

Abstract
Beige adipocytes gained much attention as an alternative cellular target in anti-obesity therapy. While recent studies have identified a number of regulatory circuits that promote beige adipocyte differentiation, the molecular basis of beige adipocyte maintenance remains unknown. Here, we demonstrate that beige adipocytes progressively lose their morphological and molecular characteristics after withdrawing external stimuli, and directly acquire white-like characteristics bypassing an intermediate precursor stage. The beige-to-white adipocyte transition is tightly coupled to a decrease in mitochondria, increase in autophagy, and activation of MiT/TFE transcription factor-mediated lysosome biogenesis. The autophagy pathway is crucial for mitochondrial clearance during the transition; inhibiting autophagy by UCP1+-adipocyte-specific deletion of Atg5 or Atg12 prevents beige adipocyte loss after withdrawing external stimuli, maintenaning high thermogenic capacity and protecting against diet-induced obesity and insulin resistance. The present study uncovers a fundamental mechanism by which autophagy-mediated mitochondrial clearance controls beige adipocyte maintenance, thereby providing new opportunities to counteract obesity.
Keywords: Beige adipocytes, obesity, diabetes, mitophagy, mitochondria
eTOC
When exposed to thermogenic stimuli, beige adipocytes transiently express UCP1 but lose that expression upon stimuli withdrawal. XXX et al investigate beige adipocyte maintenance and show that autophagy-mediated mitochondrial clearance is needed for beige to white adipocyte reversal. Inhibition of autophagy maintains functional beige adipocytes even after stimuli withdrawal.
Introduction
Brown adipose tissue (BAT) contains thermogenic adipocytes that dissipate energy in the form of heat as an evolutionally conserved defense mechanism against hypothermia. Recent works have uncovered that humans and rodents possess two distinct forms of thermogenic adipocytes; namely classical brown adipocytes and beige (or brite) adipocytes. Brown and beige adipocytes are both competent for thermogenesis via the brown/beige-specific protein uncoupled protein 1 (UCP1), and possess similar morphological characteristics, such as multilocular lipid droplets and highly abundant mitochondria (reviewed in (Kajimura et al., 2015). Nevertheless, brown and beige adipocytes arise from a distinct developmental origin. For instance, Kozak and colleagues demonstrated that the genetic variability in controlling Ucp1 expression was observed only in the subcutaneous WAT of mice but not in the interscapular BAT, suggesting that brown and beige adipocytes are under different regulation and may belong to distinct developmental lineages (Guerra et al., 1998; Xue et al., 2007). Furthermore, classical brown adipocytes develop prenatally from a dermomyotome population marked by Engrailed-1, Myf5, and Pax7, whereas beige adipocytes arise postnatally from progenitor populations in WAT expressing Ebf2, Pdgfra, and Sca1 (Atit et al., 2006; Lee et al., 2012b; Sanchez-Gurmaches et al., 2012; Schulz et al., 2011; Seale et al., 2008; Wang et al., 2014).
Of note, adult human BAT depots from the supraclavicular and other regions contain UCP1-positve adipocytes that exhibit molecular signatures resembling murine beige adipocytes (Lidell et al., 2013; Sharp et al., 2012; Shinoda et al., 2015a; Wu et al., 2012). Importantly, the selective activation of beige adipocyte biogenesis by genetic and pharmacological approaches leads to a protection from diet-induced obesity and insulin resistance (Cederberg et al., 2001; Ohyama et al., 2016; Seale et al., 2011; Shinoda et al., 2015b). In adult human BAT, glucose uptake activity is highly induced after prolonged cold exposure, in parallel with an increase in non-shivering thermogenesis and/or an improvement in insulin sensitivity, even in subjects who had previously lacked detectable BAT depots before chronic cold exposure (Lee et al., 2014b; van der Lans et al., 2013; Yoneshiro et al., 2013). These data all support the potential significance of beige adipocytes in human obesity and diabetes, and illuminate the importance of better understanding the molecular basis of beige adipocyte development and maintenance in order to pioneer future anti-obesity interventions.
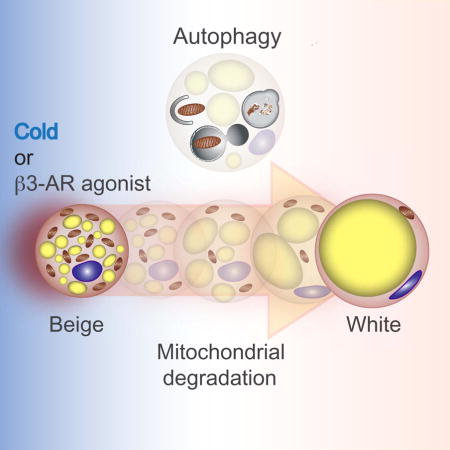
An essential characteristic of beige adipocytes is its dynamic regulation of the thermogenic gene program by external stimuli. Beige adipocytes can express high levels of UCP1 in response to chronic cold exposure or β3-AR agonists, whereas classical brown adipocytes constitutively express high levels of UCP1 (Kajimura et al., 2015). Intriguingly, UCP1 expression in the inguinal WAT became undetectable in mice within 2–3 weeks following transfer from cold environment to ambient or thermoneutral conditions (Gospodarska et al., 2015; Rosenwald et al., 2013). The cold-induced UCP1-positive beige cells became unilocular adipocytes that expressed several white adipocyte-enriched genes when re-acclimated to ambient temperature, some of which re-activate UCP1 expression in response to a subsequent bout of cold exposure (Rosenwald et al., 2013). However, the molecular mechanism of beige adipocyte maintenance remains poorly understood. Here, we demonstrate that beige adipocytes directly acquire a “white-like” phenotype after withdrawal of β3-AR agonist bypassing an intermediate precursor stage. We further show that the beige-to-white adipocyte transition is initiated by active mitochondrial clearance via autophagy. Accordingly, inhibition of autophagy by pharmacological or genetic approaches maintains thermogenically functional beige adipocytes for prolonged period of time even after withdrawal of β3-AR agonist or cold stimulus. Overall, we uncover a fundamental mechanism by which autophagy-mediated mitochondrial turnover controls beige adipocyte maintenance and whole-body energy homeostasis.
Results
Beige adipocytes directly acquire a white-like phenotype
We first utilized Ucp1Cre/+;Rosa26-GFP reporter mice and examined morphological and molecular changes of UCP1-positive beige adipocytes in vivo. Seven-day-treatment with the β3-AR agonist CL316,243 profoundly increased the number of GFP-positive beige adipocytes in the inguinal WAT (Fig. 1A and B). These adipocytes contained multilocular lipids and co-expressed UCP1. Of note, GFP-positive beige adipocytes were undetected in the inguinal WAT of the reporter mice under thermoneutrality (30°C), but highly induced in response to the β3-AR agonist treatment (Fig. S1A). Additionally, all GFP-positive adipocytes expressed endogenous UCP1 protein immediately following the β3-AR agonist treatment, further validating the experimental system (Fig. 1C and Fig. S1B). Notably, 15 to 20 days following β3-AR agonist withdrawal, GFP-positive adipocytes exhibited near-complete loss of multilocular lipids and endogenous UCP1 expression (Fig. 1B and C). A similar time-dependent decline in UCP1 protein expression was observed in the inguinal WAT depots after mice were acclimated from cold (6°C) to ambient temperature (Fig. S1C). In contrast, classical brown adipocytes in the interscapular BAT retained multilocular lipid droplets and expressed constitutively high levels of UCP1 even 30 days even after withdrawing β3-AR agonist (Fig. 1D and E).
Figure 1. Beige adipocytes directly acquire morphological characteristics of white adipocytes after withdrawing external stimuli.

(A) Schematic illustration of experiments to track beige adipocytes in vivo. Ucp1Cre/+;Rosa26-GFP reporter mice were treated with the β3-AR agonist CL316,243 at 1mg kg−1 for seven consecutive days. Interscapular BAT and inguinal WAT depots were harvested for morphological and molecular analyses at the indicated time points after β3-AR agonist withdrawal.
(B) Immunohistochemistry for GFP and endogenous UCP1 expression in the inguinal WAT from Ucp1Cre/+;Rosa26-GFP reporter mice. Inguinal WAT depots were harvested at indicated time points after β3-AR agonist withdrawal. Scale bar, 70 μm.
(C) Quantification of GFP-positive adipocytes that express endogenous UCP1 in (B). n = 150 cells or more per group.
(D) Immunohistochemistry for GFP and endogenous UCP1 expression in the interscapular BAT from Ucp1Cre/+; Rosa26-GFP reporter mice. BAT depots were harvested at indicated time points after β3-AR agonist withdrawal. Scale bar, 40 μm.
(E) Quantification of GFP-positive adipocytes that express endogenous UCP1 in (D). n = 127 cells or more per group.
(F) Morphological changes of beige adipocytes (top panel) and classical brown adipocytes (bottom panel) using the single-cell monitoring system. GFP-positive beige or brown adipocytes were isolated from Ucp1Cre/+;Rosa26-GFP reporter mice treated with the β3-AR agonist CL316,243 for seven days. Morphology of the individual GFP-positive adipocytes was monitored for 10 consecutive days. Scale bar, 70 μm.
(G) Quantification of GFP-positive beige adipocytes in (F, top panel). Stage of each cell was estimated based on the criteria shown in Figure S2B and C. n = 57 cells.
(H) Quantification of GFP-positive classical brown adipocytes in (F, bottom panel). n = 55 cells.
We postulate two potential explanations for the above results: 1) beige adipocytes de-differentiate to an intermediate precursor state, and subsequently re-differentiate into unilocular adipocytes, or 2) beige adipocytes directly acquire unilocular adipocyte characteristics without going through an intermediate precursor stage. To distinguish the above two possibilities, we developed a single-cell monitoring system and tracked morphological changes of the individual beige adipocytes ex vivo for 10 days following β3-AR agonist withdrawal (Fig. S2A). As shown in Fig. 1F and G, all of the freshly isolated beige adipocytes contained multilocular lipids and began to change morphology as early as day 3, eventually becoming unilocular adipocytes. By day 10, more than 80% of the GFP-positive beige adipocytes exhibited the unilocular lipid state (Stage III, Fig. 1G and Fig. S2B for defining adipocyte stages). Importantly, throughout these assays, we did not observe any GFP-positive fibroblast-like cells reminiscent of precursors (Fig. S2C). Consistent with the observations in vivo, the cultured beige fat progressively lost its thermogenic properties in parallel with these morphological changes (Fig. S2D). In stark contrast, classical brown adipocytes retained their multilocular lipid morphology up to 10 days under the same culture conditions, although an increase in lipid size was observed in some adipocytes (Fig. 1F and H). These data indicate that the beige adipocyte state is distinctly transient, and that there is a cell-intrinsic difference between beige adipocytes and classical brown adipocytes in maintaining the multilocular lipid state following β3-AR agonist withdrawal.
Since the unilocular lipid droplet is a morphological characteristic of white adipocytes, we employed global gene expression analyses to address whether the beige adipocyte-derived unilocular adipocytes indeed acquired the molecular characteristics of white adipocytes. To this end, we performed RNA-sequencing analysis of the following cell populations directly isolated from mice. First, we isolated GFP-positive adipocytes from the inguinal WAT of Ucp1Cre/+;Rosa26-GFP reporter mice by FACS, which were subject to RNA-sequencing analyses at 1, 5, 10, 15, and 30 days post β3-AR agonist withdrawal (Fig. 2A). As bona fide white adipocytes, we isolated GFP-positive adipocytes from the inguinal WAT of age-matched Adiponectin Cre/+;Rosa26-GFP reporter mice. Lastly, to obtain undifferentiated adipocyte precursors, we isolated Lin−/CD34+/CD29+/Sca1+ cells from the stromal vascular fraction (SVF) of inguinal WAT of age-matched wild-type mice. As shown in Fig. 2B, mRNA expression of the WAT-enriched genes, such as Resistin, Wfdc21, Spi2, Ednra, and Psat1 (Kajimura et al., 2008), were low in beige adipocytes at day 1 and day 5 following withdrawal of β3-AR agonist, however, began to increase 10 days post β3-AR agonist withdrawal. At day 30 of withdrawal, the WAT-enriched gene expression reached levels similar to bona fide white adipocytes. In parallel to this increase, we observed a concomitant progressive decline in mRNA expression of the brown/beige-selective thermogenic genes, such as Ucp1, Cidea, Cox8b, and Elovl3 (Fig. 2B).
Figure 2. Beige adipocytes directly acquire molecular characteristics of white adipocytes after withdrawing external stimuli.

(A) Top panel: Schematic illustration for isolating GFP-positive adipocytes by FACS at the indicated time points in the inguinal WAT of Ucp1Cre/+;Rosa26-GFP reporter mice. Bottom panel: Gating strategy for isolating GFP-positive adipocytes. GFP positive adipocytes were visualized after sorting at day 1 of β3-AR agonist withdrawal. Note that all the FACS-isolated cells (bright-field) express GFP and that all of the GFP positive cells from day 1 of β3-AR agonist withdrawal contained multilocular lipids.
(B) Expression profiles of the WAT-enriched genes and brown/beige fat-enriched genes in the GFP-positive FACS-isolated beige adipocytes at indicated time points after β3-AR agonist withdrawal as described in (A). The color scale shows z-scored FPKM representing the mRNA level of each gene in blue (low expression)-white-red (high expression) scheme. Gene expression in the white adipocytes FACS-isolated from the inguinal fat pad of age-matched AdiponectinCre/+;Rosa26-GFP reporter mice is shown in the right column.
(C) Principal component analysis (PCA) of transcriptome in FACS-isolated beige adipocytes (Ucp1Cre/+;Rosa26-GFP), FACS-isolated white adipocytes (AdiponectinCre/+;Rosa26-GFP), and undifferentiated adipocyte precursors (Lin−/CD34+/CD29+/Sca1+) from the SV fraction of inguinal WAT of age-matched wild-type mice. The number in parentheses represents the proportion of data variance explained by each PC.
(D) Hierarchical clustering of beige adipocytes, white adipocytes, and undifferentiated adipocyte precursors. The clustering was generated based on the RNA-sequencing data of GFP-positive beige adipocytes at day 1 of β3-AR agonist withdrawal (multi-locular state), at days 5, 10, and 15 of withdrawal (transition phase), and at day 30 of withdrawal (unilocular state). White adipocytes and undifferentiated precursors are shown in white and purple circles, respectively. The clustering was visualized by MeV. The horizontal distance represents similarities among each cluster.
Principal Component Analysis (PCA) during the transition phase indicates that the gene expression profiles of the GFP-positive adipocytes at day 30 of withdrawal exhibited a molecular signature resembling white adipocytes. Most importantly, the beige adipocytes at day 5, 10, and 15 of withdrawal progressively acquired the gene signature of white adipocytes, whereas all the beige adipocytes during the transition phase were far remote from the precursors (Fig. 2C). As an independent approach, hierarchical clustering based on the global gene signatures found that GFP-positive adipocytes at day 30 following β3-AR agonist withdrawal formed a cluster together with white adipocytes, which was clearly distinct from the beige adipocyte cluster at day 1 (Fig. 2D). The cluster analysis demonstrated that beige adipocytes during the transition phase (day 5, 10, and 15 of withdrawal) were truly distinct from that of preadipocytes. Altogether, our data provide evidence that beige adipocytes directly acquire both the morphological and molecular characteristics resembling white adipocytes after β3-AR agonist withdrawal, bypassing an intermediate precursor stage.
The beige-to-white adipocyte transition is coupled to mitochondrial clearance
To understand the mechanism by which beige-to-white adipocyte transition is regulated in vivo, we performed the Fuzzy C-Means (FCM) clustering analysis based on the obtained RNA-sequencing dataset and identified 9 distinct gene expression patterns during the beige-to-white adipocyte transition (Fig. S3A). The most frequently observed expression profile (cluster I) contained 1517 genes that were expressed highly in beige adipocytes immediately after the chronic treatment with β3-AR agonist (day 1), and progressively declined during the transition phase (Fig. 3A). This cluster contained brown/beige fat-selective mitochondrial genes, including Cox7a and Cox4i1, and key transcriptional regulators of mitochondrial biogenesis, such as Pgc1a, Pgc1b, Nrf1/2 and Tfam (Fig. 3B and Supplementary Table 1).
Figure 3. Beige-to-white adipocyte transition is accompanied by mitochondrial clearance.

(A) Gene expression profile of 1,517 genes that belongs to Cluster I during the beige-to-white adipocyte transition. Y-axis represents expression changes in the expression level (z-scored FPKM) of each gene. Gene expression profiles of other clusters are shown in Figure S3A.
(B) Expression profiles of brown/beige-enriched mitochondrial genes (Cox7a and Cox4i1) and key transcriptional regulators of mitochondrial biogenesis (Pgc1a, Pgc1b, Nrf1/2, and Tfam) in the GFP-positive adipocytes at indicated time points after β3-AR agonist withdrawal. The color scale shows z-scored FPKM representing the mRNA level of each gene in blue (low expression)-white-red (high expression) scheme. Gene expression in the white adipocytes isolated from AdiponectinCre/+;Rosa26-GFP reporter mice is shown in the right column. n = 3 for each time point of beige-to-white transition.
(C) GO analysis (cellular component) of the genes in Cluster I (GO FAT).
(D) GO analysis (biological process) of the genes in Cluster I (GO FAT).
(E) Inguinal WAT and BAT depots (3–5mm diameter) at indicated time points after β3-AR agonist withdrawal were fixed in 4% PFA and cleared for optical imaging.
(F) Immunoblotting for UCP1 and the indicate mitochondrial complex components in the inguinal WAT depots of wild-type mice under thermoneutrality and at indicated time points (days 0 – 30) following β3-AR agonist withdrawal. β-actin was used as a loading control. Molecular weight (MW) is shown on the right.
(G) Immunoblotting for UCP1 and the indicate mitochondrial complex components in the interscapular BAT depots of wild-type mice in (F).
The gene-annotation enrichment analysis found that the majority of the cluster I genes were related to mitochondrial components and function including electron transport chain and oxidative phosphorylation (Fig. 3C and D). Furthermore, mitochondria in the inguinal WAT depots, as visualized by optical tissue clearing, were abundant immediately after the chronic treatment with β3-AR agonist, but gradually became undetected at day 15 following β3-AR agonist withdrawal or thereafter (Fig. 3E, upper panel). In contrast, the interscapular BAT depots maintained high amounts of mitochondria even at day 15 or thereafter (Fig. 3E, lower). Consistent with this result, protein expression of multiple mitochondrial respiratory chain components in complex I, II and IV, followed the pattern of UCP1 expression in inguinal WAT: highly induced upon chronic treatment with β3-AR agonist and progressively declined during the transition, reaching basal levels at 15 days post β3-AR agonist withdrawal (Fig. 3F). In contrast, mitochondrial components in interscapular BAT were highly expressed and remained relatively unchanged even after β3-AR agonist withdrawal (Fig. 3G). This is likely due to active mitochondrial biogenesis, since transcriptional regulators of mitochondrial biogenesis, such as Pgc1a and Tfam, persist at high levels in the BAT following β3-AR agonist withdrawal (Fig. S3B). These data indicate that the beige-to-white adipocyte transition in inguinal WAT is tightly coupled to a progressive decline in mitochondria.
Activation of autophagy during the beige-to-white adipocyte transition
Mitochondrial content is tightly maintained by the balance between mitochondrial biogenesis and clearance. In fact, transcriptional regulators of mitochondrial biogenesis, including Pgc1a, Nrf1/2 and Tfam, were quickly down-regulated in the early phase of beige-to-white adipocyte transition (Fig. 3B). On the other hand, mitochondrial degradation is mediated by a form of autophagy, termed mitophagy (Klionsky et al., 2016). Notably, our RNA-sequencing analysis indicated an up-regulation of numerous core components of the autophagy machinery, including Atg4b, Atg12, and Atg16, during the beige-to-white adipocyte transition (Fig. 4A). In addition, many of the autophagy-related components and lysosomal enzymes, including Cts genes, Arsg, and Naga (Perera et al., 2015), were highly increased during the transition and remained high in unilocular adipocytes. The gene enrichments in the autophagy pathway and the lysosome pathway were highly significant, as Kurtosis (an indicator of peakedness of a distribution) in both pathways was platykurtic (K= −0.03 and −0.24, respectively), whereas that in randomly selected genes exhibited normal distribution (mesokurtic, K= 1.07) (Fig. 4B). Importantly, electron microscopic (EM) analyses of beige adipocytes during the transition phase identified a number of autophagic vacuoles containing remnant mitochondrial cristae structures (Fig. 4C and Fig. S4A–F), morphologically consistent with the induction of mitophagy (Klionsky et al., 2016).
Figure 4. Activation of autophagy during the beige-to-white adipocyte transition.

(A) Expression profile of the autophagy-related genes during the beige-to-white adipocyte transition. The color scale shows z-scored FPKM representing the mRNA level of each gene in blue (low expression)-white-red (high expression) scheme. n = 3 for each time point.
(B) Kurtosis of the autophagy and lysosomal genes in (A). Note that the autophagy and lysosome component genes were platykurtic (K= −0.03 and −0.24, respectively), while randomly selected genes showed mesokurtic distribution (K= 1.07).
(C) Electron microscopy images of beige adipocytes during the transition (days 5 – 30 following β3-AR agonist withdrawal). Black arrowheads indicate the autophagic vesicles containing mitochondrial remnants, as identified by remaining cristae (red arrowheads). Scale bar, 500 nm.
(D) Confocal microscopy images of beige adipocytes from GFP-LC3 mice. GFP-LC3 mice were treated with saline or the β3-AR agonist CL316,243 for seven consecutive days. The inguinal WAT depots were harvested at indicated time points (days 0 – 15) following β3-AR agonist withdrawal. Mitochondria and GFP-LC3-labelled autophagosomes were visualized by immunohistochemistry for Tom20 (red) and GFP (green), respectively. Nuclei are labeled with Hoechst (grey). The image in inset shows co-localization of GFP-LC3 and mitochondria. Scale bar, 12 μm.
(E) Quantification of the GFP-LC3 puncta in (A) at indicated time points. *** P <0.001 by Mann-Whitney U test. n = 20–30 cells per condition.
(F) Autophagic flux in adipocytes from GFP-LC3 mice at indicated time points (days 0 – 30) following β3-AR agonist withdrawal (beige adipocytes) and from GFP-LC3 mice treated with saline (white adipocytes). X-axis represents GFP-LC3 fluorescence intensity and Y-axis represents the number of adipocytes normalized to mode. Data are representatives of two independent experiments.
(G) Immunoblotting for NBR1, p62/SQSTM1, and LC3 (LC3-I and LC3-II) from lysates of adipocytes isolated from the inguinal WAT of wild-type mice treated with saline or the β3-AR agonist CL316,243 (day 0 and 30 following β3-AR agonist withdrawal). β-actin was used as a loading control. Data are representatives of 3 independent experiments. Molecular weight (MW) is shown on the right.
Based on these results, we sought to confirm whether autophagy was indeed occurring in vivo during the beige-to-white adipocyte transition. To this end, we used GFP-LC3 mice to assess the levels of punctate LC3, an indicator of autophagosome formation (Mizushima et al., 2004). We observed that these GFP-LC3 puncta were frequently co-localized with mitochondria, consistent with our findings by EM and suggestive of active mitophagy (Fig. 4D). To examine whether autophagy activity is regulated during the beige-to-white adipocyte transition, we performed the following experiments. First, we quantified the number of GFP-LC3 puncta in beige adipocytes during the transition; we observed a significantly lower number of GFP-LC3 puncta in beige adipocytes residing in the inguinal WAT of mice chronically treated with the β3-AR agonist (day 0), as compared to adipocytes in the inguinal WAT of saline-treated GFP-LC3 mice (Fig. 4D and E). The number of GFP-LC3 puncta was significantly increased at 5 days post β3-AR agonist withdrawal, and remained high 15 days following withdrawal. Second, we employed flow cytometric quantification of GFP-LC3 fluorescence levels to assess autophagic flux, as previously described (Shvets et al., 2008) (Fig. S4G). We observed a clear increase in GFP-LC3 levels in day 0 beige adipocytes treated with β3-AR agonist, as compared to white adipocytes from the saline-treated mice (Fig. 4F). After β3-AR agonist withdrawal, GFP-LC3 levels in beige adipocytes gradually decreased, indicative of increased autophagic flux in vivo, eventually reaching the levels seen in white adipocytes from the saline-treated mice (day 5, 15, and 30 in Fig. 4F). Lastly, we found that LC3-II was reduced in the beige adipocytes at day 0, which correlated with increased protein accumulation of the autophagy cargo receptors, NBR1 and p62/SQSTM1 (Fig. 4G), both of which are selectively degraded via autophagy (Klionsky et al., 2016). These markers subsequently returned to the basal levels observed in the saline-treated WAT at 30 days after β3-AR agonist withdrawal (Fig. 4G). Notably, LC3-II was reduced upon forskolin treatment in beige adipocytes in the presence and absence of the lysosomal inhibitor Bafilomycin A1 (BafA1) (Fig. S4H). These data collectively suggest that autophagy activity is low in beige adipocytes, whereas it is transiently re-activated during the beige-to-white adipocyte transition following β3-AR agonist withdrawal.
Autophagy in beige adipocytes is regulated by the cAMP-PKA pathway and the MITF transcription factor
Next, we aimed to identify the upstream regulatory circuits controlling autophagy during the beige-to-white adipocyte transition. DAVID (the Database for Annotation, Visualization and Integrated Discovery) analysis identified a “lysosome” gene ontology signature (P = 7.0 × 10−4 after Bonferroni correction) as the top biological pathway that was transiently elevated during the transition phase (Fig. 5A). Recent studies have highlighted the importance of transcriptional regulation in autophagosome formation and lysosome biogenesis by the MiT/TFE family of transcription factors (MITF, TFEB, and TFE3) (Perera et al., 2015; Sardiello et al., 2009; Settembre et al., 2011) as well as by other transcriptional regulators, such as FOXK (Bowman et al., 2014), FOXO3 (Warr et al., 2013), FXR/CREB (Lee et al., 2014a; Seok et al., 2014), and ZKSCAN3 (Chauhan et al., 2013). Therefore, we employed the Hypergeometric Optimization of Motif EnRichment analysis (HOMER) (Heinz et al., 2010) to identify conserved transcription factor binding motifs on the regulatory regions of the autophagy genes that were activated during the beige-to-white adipocyte transition. We found that the most enriched sequence motif from this analysis was the “CLEAR” consensus sequence (GTCACGTGAC) to which the MiT/TFE family of transcription factors (MITF, TFEB, and TEF3) are known to bind (P = 1.0 × 10−12) (Fig. 5B) (Sardiello et al., 2009; Settembre et al., 2011). The HOMER analysis also identified a FOXO-binding motif, however, this was much less enriched than the CLEAR binding element (P = 1.0 × 10−2) (Fig. S5A). In a completely independent unbiased analysis from our previous RNA-sequencing dataset (Shinoda et al., 2015a), we found that 91.6% (121 out of 132 genes) of the autophagy-related lysosome genes (Perera et al., 2015) were significantly down-regulated in the inguinal WAT by chronic cold exposure for 5 days (Fig. 5C). Importantly, 78.8% of the autophagy-related lysosome genes (104 out of 132 genes) were decreased both by chronic cold exposure and chronic administration of β3-AR agonist (Fig. 5D), indicating that cold exposure and β3-AR agonist similarly repress lysosome biogenesis in vivo. The HOMER-based motif analysis on the cold/β3-AR agonist-regulated lysosome gene signature similarly identified the CLEAR sequence as the most enriched transcription factor-binding site (not shown).
Figure 5. Regulation of autophagy-related lysosome biogenesis through the MiT/TFE transcription factors during the beige-to-white adipocyte transition.

(A) GO analysis (cellular component) of the genes that were transiently activated during the beige-to-white adipocyte transition (Cluster 2).
(B) The HOMER-based motif analysis of lysosome genes in (A).
(C) Expression of lysosome marker genes in the inguinal WAT of mice housed under cold or ambient temperature for 5 days. FPKM values were converted to z-score and visualized in blue (low)–white (no change)–red (high) color scheme. n=5.
(D) Regulation of the autophagy-related lysosome genes by cold exposure (shown in C) and by chronic β3-AR agonist treatment for 5 days. Note that 78.8% of the autophagy-related lysosome genes (104 out of 132 genes) were down-regulated both by cold exposure and β3-AR agonist.
(E) Relative expression of MiT/TFE members of transcription factors (Mitf, Tfe3, and Tfeb) during the beige-to-white adipocyte transition. * P <0.05, ** P <0.01 by two-tailed Student’s t-test. n = 3. Data are expressed as means ± s.e.m as compared to day 1 after β3-AR agonist treatment.
(F) Regulation of Mitf mRNA expression in response to cAMP in the presence or absence of a PKA inhibitor H89. Differentiated beige adipocytes were treated with by 10 μM forskolin (cAMP) for 4hr in the presence or absence of H89 at a dose of 10 μM. H89 was added 1 hr prior to forskolin treatment. * P <0.05, by two-tailed Student’s t-test. n = 3. Data are expressed as means ± s.e.m.
(G) mRNA expression of autophagy components that are known targets of MiT/TFE transcription factors. * P <0.05, ** P <0.01, *** P <0.001 by two-tailed Student’s t-test. n = 3. Data are expressed as means ± s.e.m.
(H) Regulation of Mitf mRNA expression in response to cAMP in a regular medium or amino acid depletion medium (starved). Differentiated beige adipocytes were cultured in amino acid free medium supplemented with 10% dialyzed serum for 4hr prior to forskolin (cAMP) treatment (10 μM, 4hr). ** P <0.01, *** P <0.001 by two-tailed Student’s t-test. n = 3. Data are expressed as means ± s.e.m.
(I) mRNA expression of the MI/TFE-target autophagy-related genes in response to cAMP under a fed or fasted state. * P <0.05, ** P <0.01, *** P <0.001 by two-tailed Student’s t-test. n = 3. Data are expressed as means ± s.e.m.
Of the three MiT/TFE family transcription factors, we found that Mitf expression was significantly induced during the initiation of beige-to-white adipocyte transition. In contrast, expression of Tfeb and Tef3 remained unchanged during the transition (Fig. 5E). Notably, previous studies (Perera et al., 2015; Sardiello et al., 2009; Settembre et al., 2011) have shown that all the autophagy-related lysosome genes, including Cts genes (Cathepsin gene family), and several autophagy components activated during the beige-to-white adipocyte transition (as listed in Fig. 4A) are direct targets of MiT/TFE transcription factors.
We further investigated the extent to which MITF and its downstream autophagy-lysosome signature are regulated by β-AR signaling in beige adipocytes. Protein kinase A (PKA) is well known to negatively regulate autophagy either by phosphorylation of LC3 or by activating mTORC1 which inhibits autophagy (He and Klionsky, 2009). On the other hand, activation of PKA in response to stimulation of β-AR positively promotes beige adipocyte development through transcriptional activation of the thermogenic gene program and mTORC1 (Liu et al., 2016). Thus, we hypothesized that activation of the PKA pathway via β-AR stimulation represses the autophagy network in beige adipocytes, whereas removal of the β-AR agonist leads to autophagy activation during the beige-to-white adipocyte transition. Accordingly, when differentiated beige adipocytes in culture were treated with forskolin (cAMP), Mitf expression was significantly decreased (Fig. 5F). Co-treatment with the PKA inhibitor, H89, largely alleviated both cAMP-mediated repression of Mitf levels (Fig. 5F) and LC3-II turnover (Fig. S4H), thereby corroborating a critical role of the PKA pathway for inhibiting autophagy in beige adipocytes. Consistent with the modest enrichment of a FOXO3 binding motif, Foxo3 expression was transiently activated during the beige-to-white adipocyte transition and repressed by cAMP through the PKA pathway (Fig. S5B and C). Importantly, the cAMP-PKA-mediated repression of Mitf and Foxo3 was accompanied by a transcriptional repression of their target genes encoding components of autophagy machinery, such as Wipi, Bnip, Bnip3l, and autophagy-related lysosome genes (Fig. 5G). Of note, the cAMP pathway was able to repress Mitf and autophagy-related lysosome gene expression in beige adipocytes even under starvation conditions, suggesting that the cAMP-mediated repression on autophagy occurs independently of nutritional cues (Fig. 5H and I). Taken together, these results indicate that autophagy in beige adipocytes is regulated by the cAMP-PKA pathway and the MITF transcription factor.
Autophagy-mediated mitochondrial clearance controls beige adipocyte maintenance
The results above motivate the hypothesis that autophagy-induced mitochondrial clearance is functionally required for beige adipocyte maintenance. Previous studies showed that genetic deletion of Atg7 via Fabp4-Cre resulted in increased beige adipocyte differentiation in vivo (Singh et al., 2009; Zhang et al., 2009). However, as Fabp4-Cre is active in brown, beige, and white adipocytes as well as some non-adipose tissues including skeletal muscle (Mullican et al., 2013), and because Atg7 deletion in skeletal muscle promotes beige adipocyte differentiation (Kim et al., 2013), the Fabp4-Cre model is not suitable to test the specific requirement of autophagy for “maintenance” of beige adipocytes. Thus, we used Ucp1-Cre mice (Kong et al., 2014) to generate mature brown/beige adipocyte-specific deletion of Atg5 or Atg12, two core autophagy regulators that are essential for the early steps of autophagosome formation (Mizushima and Komatsu, 2011). Although no Cre line currently exists to specifically target mature beige adipocytes without affecting classical brown adipocytes, this model allows us to test the requirement of autophagy for maintenance of newly developed beige adipocytes in response to cold or β3-AR agonist, given the specific expression of Ucp1 in mature brown and beige adipocytes.
The ATG12-ATG5 complex was deleted selectively in the BAT but not in the liver, using two systems: Ucp1Cre/+;Atg12flox/flox mice (Atg12Ucp1 KO), and Ucp1Cre/+; Atg5flox/flox mice (Atg5Ucp1 KO) (Fig. S6A). Atg5Ucp1 KO, Atg12Ucp1 KO mice or control mice (Atg5flox/flox or Atg12flox/flox, respectively) were treated with the β3-AR agonist CL316,243 for seven consecutive days (day 0 of β3-AR agonist withdrawal), and subsequently rested for 15 days after withdrawing β3-AR agonist (day 15 of withdrawal) (Fig. 6A). We further confirmed that Atg12 and Atg5 were significantly reduced in beige adipocytes from Atg12Ucp1 KO and Atg5Ucp1 KO mice, respectively (Fig. S6B).
Figure 6. Genetic ablation of Atg12 or Atg5 maintains beige adipocyte characteristics after removal of β3-AR agonist.

(A) Schematic illustration of experiments. Control (Atg12flox/flox or Atg5flox/flox), Atg12Ucp1 KO (Ucp1Cre/+;Atg12flox/flox), and Atg5Ucp1 KO (Ucp1Cre/+;Atg5flox/flox) mice were treated with the β3-AR agonist CL316,243 for seven consecutive days. Interscapular BAT and inguinal WAT depots were harvested for molecular analyses at day 0 and 15 following β3-AR agonist withdrawal.
(B) Immunoblotting for UCP1 and mitochondrial complexes (as indicated) in the inguinal WAT depots of control (Atg12flox/flox) and Atg12Ucp1 KO mice at day 0 and day 15 following β3-AR agonist withdrawal. Inguinal WAT depots from control and Atg12Ucp1 KO mice treated with saline were included as a reference of basal expression of UCP1 and mitochondrial complexes. β-actin was used as a loading control. Molecular weight (MW) is shown on the right.
(C) Immunoblotting for UCP1 and mitochondrial complexes (as indicated) in the inguinal WAT depots of control (Atg5flox/flox) and Atg5Ucp1 KO mice. Samples were harvested as illustrated in (B)
(D) Left; Mitochondrial DNA (mtDNA) transcripts (as indicated) were quantified in the inguinal WAT depots of control and Atg12Ucp1 KO mice at day 15 following β3-AR agonist withdrawal. Right; mRNA levels of nuclear-coded beige-enriched markers (as indicated) are shown. * P <0.05 by two-tailed Student’s t-test. n = 5. Data are expressed as means ± s.e.m.
(E) Top panel: Wild-type mice were housed at 6°C for 7 days and subsequently kept under thermoneutrality (30°C) for 15 days. During the re-warming period, the mice were treated with chloroquine at a dose of 60 mg kg−1 or saline. Inguinal WAT depots were harvested for molecular analysis. Bottom panel; Immunoblotting for UCP1 and mitochondrial complexes (as indicated) in the Inguinal WAT of mice. Molecular weight (MW) is shown on the right.
(F) Oxygen consumption rate (OCR) in the inguinal WAT depots of control and Atg12Ucp1 KO mice at day 15 following β3-AR agonist withdrawal. The isolated tissues were treated with isoproterenol or vehicle (basal). OCR data were shown per 1 mg of tissue. * P < 0.05, ** P < 0.01 by two-tailed Student’s t-test. n = 4. Data are expressed as means ± s.e.m.
(G) Quantification of whole-body oxygen consumption rate (VO2) of control and Atg12Ucp1 KO mice during day 17–18 following β3-AR agonist withdrawal. VO2 was measured by CLAMS during day and night time. ** P <0.01 by two-tailed Student’s t-test. n = 5 per genotype. Data are expressed as means ± s.e.m.
As shown in Fig. 6B, beige adipocyte biogenesis was highly induced both in control and Atg12Ucp1KO mice at day 0, as assessed by protein expression of UCP1, COX IV, and mitochondrial respiratory chain complexes. Mitochondrial DNA (mtDNA) transcripts and mRNA expression of nuclear-coded beige-enriched genes were significantly increased by chronic β3-AR agonist treatment both in control and Atg12Ucp1KO mice (Fig. S6C). No difference was found in the basal expression levels of UCP1 and mitochondrial components in inguinal WAT between control and Atg12Ucp1KO mice without β3-AR agonist treatment. These results indicate that beige adipocyte differentiation per se is intact in Atg12Ucp1KO mice in response to chronic β3-AR agonist treatment. However, at 15 days post β3-AR agonist withdrawal, we observed a striking difference between control and Atg12Ucp1 KO mice; the inguinal WAT from the Atg12Ucp1 KO mice expressed higher levels of UCP1, COX IV, and mitochondrial respiratory chain complexes, as compared to that from control mice (Fig. 6B). Importantly, a similar trend in UCP1 and mitochondrial protein expression was observed in the inguinal WAT from Atg5Ucp1 KO mice (Fig. 6C). Furthermore, mtDNA transcripts, such as Nd2, Cox2, and Cox3, were significantly higher in the inguinal WAT of Atg12Ucp1 KO mice at day 15 following β3-AR agonist withdrawal (Fig. 6D, left), whereas no significant differences in the mRNA expression of nuclear coded beige-enriched genes, such as Pgc1a, Ucp1, and Cox7a, were present between control and Atg12Ucp1 KO mice (Fig. 6D, right). The higher expression of UCP1 and mitochondrial proteins were preferentially found in beige adipocytes, with no major changes observed in classical brown adipocytes residing in interscapular BAT depots; rather, the interscapular BAT of Atg12Ucp1 KO mice expressed similar levels of UCP1 and mitochondrial proteins as compared to control mice after β3-AR agonist withdrawal (Fig. S6D). We also observed comparable, and in certain instances, lower levels of nuclear coded transcripts in the interscapular BAT of Atg12Ucp1 KO mice as compared to that of control mice at day 0 and day 15 after β3-AR agonist withdrawal (not shown). Similarly, no major difference was observed in the expression of UCP1 and mitochondrial contents in the interscapular BAT depots between control and Atg5Ucp1 KO mice at day 0 and day 15 after β3-AR agonist withdrawal (Fig. S6E).
Next we asked whether Ucp1-specific deletion of Atg12 similarly lead to high levels of mitochondria in the inguinal WAT after re-warming period following cold exposure. Consistent with the findings after β3-AR agonist withdrawal, we found that the inguinal WAT from the Atg12Ucp1 KO mice expressed higher levels of UCP1 and mitochondrial respiratory chain complexes, as compared to that from control mice at 15 days after re-warming (Fig. S6F). We further asked whether pharmacological inhibition of autophagy was able to retain high levels of UCP1 and mitochondrial contents. To this end, we treated mice with chloroquine (CQ) at a dose of 60 mg kg−1 or saline for 15 consecutive days during the re-warming period following cold exposure (Fig. 6E, top). We found that pharmacological inhibition of autophagy led to a significant retention of higher UCP1 levels and mitochondrial proteins in the inguinal WAT after re-warming (Fig. 6E, bottom). Moreover, chloroquine treatment following β3-AR agonist withdrawal significantly induced LC3 accumulation in beige adipocytes (Fig. S6G) and maintained higher levels of UCP1 and mitochondrial proteins in the inguinal WAT after β3-AR agonist withdrawal (Fig. S6H). These data indicate that autophagy-mediated mitochondrial clearance via Atg5 and Atg12 is required for efficient beige-to-white adipocyte transition.
The distinct effects of autophagy deletion on beige adipocyte maintenance motivated the intriguing hypothesis that Atg12Ucp1 KO mice would exhibit higher thermogenic capacity after the removal of external cues. To test this hypothesis, we first measured oxygen consumption rate (OCR) of the inguinal WAT from control and Atg12Ucp1 KO mice at 15 days post β3-AR agonist withdrawal. As shown in Fig. 6F, OCR was significantly higher in the inguinal WAT of Atg12Ucp1 KO mice than control mice when the tissues were treated with isoproterenol. In contrast, no significant difference was observed without isoproterenol treatment (at basal state). Thus, the beige adipocytes that persisted in Atg12Ucp1 KO were thermogenically active in response to cAMP stimulation.
To examine the metabolic significance of retaining thermogenic beige adipocytes in vivo, we next measured whole-body energy expenditure (VO2) of control and Atg12Ucp1 KO mice during 17 to 18 days post β3-AR agonist withdrawal. As shown in Fig 6G, Atg12Ucp1 KO mice exhibited significantly higher VO2 levels compared to control mice during the night phase. On the other hand, no significant difference was found in locomotor activity and food intake between control and Atg12Ucp1 KO mice (Fig. S7A).
Prolonged maintenance of beige adipocytes prevents diet-induced obesity and insulin resistance
Obesity is known to impair beige adipocyte biogenesis, partly through activation of TGFβ and Notch signals in WAT (Bi et al., 2014; Yadav et al., 2011). Here, we determine the extent to which obesity also affects the kinetics of beige-to-white adipocyte transition. To this end, we examined the morphological change of beige adipocytes in the inguinal WAT using Ucp1-Cre; mT/mG reporter mice under a regular diet (body weight, 29.5 ± 1.4 g) and age-matched obese mice under a high-fat diet for 12 weeks (body weight, 49.8 ± 0.8 g). We found that beige adipocytes (i.e., UCP1+/GFP+ multilocular adipocytes) in the inguinal WAT of obese mice acquired a “white-like” state (i.e., unilocular lipids and loss of UCP1 expression) at a faster rate than age-matched lean mice (Fig. 7A and B). On the other hand, no major change was found in the morphology of UCP1+ brown adipocytes between obese and lean mice (Fig. S7B and C). This observation is intriguing because recent studies indicate that autophagy is altered in the adipose tissues of obese and type 2 diabetes patients (Jansen et al., 2012; Kosacka et al., 2015; Kovsan et al., 2011; Nunez et al., 2013; Ost et al., 2010). For instance, obesity-induced insulin resistance and type 2 diabetes impair mTOR signaling, thereby leading to autophagy activation in human adipose tissues (Kosacka et al., 2015; Ost et al., 2010). These results indicate that obesity not only impairs beige adipocyte differentiation but also accelerates the beige-to-white adipocyte transition, at least in part, through the activation of autophagy-lysosome biogenesis.
Figure 7. Prolonged maintenance of beige adipocytes by autophagy inhibition protects animals from diet-induced obesity and insulin resistance.

(A) Confocal images of fixed inguinal WAT sections from Ucp1Cre/+;mT/mG reporter mice. Inguinal WAT depots from lean mice under a regular diet (top panel) and age-matched obese mice under a high-fat diet (bottom panel) were immunostained for endogenous UCP1 (Red). Note that the cellular membranes of beige adipocytes were visualized by membrane-targeted GFP (mGFP, Green) of the mT/mG reporter mice. Scale bar, 57 μm.
(B) Quantification of mGFP-positive adipocytes in lean and obese mice that express endogenous UCP1 in (A). n = 100 cells or more per group. ** P <0.01, *** P <0.001 by two-tailed Student’s t-test.
(C) Schematic of the metabolic experiment in control (Atg12flox/flox) and Atg12Ucp1 KO mice. Control and Atg12Ucp1 KO mice were treated with CL316,243 for seven days to induce beige adipocyte biogenesis. Subsequently, the mice were acclimated to thermoneutrality (30 °C) under a high-fat diet for 8 weeks.
(D) Body weight of control (Atg12flox/flox) and Atg12Ucp1 KO mice under a high-fat diet. Body weight was measured twice a week. * P <0.05, ** P <0.01. n = 8 – 10 per genotype. The graph in the inset shows body weight gain of control and Atg12Ucp1 KO mice. Significance was determined by two-way repeated-measures ANOVA followed by Fisher’s LSD test. Data are expressed as means ± s.e.m.
(E) Body composition of control (Atg12flox/flox) and Atg12Ucp1 KO mice from (D) at the end of 8 weeks of high-fat diet. * P <0.05 by two-tailed Student’s t-test. Data are expressed as means ± s.e.m.
(F) Tissue weight of inguinal WAT, epididymal WAT, and liver from control (Atg12flox/flox) and Atg12Ucp1 KO mice from (D) after 9 weeks of high fat diet. * P <0.05, ** P <0.01. Data are expressed as means ± s.e.m.
(G) Liver triglyceride levels in control (Atg12flox/flox) and Atg12Ucp1 KO mice after 9 weeks of high fat diet. *** P <0.001. Data are expressed as means ± s.e.m.
(H) After 8 weeks of high-fat diet, control (Atg12flox/flox) and Atg12Ucp1 KO mice were fasted for 12 hours and injected with 1.5g kg−1 glucose. Whole-body glucose was measured at 15, 30, 60, 90, 120, and 150 min. * P <0.05, n = 6 – 8 per genotype. Significance was determined by two-way repeated-measures ANOVA followed by Fisher’s LSD test. Data are expressed as means ± s.e.m.
(I) After 8.5 weeks of high fat diet diet, control (Atg12flox/flox) and Atg12Ucp1 KO mice were fasted for 3 hours and injected with 0.75 U kg−1 insulin. Whole-body glucose was measured at 15, 30, 45, 60, 75, and 90 min. * P <0.05, ** P <0.01, *** P <0.001, n = 7–8 per genotype. Significance was determined by two-way repeated-measures ANOVA followed by Fisher’s LSD test. Data are expressed as means ± s.e.m.
The above results motivated us to ask whether the persistence of thermogenic beige adipocytes in Atg12Ucp1 KO mice impacts weight gain in response to an obesogenic diet. Based on the previous observation that Ucp1 deletion induces obesity specifically under conditions of thermoneutrality (Feldmann et al., 2009), individually-housed control and Atg12Ucp1 KO mice were chronically treated with the β3-AR agonist CL316,243 for seven consecutive days to induce beige adipocyte development, and the mice were subsequently fed a high-fat diet for 8 weeks under thermoneutrality (Fig. 7C). While there was no significant difference in body weight between control and Atg12Ucp1 KO mice immediately after β3-AR agonist treatment (day 0), Atg12Ucp1 KO mice gained significantly less body weight than control mice after acclimation to thermoneutrality (Fig. 7D). The difference in body weight between control and Atg12Ucp1 KO was due to a significantly reduced adipose mass, but not due to changes in lean mass (Fig. 7E). Consistent with this result, white adipose tissue mass (inguinal WAT and epididymal WAT) in Atg12Ucp1 KO mice was lower than control mice (Fig. 7F). Liver mass was slightly but significantly lower in Atg12Ucp1 KO mice, likely due to reduced hepatic triglyceride (TG) contents in Atg12Ucp1 KO mice (Fig. 7G). Importantly, after 8 weeks of high-fat diet feeding, Atg12Ucp1 KO mice exhibited significantly improved systemic glucose homeostasis compared to control mice, as assessed by glucose-tolerance test (Fig. 7H) and insulin-tolerance test (Fig. 7I). In contrast, such metabolic phenotypes were not observed in the absence of β3-AR agonist treatment (Fig. S7D–F). Thus, the metabolic phenotypes, i.e., reduced body-weight gain and improved glucose homeostasis, found in Atg12Ucp1 KO mice after β3-AR agonist treatment, are largely due to retention of thermogenically active beige adipocytes that are recruited by chronic β3-AR agonist treatment. These observations are consistent with the above finding that Atg12Ucp1 KO mice maintain higher amounts of UCP1 and other mitochondrial proteins in the inguinal WAT for prolonged periods compared to autophagy-competent controls, specifically following withdrawal of β3-AR agonist. Altogether, these data indicate that prolonged maintenance of thermogenically active beige fat is sufficient to increase whole-body energy expenditure and protect mice from diet-induced obesity and insulin resistance.
Discussion
The present study demonstrates that autophagy-induced mitochondrial turnover is crucial for beige adipocyte maintenance and energy expenditure in vivo. Accumulating evidence shows that beige adipocyte biogenesis is induced by a variety of external stimuli, such as chronic cold exposure, exercise, long-term treatment of PPARγ agonists, cancer cachexia, and environmental enrichment (reviewed in (Kajimura et al., 2015). The induced beige adipocytes appear to arise from de novo differentiation of beige precursors (Wang et al., 2013) or direct conversion from mature white adipocytes (Barbatelli et al., 2010; Himms-Hagen et al., 2000; Lee et al., 2015). Regardless of cellular origin, the newly recruited beige adipocytes gradually lose their morphological and molecular characteristics upon removal of external cues. Given the nature of lineage-tracing experiments, however, the prior studies were not able to determine whether this transition is mediated through de-differentiation of beige adipocytes to an intermediate precursor state and subsequent re-differentiation into white adipocytes, or through a direct conversion. Moreover, the inducible Cre-ER system used for the previous work may have certain technical limitations for lineage tracing; a recent study showed that newly recruited beige adipocytes during the chase phase (i.e., after tamoxifen withdrawal) may be unintentionally labeled because the hydrophobic properties of tamoxifen make it difficult to “wash-out” in adipose tissues (Ye et al., 2015). Our data provide direct evidence that beige adipocytes possess cell-intrinsic capacity to acquire a white-like state bypassing an intermediate precursor stage. Future analysis of chromatin reorganization and epigenetic regulation during this transition will additionally uncover the fundamental mechanisms by which environmental cues control beige adipocyte maintenance.
While recent studies reported a variety of external and internal cues that promote beige adipocyte differentiation, the molecular mechanism of beige adipocyte “maintenance” remains unknown. Genetic knockout of ATG7, the E1-like enzyme required for autophagosome formation, results in increased beige adipocytes in WAT, indicating a role of autophagy in beige adipocyte differentiation (Singh et al., 2009; Zhang et al., 2009). However these studies used Fabp4-Cre system, leading to knockout of ATG7 in all types of adipocytes and non-adipose tissues such as skeletal muscle and brain (Mullican et al., 2013). Thus it remained unclear whether autophagy was involved in the specific differentiation of beige adipocyte from precursors, or maintenance of mature beige adipocytes. In addition, because ATG7 controls p53-dependent transcription and cell cycle progression independently of its E1-like enzymatic activity, it is difficult to ascertain a general role for autophagy in those previous studies (Lee et al., 2012a). Thus, we selectively deleted either Atg5 or Atg12 in differentiated beige/brown adipocytes using Ucp1-Cre in order to test the specific requirement of autophagy for beige adipocyte maintenance per se. While ATG5 and ATG12 each possess unique functions in other cell types (Kimmey et al., 2015; Malhotra et al., 2015), we demonstrate that genetic deletion of Atg5 or Atg12 in beige adipocytes exhibit highly concordant phenotypes, most notably, the substantial retention of UCP1 and mitochondrial proteins in the subcutaneous WAT after withdrawing β3-AR agonist (Fig. 6). These results strongly argue against any individual effects exerted by these ATGs. In further support, pharmacological autophagy inhibition with the anti-malarial chloroquine retains high levels of UCP1 and mitochondrial proteins after re-warming following cold exposure as well as after β3-AR agonist withdrawal. Taken together, these results, obtained using genetic and pharmacological approaches, corroborate a critical requirement for the autophagy pathway in clearance of beige adipocyte mitochondria during the beige-to-white transition in vivo, thereby intimating a specific role of mitophagy in beige adipocyte maintenance.
Recent studies reported that adult human BAT from supraclavicular regions displays molecular signatures that resemble beige adipocytes (Lidell et al., 2013; Sharp et al., 2012; Shinoda et al., 2015a; Wu et al., 2012) and that chronic cold acclimation increases glucose uptake in the BAT of adult humans who do not possess detectable BAT before cold treatment (Lee et al., 2014b; van der Lans et al., 2013; Yoneshiro et al., 2013). These studies indicate that adult humans possess beige-like “recruitable” thermogenic adipocytes. Notably, the prevalence of human BAT is inversely correlated with BMI and adiposity (Cypess et al., 2009; Saito et al., 2009; van Marken Lichtenbelt et al., 2009), whereas autophagy is up-regulated in adipose tissue of obese subjects, exhibiting a positive correlation with the degree of obesity and visceral fat distribution (Jansen et al., 2012; Kosacka et al., 2015; Kovsan et al., 2011; Nunez et al., 2013; Ost et al., 2010). Our studies in rodents also indicate that obesity accelerates the beige-to-white adipocyte transition. It is conceivable that the altered kinetics of the transition under obesity is due partly to the activation of autophagy-related lysosome biogenesis; thus, autophagy/lysosome inhibition can be an effective approach to retain high thermogenically active beige adipocytes for prolonged period.
It has been appreciated that classical brown adipocytes in the interscapular BAT can acquire a “white-like” unilocular morphology in morbidly obese mice, such as ob/ob mice, or in aged mice (Cinti, 1999; Sellayah and Sikder, 2014). Our experiments, on the other hand, were performed in young mice under a relatively short-term high fat diet (4–8 weeks) in which mitochondrial biogenesis remained active in the BAT. It is likely that mitochondrial biogenesis rather than mitochondrial clearance largely contributes to the maintenance of high mitochondrial contents in BAT, whereas mitochondrial clearance plays a critical role in the mitochondrial homeostasis of beige fat particularly when external cues are withdrawn. Our data, however, do not exclude the possibility that autophagy-mediated mitochondrial clearance also play a role in the maintenance of classical brown adipocytes in morbidly obese mice or aged mice.
In summary, the present study identified autophagy-mediated mitochondrial clearance as a previously unappreciated mechanism that controls beige adipocyte maintenance and whole-body energy homeostasis. This may offer a new therapeutic opportunity to combat obesity and insulin resistance through prolonged maintenance of thermogenic beige adipocytes.
Experimental Procedures
Animals
All animal experiments were performed under the guidelines established by the UCSF Institutional Animal Care and Use Committee. Details of the transgene design can be found in the Supplemental Experimental Procedures. To visualize brown and beige adipocytes in vivo, Ucp1Cre/+ mice were crossed with Rosa26-GFP or mT/mG mice (The Jackson Laboratory). To induce beige adipocyte biogenesis, the β3-AR agonist CL316,243 (Sigma) was administered intraperitoneally to male mice at a dose of 1mg kg−1 for seven consecutive days. To assess the basal amount of GFP-positive cells present in adipose tissues prior to β3-AR agonist treatment, mice were bred and treated with β3-AR agonist at thermoneutrality (30 °C). For cold exposure experiments, animals were single-caged and exposed to 6 °C for five to seven days.
Ex vivo monitoring of beige and brown adipocytes
Mature adipocytes from the inguinal WAT and interscapular BAT depots of CL316,243-treated Ucp1Cre/+;Rosa26-GFP mice were isolated by fractionation, embedded in collagen gel containing 2.5 mg ml−1 Collagen I (BD354236), 1 μg ml−1 Fibronectin (Millipore), and 0.1% BSA. The cell-containing gel was allowed to solidify at 37°C for 30 min and cultured in DMEM supplemented with 10% FBS. Culture medium did not contain any stimuli, such as β3-AR agonist, that control beige adipocyte differentiation. Individual live GFP+ adipocytes were traced daily for ten consecutive days using the In Cell Analyzer 2000 to define and re-image the position of interest over time. Images were processed using In Cell Developer Toolbox V1.8 (GE Healthcare Life Sciences) and Volocity 6.1.1 software (Improvision).
Flow Cytometry
To isolate GFP-positive adipocytes, inguinal WAT depots from Ucp1Cre/+;Rosa26-GFP or AdiponectinCre/+;Rosa26-GFP mice were digested to single cells according to the previous study (Ohno et al., 2012). Adipocytes were first gated based on size and granularity. Live GFP-positive cells were gated based on GFP-negative control cells from wild-type mice not expressing GFP.
RNA-sequencing and Bioinformatics
Total RNA was extracted from FACS-isolated GFP-positive adipocytes or preadipocytes using the RNeasy Micro Kit (Qiagen). Libraries were constructed from minimum of 22 ng of total RNA as previously described (Shinoda et al., 2015a). High-throughput sequencing was performed using a HiSeq 2500 instrument (Illumina) at the UCSF Institute for Human Genetics core facility. Reproducibility of the RNA-sequencing data was confirmed by sequencing the identical RNA-samples (R2 = 0.983). The data were deposited in ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession numbers E-MTAB-3978.
Clustering of genes was performed as follows: the FPKM profiles of genes at several time points (day 0 – 30 of β3-AR agonist withdrawal) were z-transformed, as such, the mean for each gene was zero and standard deviation was one. The Fuzzy C-Means (FCM) clustering analysis was described elsewhere (Olsen et al., 2006). The top 10,000 highly expressed genes were used for the clustering. The FCM parameters were c = 9 and m = 1.15, respectively. For principal component analysis (PCA), preadipocytes and white adipocytes were first mapped onto two-dimensional (PC1 and PC2) space, based on the expression of 10,138 genes that showed differences between the two groups by 2-fold or more. The transcriptome of GFP-positive adipocytes at each time point (day 0–30) was mapped on the same PC plot, based on the expression levels of 10,138 genes. Similar PC plots were reproducibly found when different number of genes (2,000, 1,000, or 500) were applied.
Metabolic studies
Ten-week-old Atg12Ucp1 KO mice (Ucp1Cre/+;Atg12flox/flox) and littermate control mice (Atg12 flox/flox) were treated with β3-AR agonist for 7 days and maintained at thermoneutrality during β3-AR agonist withdrawal. Whole-body energy expenditure was measured between 17 and 18 days after β3-AR agonist withdrawal using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments). Locomotor activity was simultaneously monitored by the CLAMS. For diet-induced obesity study, 9-week-old Atg12Ucp1 KO and control (Atg12 flox/flox) male mice were fed a high-fat diet. Following seven-day β3-AR agonist treatment, mice were transferred to thermoneutrality (30 °C) for 8 weeks. Body weight was measured twice per week. Glucose tolerance test experiments were performed at 8 weeks or 12 weeks of high-fat diet. Insulin tolerance test was performed after 8.5 weeks of high-fat diet.
Supplementary Material
Highlights.
Beige adipocytes directly acquire a “white-like” state after withdrawing stimuli.
Autophagy is activated during the beige-to-white fat transition.
Genetic and pharmacological inhibition of autophagy retains beige adipocytes.
Prolonged maintenance of beige fat ameliorates obesity and glucose intolerance.
Acknowledgments
We are grateful to Dr. Luke Cassereau and Dr. Valerie Weaver at University of California, San Francisco for their help in developing the single-cell monitoring system, Anthony Jose from the FACS Core for his help in isolating mature adipocytes, Dr. Noboru Mizushima at the University of Tokyo for providing GFP-LC3 mice, Dr. Evan Rosen in Beth Israel Deaconess Medical Center and Harvard Medical School for providing Ucp1Cre/+ mice, Dr. Christophe Paillart for his help in the CLAMS studies, and Larry Ackerman for his help with EM. We acknowledge support from the NIH DK97441 and DK108822, the DERC center grant DK63720, the Pew Charitable Trust, and Japan Science and Technology Agency to S.K., and NIH CA126792 to JD and the American Cancer Society, and the Pancreatic Cancer Action Network–AACR Career Development award to R.M.P. S.A-K. is supported by AHA 15PRE23050029 and CIRM TG2-01153. K.S. is supported by LLHF 2014-D-025-FEL. Y.H. and K.I. are supported by the Manpei Suzuki Diabetes Foundation. Q.K. is supported by CSC No.201506350063.
Footnotes
AUTHOR CONTRIBUTIONS
S. A-K and S.K. conceived the study, designed experiments, analyzed and interpreted the data. K.S., performed bioinformatics analyses. S.A-K., Y.H., K.I., H.H., Q.K. performed experiments and interpreted the data. Y.Y. provided technical help. R. P. and J.D. provided reagents and expertise in the field of autophagy. S. A-K. and S.K. wrote the manuscript. S. A-K., R.P., J.D., and S.K. edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL, Niswander L, Conlon RA. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Developmental biology. 2006;296:164–176. doi: 10.1016/j.ydbio.2006.04.449. [DOI] [PubMed] [Google Scholar]
- Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristiansen K, Giacobino JP, De Matteis R, Cinti S. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte trans differentiation. American journal of physiology. 2010;298:E1244–1253. doi: 10.1152/ajpendo.00600.2009. [DOI] [PubMed] [Google Scholar]
- Bi P, Shan T, Liu W, Yue F, Yang X, Liang XR, Wang J, Li J, Carlesso N, Liu X, et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nature medicine. 2014;20:911–918. doi: 10.1038/nm.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CJ, Ayer DE, Dynlacht BD. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nature cell biology. 2014;16:1202–1214. doi: 10.1038/ncb3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell. 2001;106:563–573. doi: 10.1016/s0092-8674(01)00474-3. [DOI] [PubMed] [Google Scholar]
- Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, Boyd DD. ZKSCAN3 is a master transcriptional repressor of autophagy. Molecular cell. 2013;50:16–28. doi: 10.1016/j.molcel.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti S. The adipose organ. Editrice Kurtis; Milano, Italy: 1999. [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. Identification and importance of brown adipose tissue in adult humans. The New England journal of medicine. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell metabolism. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Gospodarska E, Nowialis P, Kozak LP. Mitochondrial turnover: a phenotype distinguishing brown adipocytes from interscapular brown adipose tissue and white adipose tissue. The Journal of biological chemistry. 2015;290:8243–8255. doi: 10.1074/jbc.M115.637785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. The Journal of clinical investigation. 1998;102:412–420. doi: 10.1172/JCI3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S. Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. American journal of physiology Cell physiology. 2000;279:C670–681. doi: 10.1152/ajpcell.2000.279.3.C670. [DOI] [PubMed] [Google Scholar]
- Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology. 2012;153:5866–5874. doi: 10.1210/en.2012-1625. [DOI] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes & development. 2008;22:1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, Seale P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell metabolism. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nature medicine. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Banks A, Liu T, Kazak L, Rao RR, Cohen P, Wang X, Yu S, Lo JC, Tseng YH, et al. IRF4 is a key thermogenic transcriptional partner of PGC-1alpha. Cell. 2014;158:69–83. doi: 10.1016/j.cell.2014.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosacka J, Kern M, Kloting N, Paeschke S, Rudich A, Haim Y, Gericke M, Serke H, Stumvoll M, Bechmann I, et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Molecular and cellular endocrinology. 2015;409:21–32. doi: 10.1016/j.mce.2015.03.015. [DOI] [PubMed] [Google Scholar]
- Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schon MR, et al. Altered autophagy in human adipose tissues in obesity. The Journal of clinical endocrinology and metabolism. 2011;96:E268–277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science (New York, N Y. 2012a;336:225–228. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014a;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Smith S, Linderman J, Courville AB, Brychta RJ, Dieckmann W, Werner CD, Chen KY, Celi FS. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes. 2014b;63:3686–3698. doi: 10.2337/db14-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Konkar AA, Granneman JG. Cellular origins of cold-induced brown adipocytes in adult mice. FASEB J. 2015;29:286–299. doi: 10.1096/fj.14-263038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Mottillo EP, Granneman JG. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell metabolism. 2012b;15:480–491. doi: 10.1016/j.cmet.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidell ME, Betz MJ, Leinhard OD, Heglind M, Elander L, Slawik M, Mussack T, Nilsson D, Romu T, Nuutila P, et al. Evidence for two types of brown adipose tissue in humans. Nature medicine. 2013;19:631–634. doi: 10.1038/nm.3017. [DOI] [PubMed] [Google Scholar]
- Liu D, Bordicchia M, Zhang C, Fang H, Wei W, Li JL, Guilherme A, Guntur K, Czech MP, Collins S. Activation of mTORC1 is essential for beta-adrenergic stimulation of adipose browning. The Journal of clinical investigation. 2016;126:1704–1716. doi: 10.1172/JCI83532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R, Warne JP, Salas E, Xu AW, Debnath J. Loss of Atg12, but not Atg5, in pro-opiomelanocortin neurons exacerbates diet-induced obesity. Autophagy. 2015;11:145–154. doi: 10.1080/15548627.2014.998917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular biology of the cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullican SE, Tomaru T, Gaddis CA, Peed LC, Sundaram A, Lazar MA. A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Molecular endocrinology (Baltimore, Md. 2013;27:127–134. doi: 10.1210/me.2012-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez CE, Rodrigues VS, Gomes FS, Moura RF, Victorio SC, Bombassaro B, Chaim EA, Pareja JC, Geloneze B, Velloso LA, et al. Defective regulation of adipose tissue autophagy in obesity. International journal of obesity (2005) 2013;37:1473–1480. doi: 10.1038/ijo.2013.27. [DOI] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARgamma agonists Induce a White-to-Brown Fat Conversion through Stabilization of PRDM16 Protein. Cell metabolism. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama K, Nogusa Y, Shinoda K, Suzuki K, Bannai M, Kajimura S. A Synergistic Antiobesity Effect by a Combination of Capsinoids and Cold Temperature Through Promoting Beige Adipocyte Biogenesis. Diabetes. 2016;65:1410–1423. doi: 10.2337/db15-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Ost A, Svensson K, Ruishalme I, Brannmark C, Franck N, Krook H, Sandstrom P, Kjolhede P, Stralfors P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Molecular medicine. 2010;16:235–246. doi: 10.2119/molmed.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nature cell biology. 2013 doi: 10.1038/ncb2740. [DOI] [PubMed] [Google Scholar]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Hung CM, Sparks CA, Tang Y, Li H, Guertin DA. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell metabolism. 2012;16:348–362. doi: 10.1016/j.cmet.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science (New York, N Y. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Huang TL, Tran TT, Zhang H, Townsend KL, Shadrach JL, Cerletti M, McDougall LE, Giorgadze N, Tchkonia T, et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:143–148. doi: 10.1073/pnas.1010929108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. The Journal of clinical investigation. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellayah D, Sikder D. Orexin restores aging-related brown adipose tissue dysfunction in male mice. Endocrinology. 2014;155:485–501. doi: 10.1210/en.2013-1629. [DOI] [PubMed] [Google Scholar]
- Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science (New York, N Y. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp LZ, Shinoda K, Ohno H, Scheel DW, Tomoda E, Ruiz L, Hu H, Wang L, Pavlova Z, Gilsanz V, et al. Human BAT possesses molecular signatures that resemble beige/brite cells. PloS one. 2012;7:e49452. doi: 10.1371/journal.pone.0049452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda K, Luijten IH, Hasegawa Y, Hong H, Sonne SB, Kim M, Xue R, Chondronikola M, Cypess AM, Tseng YH, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nature medicine. 2015a;21:389–394. doi: 10.1038/nm.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda K, Ohyama K, Hasegawa Y, Chang HY, Ogura M, Sato A, Hong H, Hosono T, Sharp LZ, Scheel DW, et al. Phosphoproteomics Identifies CK2 as a Negative Regulator of Beige Adipocyte Thermogenesis and Energy Expenditure. Cell metabolism. 2015b;22:997–1008. doi: 10.1016/j.cmet.2015.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvets E, Fass E, Elazar Z. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy. 2008;4:621–628. doi: 10.4161/auto.5939. [DOI] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. The Journal of clinical investigation. 2009;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ, Hansen J, Jorgensen JA, Wu J, Mottaghy FM, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. The Journal of clinical investigation. 2013;123:3395–3403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. The New England journal of medicine. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nature medicine. 2013;19:1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Kissig M, Rajakumari S, Huang L, Lim HW, Won KJ, Seale P. Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:14466–14471. doi: 10.1073/pnas.1412685111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegue E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323–327. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Rim JS, Hogan JC, Coulter AA, Koza RA, Kozak LP. Genetic variability affects the development of brown adipocytes in white fat but not in interscapular brown fat. Journal of lipid research. 2007;48:41–51. doi: 10.1194/jlr.M600287-JLR200. [DOI] [PubMed] [Google Scholar]
- Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, Zerfas P, Zhigang D, Wright EC, Stuelten C, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell metabolism. 2011;14:67–79. doi: 10.1016/j.cmet.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Wang QA, Tao C, Vishvanath L, Shao M, McDonald JG, Gupta RK, Scherer PE. Impact of tamoxifen on adipocyte lineage tracing: Inducer of adipogenesis and prolonged nuclear translocation of Cre recombinase. Molecular metabolism. 2015;4:771–778. doi: 10.1016/j.molmet.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, Iwanaga T, Saito M. Recruited brown adipose tissue as an antiobesity agent in humans. The Journal of clinical investigation. 2013;123:3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.