cases with NTRK1-related fusions by next generation sequencing among 1272 soft tissue sarcomas using the Foundation One Heme CGP Test. The fusions included LMNA-NTRK1 in 3 cases, SQSTM1-NTRK1 and TPM3-NTRK1, in one case each. No description of the morphologic diagnosis was provided, but 3 of the 5 patients were younger than 5 years of age. Furthermore, one of the patients with an undifferentiated sarcoma harboring an LMNA-NTRK1 fusion responded to LOXO1, a small molecule that targets the ATP binding site of the TRK receptor family. All the 3 LMNA-NTRK1 fusion cases in the study showed CDKN2A/B deletions. Interestingly, Wong et al32 recently described an infant patient with a congenital fibrosarcoma harboring an LMNA-NTRK1 gene fusion, who responded to crizotinib. This may possibly represent an example of LPF-like NT.
In a more recent study, Haller et al 33 described 4 patients (2 children and 2 adults) with soft tissue sarcomas with NTRK1 associated gene fusions, including TPM3-NTRK1 in 2 cases, LMNA-NTRK1 in 1 case and no identified gene partner in the remaining case. The NTRK1-rearranged tumors had a variable morphology and immunophenotype, including infantile hemangiopericytoma, myofibroblastic sarcoma and myopericytoma-like. Mitotic activity reported in these 4 cases ranged from 12 to 17 per 10 high power fields. Immunohisto-chemically, 2 of the cases showed focal SMA staining and CD34 positivity. In comparison, none of our study cases showed a hemangiopericytoma-like or myopericytoma-like morphology. Although cytologic atypia was identified in our cases, most of the cases had a low mitotic activity of less than 2 per 10 high power fields. The immunoprofile of our cases was also distinct with all 14 cases being positive for S100 and 10 of the 11 cases tested showing CD34 positivity. We have additionally performed NTRK1 FISH on 20 cases of myofibroma /myopericytoma, but no NTRK1 genetic abnormalities were found (data not shown). We believe that our S100-positive LPF-NTs represent a different pathologic entity from those reported by Haller et al. and therefore this report expands the spectrum of soft tissue tumors with NTRK1 related gene fusions.
The differential diagnosis of LPF-NT, either based on morphology or immunoprofile, includes many entities that involve superficial soft tissue and have an infiltrative pattern such as classic lipofibromatosis, fibrous hamartoma of infancy, calcifying aponeurotic fibroma, dermatofibrosarcoma protuberans, fibrosarcoma, malignant peripheral nerve sheath tumors and lesions in the myofibroma/ myopericytoma family. Classic LPF typically occurs in children with a predilection for the hands and feet and shows bland monomorphic spindle cells in fascicles intimately admixed with and infiltrating adipose tissue. The interface with adipose tissue shows characteristic pseuodolipoblast-like cells. Immunohistochemically, they show variable staining for CD34 and SMA and are negative for S100. Calcifying aponeurotic fibroma is composed of a bland appearing fibroblastic component with an infiltrative pattern resembling LPF, but showing distinctive foci of calcification and often osteoclast-like giant cells. Another pediatric fibroblastic proliferation, fibrous hamartoma of infancy, also shows overlapping features with LPF-NT, with the mature fibroblastic component intimately admixed with adipose tissue. However, its distinctive feature is the immature mesenchymal component of primitive spindle, stellate or ovoid cells and its negativity for S100 protein is a distinguishing feature from LPF-NT. Dermatofibrosarcoma protuberans with or without fibrosarcomatous transformation may show a similar morphology to LPF-NT of a dermal to subcutaneous lesion with bland spindle cells infiltrating the adipose tissue in a honeycomb pattern. Although they share CD34 immunoreactivity, DFSPs are consistently negative for S100. Furthermore, DFSPs are characterized by COL1A1-PDGFB gene fusion which can be tested by using PDGFB FISH probes. One of the lesions that LPF-NT can be difficult to differentiate from is low grade malignant peripheral nerve sheath tumor (MPNST). The similarities include atypical cells in fascicles in an infiltrative pattern of growth and the immunohistochemical co-expression of S100 and CD34. About half of MPNST arise in a background of a pre-existing neurofibroma in the setting of NF1. Clinical information available in 10 of our LPF-NT cases, showed no clinical evidence of NF1 and none of our cases showed a background of neurofibroma. Immunohistochemically, SOX10 was negative in all of our cases and H3K27me3 expression was not lost in any of our study cases, features that may further distinguish our LPF-NT cases from MPNSTs. One other differential, especially in the pediatric age group, are lesions in the myofibroma family of tumors. Such lesions are typically superficial and show cellular spindle cells arranged in bundles or whorls. These lesions are immunohistochemically positive for SMA, however, they do not show the characteristic infiltrative pattern of LPF-NT or positivity for S100 protein.
In conclusion, we describe a novel subset of soft tissue tumors with distinctive lipofibromatosis-like morphology, S100 immunopositivity and NTRK1-associated genetic abnormalities. Our study shows that NTRK1 immunohistochemistry is a useful diagnostic tool to identify these tumors. Although some of these tumors recurred locally, our follow-up data, albeit limited, showed no metastases or death from disease, in keeping with a benign/locally aggressive neoplasm. We propose the tentative name lipofibromatosis-like neural tumor. Given the recent therapeutic advances with targeted NTRK1 inhibitors, identifying these cases may make a significant impact on the management of these patients.
We have recently encountered an additional case that might fit the description of LPF-NT, which followed a more aggressive clinical course that is worth mentioning. The patient clinical history started 23 years ago when she was 37 years old with a superficial leg lesion, which based on imaging was thought to be a lipoma and was not excised. The mass was stable for more than 2 decades and only recently started to grow and was resected. Sections showed a cellular spindle cell neoplasm arranged in intersecting fascicles, with limited if any infiltrating pattern into adipose tissue. The tumor had clearly malignant features with an increased mitotic activity (18 MF/10 HPFs) and areas of necrosis. Immunhistochemically, the tumor was strongly positive for S100 and focally positive for CD34. Stains for SMA, desmin, SOX10, Cytokeratin, HMB45, Melan A and EMA were negative. FISH studies performed on the resection material showed evidence of LMNA-NTRK1 gene fusion. Immunohistochemical stain for NTRK1 showed focal weak positivity. Subsequent follow-up (33 months after diagnosis) revealed that the patient developed a 1 cm solitary lung nodule, which was biopsied and confirmed as metastatic disease. Although this case lacks the typical LPF pattern of infiltration seen in all the remaining cases, the presence of LMNA-NTRK1 fusion and similar immunoprofile raises the distinct possibility of a malignant counterpart of LPF-like NT. We speculate that the prolonged duration before surgical removal may have triggered additional genetic alterations leading to sarcomatous transformation and metastatic potential.
Supplementary Material
Schematic representation of the LMNA-NTRK1 gene fusion resulting from an interstitial deletion. LMNA and NTRK1 genes (inset) have similar directions of transcription and are located 0.7 Mb apart. FISH validation (case 4) showing LMNA break-apart assay (left panel, loss of the green telomeric signal); NTRK1 break-apart (center panel, loss of the centromeric red signal); while the LMNA-NTRK1 fusion assay (right panel) show the centromeric part of LMNA (red) fusing to the telomeric part of NTRK1 (green).
(A) ROS1 rearranged tumor (case 11) showing plump spindle cells in fascicles, in a partially collagenous background; (B) FISH using ROS1 break-apart probes showing a split (arrows) between the red (centromeric) and green (telomeric) signals; (C) Morphology of the ALK rearranged tumor (case 12) showing spindle to ovoid cells arranged in a fascicular pattern; (D) FISH using ALK break-apart probes showing a split with deletion of the centromeric (red) signal and retention of the telomeric (green) signal (arrows).
Acknowledgments
The authors acknowledge the referring physicians who also supplied clinical and follow-up data on the patients included in this study: Dr. M. Lemos, Lisbon, Portugal (2 cases), Drs. V. Verkarre and L. Galmiche, Paris, France, Drs. W. He and J. Wittig, Hackensack, NJ, Drs. S. Peters, S. Harawi and K. Song, Hackensack, NJ, Dr. M. Carter, Chico, CA, Dr. H. Domanski, Lund, Sweden, Dr. Thomas Klockl, Linkoping, Sweden, Drs. P. Connelly and A. Shrit, Dayton, OH, Prof. J.-M. Coindre, Bordeaux, France, Dr. A. Sassoon, Orange, CA, Dr. M. Lovell, San Antonio, TX, Dr. M. Hardt-Madsen, Svendborg, Denmark, Dr. M. Bak, Odense, Denmark.
Supported in part by: P50 CA 140146-01 (NPA, CRA), KAFC (CRA).
Footnotes
Conflict of interest: none
References
- 1.Fletcher C, Bridge JA, Hogendoorn PC, et al. WHO Classification of Tumours of Soft Tissue and Bone. IARC; Lyon: 2013. [Google Scholar]
- 2.Kenney B, Richkind KE, Friedlaender G, et al. Chromosomal rearrangements in lipofibromatosis. Cancer Genet Cytogenet. 2007;179:136–139. doi: 10.1016/j.cancergencyto.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Fetsch JF, Miettinen M, Laskin WB, et al. A clinicopathologic study of 45 pediatric soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J Surg Pathol. 2000;24:1491–1500. doi: 10.1097/00000478-200011000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Quail MA, Kozarewa I, Smith F, et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sboner A, Habegger L, Pflueger D, et al. FusionSeq: a modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol. 2010;11:R104. doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habegger L, Sboner A, Gianoulis TA, et al. RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics. 2011;27:281–283. doi: 10.1093/bioinformatics/btq643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. doi: 10.1158/2159-8290.CD-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am J Surg Pathol. 2016;40:479–489. doi: 10.1097/PAS.0000000000000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4–13. doi: 10.1038/modpathol.2015.134. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Bledsoe KL, Graham RP, et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet. 2014;46:666–668. doi: 10.1038/ng.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbacid M. Structural and functional properties of the TRK family of neurotrophin receptors. Ann N Y Acad Sci. 1995;766:442–458. doi: 10.1111/j.1749-6632.1995.tb26693.x. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan DR, Hempstead BL, Martin-Zanca D, et al. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 19.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 20.Greco A, Villa R, Tubino B, et al. A novel NTRK1 mutation associated with congenital insensitivity to pain with anhidrosis. Am J Hum Genet. 1999;64:1207–1210. doi: 10.1086/302319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Indo Y. Nerve growth factor and the physiology of pain: lessons from congenital insensitivity to pain with anhidrosis. Clin Genet. 2012;82:341–350. doi: 10.1111/j.1399-0004.2012.01943.x. [DOI] [PubMed] [Google Scholar]
- 22.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 24.Greco A, Miranda C, Pagliardini S, et al. Chromosome 1 rearrangements involving the genes TPR and NTRK1 produce structurally different thyroid-specific TRK oncogenes. Genes Chromosomes Cancer. 1997;19:112–123. [PubMed] [Google Scholar]
- 25.Greco A, Pierotti MA, Bongarzone I, et al. TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene. 1992;7:237–242. [PubMed] [Google Scholar]
- 26.Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469–1472. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J, Lee Y, Cho HJ, et al. NTRK1 fusion in glioblastoma multiforme. PLoS One. 2014;9:e91940. doi: 10.1371/journal.pone.0091940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116. doi: 10.1038/ncomms4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–242. doi: 10.1634/theoncologist.2013-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doebele RC, Davis LE, Vaishnavi A, et al. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 2015;5:1049–1057. doi: 10.1158/2159-8290.CD-15-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong V, Pavlick D, Brennan T, et al. Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv307. [DOI] [PubMed] [Google Scholar]
- 33.Haller F, Knopf J, Ackermann A, et al. Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol. 2016;238:700–710. doi: 10.1002/path.4701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of the LMNA-NTRK1 gene fusion resulting from an interstitial deletion. LMNA and NTRK1 genes (inset) have similar directions of transcription and are located 0.7 Mb apart. FISH validation (case 4) showing LMNA break-apart assay (left panel, loss of the green telomeric signal); NTRK1 break-apart (center panel, loss of the centromeric red signal); while the LMNA-NTRK1 fusion assay (right panel) show the centromeric part of LMNA (red) fusing to the telomeric part of NTRK1 (green).
(A) ROS1 rearranged tumor (case 11) showing plump spindle cells in fascicles, in a partially collagenous background; (B) FISH using ROS1 break-apart probes showing a split (arrows) between the red (centromeric) and green (telomeric) signals; (C) Morphology of the ALK rearranged tumor (case 12) showing spindle to ovoid cells arranged in a fascicular pattern; (D) FISH using ALK break-apart probes showing a split with deletion of the centromeric (red) signal and retention of the telomeric (green) signal (arrows).