

Abstract
The mitochondrial genome (mtDNA) is intimately linked to cellular and organismal health, as demonstrated by the fact that mutations in and depletion of mtDNA result in severe mitochondrial disease in humans. However, cells contain hundreds to thousands of copies of mtDNA, which provides genetic redundancy, and creates a threshold effect in which a large percentage of mtDNA must be lost prior to clinical pathogenesis. As certain pharmaceuticals and genetic mutations can result in depletion of mtDNA, and as many environmental toxicants target mitochondria, it is important to understand whether reduced mtDNA will sensitize an individual to toxicant exposure. Here, using ethidium bromide (EtBr), which preferentially inhibits mtDNA replication, we reduced mtDNA 35-55% in the in vivo model organism Caenorhabditis elegans. Chronic, lifelong, low-dose EtBr exposure did not disrupt nematode development or lifespan, and induced only mild alterations in mitochondrial respiration, while having no effect on steady-state ATP levels. Next, we exposed nematodes with reduced mtDNA to the known and suspected mitochondrial toxicants aflatoxin B1, arsenite, paraquat, rotenone or ultraviolet C radiation (UVC). EtBr pre-exposure resulted in mild sensitization of nematodes to UVC and arsenite, had no effect on AfB1 and paraquat, and provided some protection from rotenone toxicity. These mixed results provide a first line of evidence suggesting that reduced mtDNA content may sensitize an individual to certain environmental exposures.
Keywords: mtDNA depletion, Caenorhabditis elegans, DNA damage, ultraviolet C, arsenite, environmental toxicant
1. Introduction
The mitochondrial genome (mtDNA) encodes 13 subunits of the electron transport chain (ETC), 22 tRNAs, and 2 rRNAs [1], thus critically linking mtDNA to energy production via oxidative phosphorylation (OXPHOS). Furthermore, the integrity of mtDNA is paramount for cellular and organismal health, which is demonstrated by the fact that mutations, deletions, and depletion of mtDNA cause severe respiratory chain disease in humans [2, 3]. However, depending upon the age and energetic demands of a tissue, a cell can contain hundreds to thousands of copies of mtDNA [4], providing a cell with genetic redundancy that helps buffer against somatic and maternally-inherited mtDNA mutations. Thus, in the context of heteroplasmy (multiple mtDNA variants within a tissue), levels of pathogenic mutations must exceed a threshold, which generally ranges from 60–90% depending upon the severity of the mutation and the energy demands of the effected tissue, before overt pathology ensues [2, 3].
Another kind of mtDNA-related disease results from depletion of mtDNA, which can cause a severe group of disorders known as mtDNA depletion syndrome (MDS). MDS is one of the most common childhood mitochondrial disorders [5], and is primarily caused by mutations in nuclear encoded genes that play a role in mitochondrial dNTP pool maintenance (TK2, DGUOK, RRM2B, TYMP, SUCLA2, SUCLG1) or mtDNA replication (POLG1, C10orf). This genetically heterogeneous group of disorders is characterized by a severe loss of mtDNA that results in impaired ATP production. The pathogenic threshold is dependent upon the energetic demands of the effected tissue, and severity of any concurrent mutations in the affected tissues or organs, which are most frequently brain, liver, kidney or muscle [6, 7]. Mutations in the mitochondrial fusion gene (MFN2) [8, 9], and exposure to nucleoside reverse transcriptase inhibitors (NRTIs) [10–12], which are used to prevent the transmission of HIV from mother-to-child, can also causes severe tissue-specific reductions in mtDNA content.
Growing evidence has demonstrated that mitochondria and mtDNA are important targets of numerous drugs and environmental toxicants [13]. The phospholipid rich mitochondrial double membrane attracts lipophilic genotoxicants (e.g. polycyclic aromatic hydrocarbons (PAHs), aflatoxins), while proton pumping gives the mitochondrial matrix a slight negative charge that attracts cationic compounds (Cd2+, Mn2+, Pb2+, EtBr+) that can disrupt mitochondrial energy production through a number of routes. A lack of introns, protective histones, and some DNA repair pathways (e.g. nucleotide excision repair (NER)) contribute to mtDNA vulnerability to certain genotoxicants, such as PAHs, which induce irreparable mtDNA damage due to a lack of NER [14–18]. Although high mtDNA copy number provides genetic redundancy that may buffer against toxicant-induced mtDNA damage, little is known about how individuals with reduced mtDNA content will respond to environmental mitotoxicants.
Here, using the in vivo model organism Caenorhabditis elegans we tested the hypothesis that reduced mtDNA content (but not exceeding the pathogenic threshold) would increase vulnerability to secondary mitotoxicant exposure. As nematodes share highly conserved mitochondrial biology [19], energy metabolism pathways [20], and mtDNA [21] with humans, they represent an excellent in vivo model to test this hypothesis. Furthermore, availability of transgenic strains, such as the PE255 ATP-reporter strain [22, 23], allow for the rapid assessment of mitochondrial function in vivo. To test this hypothesis, we reduced mtDNA copy number 35-55% and then exposed nematodes to known and suspected mitochondrial toxicants (aflatoxin B1 (AfB1), arsenite, paraquat, rotenone, and ultraviolet C radiation (UVC)). Interestingly, the effects were not dramatic; we report mild exacerbation of mitochondrial dysfunction in the context of reduced mtDNA content in the context of some but not all exposures.
2. Material and Methods
2.1 Strains and culture conditions
Luciferase-expressing, PE255 glp-4 (bn-2) nematodes were generously provided by Dr. Christina Lagido (University of Aberdeen, Aberdeen, UK). PE255 nematodes were maintained at the permissive temperature of 15°C until experiments were initiated, at which time they were shifted to the restrictive temperature of 25°C, which eliminates development of the nematode germline [24]. Synchronous populations of larval stage one (L1) nematodes were generated via sodium hydroxide bleach treatment, followed by overnight incubation on an orbital shaker in complete K-medium (150μl 1M CaCl2, 150μl 1M MgSO4, 25μl 10mg/ml cholesterol, 50ml sterile K-medium (2.35g KCl, 3g NaCl, 1L ddH2O)) at 20°C [25]. Nematodes were then cultured on K-agar plates seeded with Escherichia coli OP50 as previously described [26, 27].
2.2 Ethidium bromide and toxicant exposure
Synchronous populations of L1 stage PE255 nematodes were cultured on OP50 seeded K-agar plates containing 0, 0.05 or 1.0μg/mL EtBr (dissolved in ddH2O, Sigma-Aldrich, St. Louis, MO). EtBr exposures were continuously maintained for the duration of all experiments to maintain mtDNA knockdown.
2.2.1 UVC exposure
Synchronous populations of control and EtBr-exposed L4 stage nematodes were transferred onto OP50-free K-agar plates containing 0 or 1.0μg/ml EtBr, allowed to disperse, and then exposed to 0, 25, or 50J/m2 UVC using an ultra violet lamp (UVLMS-38 EL Series 3UV Lamp, UVP, Upland, CA, USA) with peak emission at 254nm. Nematodes were then transferred back onto OP50 seeded control or EtBr plates for subsequent ATP determination.
2.2.2 Chronic toxicant exposures
Control and EtBr-exposed L4 nematodes were exposed to AfB1 (dissolved in 1% DMSO, Sigma-Aldrich) or arsenite (Ricca Chemical Company, dissolved in ddH2O) for 4 to 9 days using the spot dead method as previously described [28]. Briefly, 1.5ml arsenite (0, 50, 250, 500μM) or AfB1 (0, 5, 25, 50μM in 1% DMSO) was added to the surface of peptone-free, control or EtBr (1μg/ml) K-agar plates and allowed to dry. K-agar plates were then seeded with 300μl 20X concentrated UVC-killed UvrA bacteria (UV-sensitive strain, due to lack of NER [29]). Nematodes were transferred daily to freshly prepared plates.
2.2.3 Acute toxicant exposures
Approximately 3,000 control and EtBr-exposed L4 stage nematodes were exposed to AfB1 (0, 25, 50μM in 1% DMSO), arsenite (0, 50, 100μM dissolved in ddH2O), methyl viologen dichloride (0, 0.5, 1.0mM paraquat hydrate dissolved in ddH2O, Sigma-Aldrich), or rotenone (0, 0.5, or 1.0μM dissolved in 1% DMSO, Sigma-Aldrich) in complete K-medium containing UVC-killed UvrA, and 0 or 1.0μg/ml EtBr for 48h on an orbital shaker at 25°C. Following toxicant exposure, nematodes were rinsed three times with 15ml k-medium to remove excess toxicant through dilution. Nematodes were then transferred back to control or EtBr containing plates and ATP levels were measured daily for 4 consecutive days.
2.3 Copy number determination
Nuclear (nucDNA) and mtDNA genome copy number was determined as previously described [30, 31]. Briefly, 6 nematodes were added to 90μl proteinase K-containing lysis buffer using a platinum worm pick and frozen at −80°C. Samples were then thawed and lysed via a 1h incubation at 65°C. Crude nematode lysate was then used as template DNA for RT-PCR based determination of mtDNA and nucDNA copy number. Standard curves were used for absolute copy number determination. Copy number experiments were repeated 3 separate times.
2.4 Growth assay
An aliquot of nematodes grown on 0, 0.05, or 1.0μg/ml EtBr was collected every 2 days, for 12 consecutive days, and frozen at −20°C for size determination. On the day of analysis, samples were thawed and imaged at 10× magnification on a Zeiss Axioskop. Images were analyzed using NIS elements software (Nikon Inc., Melville, NY). An aliquot of live and frozen nematodes was compared to ensure that the freeze-thawing process did not affect measurements (S. Figure 2B). Approximately 20 nematodes were measured for each exposure group within each time point from 2 independent experiments.
2.5 Determination of in vivo steady-state ATP levels
In vivo steady-state ATP levels were determined as previously described [23], and as visualized in [22]. Briefly, 50 nematodes (suspended in K-medium) were loaded into each well of a white 96-well plate (four wells per treatment), and then GFP fluorescence was measured (emissions filter: 502nm; excitation filter: 485nm) using a FLUOstar Optima microplate reader (BMG Labtech, Germany). 50μl of luminescence buffer (140mM Na2PO4, 30mM citric acid (pH 6.5), 1% DMSO, 0.05% Triton X-100, 100μM D-luciferin) was then injected into each well and nematode bioluminescence was measured 3 minutes later using a luminescence optic (BMG Labtech). As PE255 nematodes express a GFP∷luciferase fusion protein, each well's luminescence value was normalized to GFP fluorescence to correct for overall luciferase content and slight discrepancies in the number and size of nematodes loaded per well. All experiments were repeated at 3-5 separate times.
2.6 Lifespan
Twenty-five L1 stage PE255 nematodes were placed on OP50 seeded K-agar plates containing 0 or 1.0μg/ml EtBr at 25°C for the duration of their lives. Nematodes were scored daily, and judged dead when they failed to move in response to probing with a platinum worm pick. Lifespan experiments were repeated 2 separate times.
2.7 Seahorse XFe analysis
The fundamental parameters of the mitochondrial respiratory chain (basal oxygen consumption rate (OCR), maximal OCR, ATP-linked respiration, spare respiratory capacity (SRC), and proton leak) were measured following 4 and 8 days of exposure to EtBr using the Seahorse XFe24 Bioanalyzer (Seahorse Bioscience, Massachusetts, USA) as previously described [32, 33]. Briefly, 50 nematodes were loaded into each well of a Seahorse utility plate and 8 basal OCR readings were taken. Nematodes were then exposed to either 25μM FCCP (mitochondrial uncoupler), 20μM DCCD (ATP synthase inhibitor), or 10mM sodium azide (cytochrome c oxidase inhibitor) and an additional 8, 16, or 4 oxygen consumption measurements were taken, respectively. SRC was calculated by subtracting a wells basal OCR from its FCCP response, ATP-linked respiration was calculated by subtracting a wells DCCD response from its basal OCR, while proton leak was calculated by subtracting a wells azide response from its DCCD response. All experiments were repeated 3 separate times.
2.8 Gene expression
mRNA levels of two cytochrome P450s (CYP35B2, CYP33C6), three mitochondrial heat shock proteins (hsp-6, hsp-60a, hsp-60b), mitochondrial DNA polymerase (polg-1), and the nematode mitochondrial transcription factor A homolog (hmg-5) were measured using real-time PCR. mRNA was converted to cDNA using the Qiagen Omniscript Reverse Transcription kit. RT-PCR was performed using a 7300 Real Time PCR System (Applied Biosystems). The fold change of each gene was calculated by comparing the Ct of each gene to that of the housekeeping gene cdc-42 [34]. RT-PCR primers for polg-1 and cdc-42 were based on the literature [35]. Unpublished primers were as follows: CYP35B2 (For–5'-GTG GGC TGA AAT GCG AAG AT-3', Rev-5'-CCA ACG GCA AGG TCA AAG AA-3'(177bp amplicon, annealing temp 60°C)); CYP33C6 (For-5'-GCT GCG GTT TGT GTA TTC CT-3', Rev-5'-GTC CAC CGT CTG AAG CAT TC-3' (164bp amplicon, annealing temp 64°C)); hsp-6 (For-5'-TCG TGT CAT CAA CGA GCC AA-3', Rev-5'- AGC GAT GAT CTT ATC TCC AGC G-3' (76bp amplicon, annealing temp 56°C)); hsp-60a (For-5'-AGG CTC TTA CCA CTC TTG TTC T-3', Rev-5'- CTC CCG TCG CAA TTC CCA TA-3' (123bp amplicon, annealing temp 56°C)); hsp-60b (For-5'-CCA AGA AGG TCA CCA TCA CC-3', Rev-5'-TCT GTT TGA TCT CCA CGC CC-3' (64bp amplicon, annealing temp 56°C)); hmg-5 (For-5'-TGT CTG GAG CTG GAA TGG AA-3', Rev-5'-GCT TCT TCG CTT CGT CTG TG-3' (108bp amplicon, annealing temp 60°C)). Samples were run in triplicate from two separate experiments.
2.9 Glycolysis assay
To detect changes in glycolysis, we exposed control, 100μM arsenite, EtBr, and EtBr and 100μM arsenite co-exposed nematodes to 2-deoxy-D-glucose (glycolysis inhibitor), and then measured the change in steady-state ATP as previously described [36, 37]. Briefly, 50 nematodes were loaded into each well (4 wells per treatment) of a white 96-well plate and exposed to 50mM 2-DG for 4.5h at 25°C. Following 2-DG exposure steady-state ATP levels were determined as described above. The effect of 2-DG on each treatment group was then normalized to percent vehicle control (EPA H2O). The experiment was repeated four separate times.
2.10 Statistics
Copy number (mtDNA, nucDNA, and their ratio), gene expression, steady-state ATP, nematode growth, and Seahorse XFe data were initially analyzed with a one, two, or three way ANOVA, and when warranted post-hoc comparisons were made via Tukey's HSD. Lifespan data was analyzed via the non-parametric Mantel-Cox test. Statistics were performed using JMP v11.0 software (SAS Institute).
3. Results
As we were primarily interested in investigating the effects of mtDNA knockdown on somatic tissues, and development of the nematode germline during the L3/L4 transition is associated with large increases in mtDNA copy number [19, 38] which would confound the interpretation of our results, we used the germline-deficient, in vivo ATP reporter strain PE255 glp-4 for all experiments [22, 23]. EtBr, a well-known inhibitor of mtDNA replication [39–41] that is frequently used to generate rho0 (mtDNA-deficient) cells, was used to reduce mtDNA copy number in nematodes.
3.1 Chronic, low dose EtBr exposure reduces mtDNA copy number without affecting nucDNA copy number, nematode lifespan, or larval development
Chronic exposure to 1.0, but not 0.05μg/ml EtBr reduced mtDNA copy number 32–51% over the first 4 days of exposure (two way ANOVA, main effect of time (p<0.0001), not EtBr (p=0.07), and their interaction (p=0.013)) (Figure 1A), whereas EtBr had no effect on nucDNA copy number (two way ANOVA, main effect of time (p<0.0001), but not EtBr (p=0.09), or their interaction (p=0.36)) (Figure 1B), demonstrating EtBr's ability to specifically inhibit mtDNA, but not nucDNA replication in C. elegans. Furthermore, a robust reduction (35-55%) in the ratio of mtDNA to nucDNA was observed over the first 8 days of EtBr exposure (two way ANOVA, main effects of time (p<0.0001), EtBr (p<0.0001), and their interaction (p=0.0073)) (Figure 1C). Interestingly, the mtDNA:nDNA ratio returned to control levels by the twelfth day of exposure, which we postulate is due to reduced feeding and/or reduced uptake of EtBr through the nematode cuticle (a collagenous barrier known to limit toxicant uptake [42–44]) in aging nematodes, resulting in a loss of inhibition of mtDNA replication.
Figure 1. Ethidium bromide reduces mtDNA copy number.

Chronic, lifelong exposure to EtBr reduced (A) mtDNA copy number (2 way ANOVA, main effect of time (p<0.0001), but not EtBr (p=0.073), and their interaction (p=0.013)), but not (B) nucDNA copy number (2 way ANOVA, main effect of time (p<0.0001), but not EtBr (p=0.089), or their interaction (p=0.36)), while also reducing the (C) mtDNA:nucDNA ratio (2 way ANOVA, main effect of time (p<0.0001), EtBr (p<0.0001), and their interaction (p=0.0073). Asterisk denotes statistical significance (p<0.05) for post-hoc comparison (Tukey's HSD) to control within each time point. N=6–9. Bars ± SEM.
As chronic exposure to higher concentrations of EtBr (≥5.0μg/ml) has previously been reported to extend nematode lifespan through an induction of the mitochondrial unfolded protein response (UPRmt) [38, 45], we tested whether lower concentrations of EtBr would elicit a similar effect. Chronic (48h) exposure to 1.0μg/ml EtBr elicited a mild, yet statistically significant induction of mitochondrial hsp-6 (one way ANOVA, p=0.0016) and hsp-60b (p=0.0056), but not hsp-60a (p=0.25) (Figure 2). However, lifelong exposure to 1.0μg/ml EtBr did not extend nematode lifespan (p=0.26) (S. Figure 1), suggesting that the low dose EtBr-induced UPRmt is not robust enough to extend lifespan. Furthermore, 1.0μg/ml EtBr caused no larval growth delay (two way ANOVA, main effect of time (p<0.0001), but not EtBr (p=0.27) or their interaction (p=0.87)) (S. Figure 2A), while 5.0μg/ml EtBr caused severe larval growth delay and L3 stage arrested (data not shown), a finding that has been previously reported [46]. Collectively, these results demonstrate that in C. elegans, chronic, low-dose EtBr exposure can effectively reduce mtDNA content, while having no observable adverse organismal level effects.
Figure 2. Ethidium bromide induces mitochondrial heat shock protein expression.
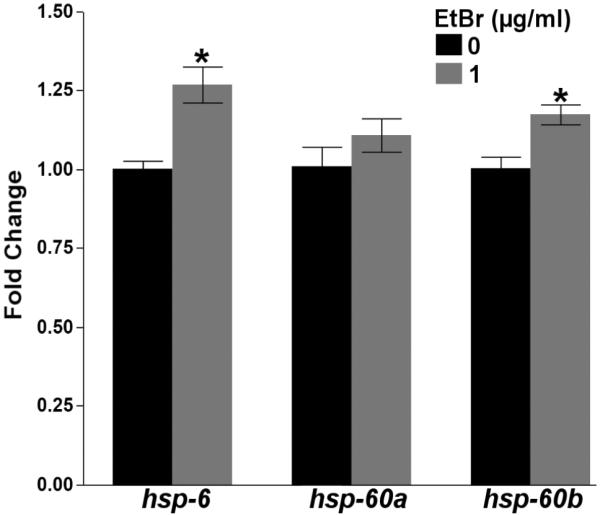
Chronic (48h) exposure to EtBr induced the expression of the mitochondrial heat shock proteins hsp-6 (one way ANOVA, p=0.0016), hsp-60b (p=0.0056), but not hsp-60a (p=0.25) in L4 PE255 glp-4 nematodes. Asterisk denotes statistical significance compared (p<0.05) to control. N=6. Bars ± SEM.
3.2 mtDNA knockdown causes mild disruption of the mitochondrial respiratory chain
As the pathogenic threshold for mtDNA depletion syndrome is frequently reported to be around 65% [6, 7], we hypothesized that a 35-55% reduction in the whole worm mtDNA:nucDNA ratio would not cause major mitochondrial dysfunction. In agreement with this, only mild alterations in basal respiration (Figure 3A), ATP-linked respiration (Figure 3B), and proton leak (Figure 3D) were detected following chronic EtBr exposure (2 way ANOVA, main effects of Time (p<0.0001 for all) and EtBr (p<0.05 for all), but not their interaction (p>0.05 for all)), while 1.0μg/ml EtBr reduced spare respiratory capacity in 4 day old nematodes (2 way ANOVA, main effects of time (p=0.0009), and time*EtBr (p=0.0016), but not EtBr (p=0.93)) (Figure 3C), and increased FCCP-uncoupled maximal OCR in 8 day old nematodes (2 way ANOVA, main effects of time (p<0.0001), and time*EtBr (p=0.0074), but not EtBr (p=0.36)) (Figure 3A). Although increased maximal OCR in 8 day old nematodes exposed to EtBr was unexpected, compensatory changes in mtDNA and function have been observed following NRTI cessation [47], and we have previously observed increased mitochondrial DNA polymerase gamma expression in response to polymerase stalling, UVC-induced photodimers, which also result in large reductions in mtDNA copy number [35]. As we observe an increase in mtDNA copy number from day 4 to day 12 (Figure 1A), which we hypothesis is due to reduced feeding in aging nematodes and thus reduced uptake of EtBr, we measured the effect of EtBr on DNA polymerase gamma (polg-1, the sole replicative mitochondrial DNA polymerase), and hmg-5 (TFAM homolog, required for mtDNA replication and transcription) expression to investigate a potential compensatory response that may explain EtBr-induced increases in maximal OCR. However, 1.0μg/ml EtBr did not affect polg-1 expression (Figure 4A), and only mildly increased hmg-5 expression (Figure 4B).
Figure 3. Ethidium bromide induces mild alterations in the mitochondrial respiratory chain.

Reduced mtDNA content induced mild alterations in (A) basal respiration (2 way ANOVA, main effects of time (p<0.0001), and EtBr (p=0.0011), but not their interaction (p=0.45)), (B) ATP-Linked OCR (2 Way ANOVA, main effects of time (p<0.0001) and EtBr (p=0.036), but not their interaction (p=0.41)), and (D) proton leak (2 way ANOVA, main effects of time (p<0.0001), and EtBr (p<0.0001), but not their interaction (p=0.21)), while an increase in (A) FCCP-induced maximal OCR was observed after 8 days of EtBr exposure (2 way ANVOA, main effect of time (p<0.0001), but not EtBr (p=0.36), and their interaction (p=0.0074)), and reduced (C) spare capacity was observed after 4 days of EtBr exposure (2 way ANOVA, main effect of time (p=0.0009), but not EtBr (p=0.93), and their interaction (p=0.0016)). Time, on the x-axis, refers to both nematode age and the length of the EtBr exposure. Asterisk denotes statistical significance (p<0.05) for post-hoc comparison (Tukey's HSD) to control within each time point. N=21–46. Bars ± SEM.
Figure 4. Ethidium bromide increases hmg-5 expression.

Chronic, lifelong exposure to EtBr does not affect (A) polg-1 expression (two way ANOVA, main effect of time (p=0.0048), but not EtBr or their interaction (p>0.05 for both)), but does induce the expression of (B) hmg-5 (two way ANOVA, main effects of time (p<0.0001) and EtBr (p=0.041), but not their interaction (p=0.26)), the nematode homolog of TFAM, which plays a role in mtDNA replication and transcription. Time, on the x-axis, refers to both nematode age and the length of the EtBr exposure. Data is normalized to fold change over the day 2 control samples. N=6. Bars ± SEM.
Although mild alterations in mitochondrial function were observed following chronic, low-dose EtBr exposure, no alterations in steady-state ATP levels were observed throughout the course of a 12 day EtBr exposure (2 way ANOVA, main effect of time (p<0.0001), but not EtBr (p=0.53), or their interaction (p=0.93)) (Figure 5), further demonstrating that the magnitude of EtBr-induced mtDNA knockdown (35-55%) did not induce major mitochondrial dysfunction.
Figure 5. Ethidium bromide does not alter steady-state ATP levels.

Chronic, lifelong exposure to EtBr does not affect steady-state ATP levels throughout the nematode lifespan (2 way ANOVA, main effect of time (p<0.0001), but not EtBr (p=0.53), or their interaction (p=0.93)). Time, on the x-axis, refers to both nematode age and the length of the EtBr exposure. Relative luminescence is a surrogate measure of in vivo steady-state ATP levels. N=2. Bars ± SEM.
3.3 Reduced mtDNA content sensitizes C. elegans to secondary exposure to some but not all mitotoxicants
Acute and/or chronic exposures to the known and suspected mitochondrial toxicants AfB1, arsenite, paraquat, rotenone, and UVC were initiated after 48h of EtBr exposure in L4 stage nematodes, as this is when the most robust reduction in mtDNA (51%) and the mtDNA:nucDNA ratio (55%) was observed. With the exception of UVC (which does not penetrate the ozone layer), these toxicants are of human health concern. AfB1, a foodborne mycotoxin and important carcinogen in the developing world, causes bulky DNA lesions when its epoxide metabolite reacts with DNA. We and others have found that AfB1 preferentially, and irreparably (due to a lack of NER) damages mtDNA [15–17]. Arsenite, a global drinking water contaminant associated with the development of skin, lung, and bladder cancer, is a well-known inhibitor of several Krebs cycle dehydrogenases [48], ETC complexes [49], and is also capable of causing metabolic shifts from OXPHOS to aerobic glycolysis, otherwise known as the Warburg effect [36, 50]. Exposure to rotenone, a pesticide and prototypical mitotoxicant that acts by inhibiting complex I of the ETC leading to superoxide production, is associated with the development of Parkinson's disease [51]. Like rotenone, the herbicide paraquat is also associated with the development of Parkinson's disease [51], but functions as a redox cycler that preferentially damages mtDNA [15]; however, paraquat-induced mtDNA damage is oxidative and largely repaired by base excision repair. Alternatively, UVC causes equal amounts of nucDNA and mtDNA photodimers; however, photodimers are only repaired in the nuclear genome [52, 53], so that UVC can be used as a tool to rapidly induce irreparable mtDNA damage.
3.3.1 Reduced mtDNA content does not sensitize nematodes to the effects of chronic arsenite or AfB1 exposure on steady-state ATP levels
Following mtDNA knockdown, nematodes were chronically (96h) exposed to arsenite or AfB1. Neither arsenite (S. Figure 3) nor AfB1 (S. Figure 4) differentially altered steady-state ATP levels between control and EtBr exposed nematodes (3 way ANOVA, p>0.05 for both). A trend in reduced steady-state ATP levels in nematodes treated with EtBr was observed following 72 and 96h of AfB1 exposure (S. Figure 4). Therefore, we chronically exposed control and EtBr treated nematodes to AfB1 for 9 days, starting at the L4 stage, to see if the observed effects on ATP would become more pronounced with a longer exposure. However, exposure to AfB1 for 9 days at this concentration failed to significantly affect ATP levels in control or EtBr treated nematodes (S. Figure 5) (3 way ANOVA, p>0.05). P-values for all effects and interaction terms are shown in Supplemental File 1.
As the nematode cuticle (a protective collagenous barrier) has been shown to limit toxicant uptake [42–44], and all chronic arsenite and AfB1 exposures were performed on K-agar plates, we next performed liquid toxicant exposures, which has been shown to help facilitate toxicant uptake in nematodes [28].
3.3.2 Reduced mtDNA content sensitizes nematodes to acute arsenite and UVC exposure, but not to AfB1, paraquat or rotenone exposure, in liquid culture
Following mtDNA knockdown nematodes were exposed to AfB1, arsenite, paraquat and rotenone in liquid for 48h, and then steady-state ATP levels were determined every 24h for four days. Neither AfB1 nor paraquat significantly altered steady-state ATP levels between control and EtBr exposed nematodes (S. Figures 6 & 7) (3 way ANOVA, p>0.05 for both). Unfortunately, higher concentrations of AfB1 could not be tested due to limited water solubility, while higher concentrations of paraquat (≥5mM) induced mortality.
Rotenone, a prototypical mitochondrial toxicant and ETC complex I inhibitor, reduced steady-state ATP levels 50% in both control and EtBr treated nematodes after a 48h rotenone exposure (Figure 6A). Unexpectedly, steady-state ATP levels remained reduced in control nematodes 24h after rotenone exposure, while ATP levels in nematodes treated with EtBr completely recovered by 24h, and then increased by 48h post-rotenone exposure (3 way ANOVA, rotenone*EtBr*Time interaction (p=0.0091)) (Figure 6A & S. Figure 8). This might suggest that nematodes with reduced mtDNA content recover more rapidly from rotenone exposure. An alternative hypothesis is that because rotenone is mainly metabolized via cytochrome P450s [54], and we have previously reported that high dose (5.0μg/ml) EtBr can induce CYP expression [35], the EtBr exposure protected against rotenone by increasing its metabolism. Therefore, we next tested whether the low doses (1.0μg/ml) of EtBr used in this study would also induce CYP expression. In agreement with our hypothesis, EtBr caused a very large (>180-fold) induction of CYP35B2 (one way ANOVA, p<0.001) (Figure 6B), but not CYP33C6 (p=0.27) (Figure 6C), two CYPs previously shown to play a role in xenobiotic metabolism in nematodes [55, 56]. Unfortunately, it is not clear which of the many nematode CYP proteins is responsible for rotenone metabolism.
Figure 6. Nematodes with reduced mtDNA content recover from rotenone faster than control nematodes.

(A) Steady-state ATP levels were reduced 50% in control and EtBr treated nematodes following a 48h liquid rotenone exposure; however, steady-state ATP levels recovered to control levels faster in nematodes co-exposed to EtBr and rotenone (3 way ANOVA, EtBr*Time*Rotenone interaction (p=0.0091), N=3). 48h exposure to EtBr induced the expression of (B) CYP35B2 (one way ANOVA, p<0.0001, N=6), but not (C) CYP33C6 (one way ANOVA, p=0.27, N=6). P-values for all effect and interaction terms are shown in Supplemental File 1. Relative luminescence values are shown in Supplemental Figure 8. Asterisk denotes statistical significance (p<0.05) for post-hoc comparison (Tukey's HSD) to control within each time point. Bars±SEM.
A persistent trend toward increased steady-state ATP levels was observed following arsenite exposure; however, reducing mtDNA copy number with EtBr prevented this trend (3-way ANOVA, EtBr*arsenite interaction (p=0.048)) (Figure 7A & S. Figure 9). As we have previously shown that similar concentrations of arsenite can induce glycolysis [36], we hypothesized that a persistent increase in glycolysis might be responsible for the increased steady-state ATP levels. As previously reported, the glycolytic inhibitor 2-deoxy-D-glucose significantly reduced steady-state ATP levels in arsenite-treated nematodes immediately after arsenite exposure, demonstrating an induction of glycolysis (one way ANOVA, p=0.045) (Figure 7B). However, this effect was not observed in EtBr, or EtBr and arsenite co-treated nematodes, nor was it persistent (one way ANOVAs, p>0.05 for 24, 48, 72hrs).
Figure 7. Reduced mtDNA content sensitizes C. elegans to arsenite.

(A) A persistent trend in increased steady-state ATP levels was observed following a 48h liquid exposure to arsenite, while reducing mtDNA content with EtBr prevented the observed trend (3 way ANOVA, EtBr*arsenite interaction (p=0.048)). P-values for all effect and interaction terms are shown in Supplemental File 1. Relative luminescence values are shown in Supplemental Figure 9. (B) Inhibition of glycolysis with 2-deoxy-D-glucose significantly reduces steady-state ATP levels in nematodes treated with 100μM arsenite immediately after (T=0) arsenite exposure demonstrating the induction of glycolysis (one way ANOVA, p=0.045). However, this effect was not observed in nematodes treated with 1.0μg/ml EtBr or co-treated with EtBr and arsenite, nor is the induction of glycolysis persistent (p>0.05 for one way ANOVAs 24, 48, 72h post arsenite). Asterisk denotes statistical significance (p<0.05) for post-hoc comparison (Tukey's HSD) to control. N=3-5. Bars±SEM.
Interestingly, a transient increase in steady-state ATP levels was observed 24h post UVC exposure in nematodes with reduced mtDNA content; however, steady-state ATP levels declined below control values 48 and 72h post-UVC, but then recovered by 96h (3 way ANOVA, time*arsenite*UVC interaction (p=0.049)) (Figure 8 & S. Figure 10).
Figure 8. Reduced mtDNA content sensitizes C. elegans to UVC.

Steady-state ATP levels were reduced 48–72h post-exposure to a single dose of 25 or 50 j/m2 UVC in nematodes with reduced mtDNA content (3 way ANOVA, EtBr*Time*UVC interaction (p=0.049)). P-values for all effect and interaction terms are shown in Supplemental File 1. Relative luminescence values are shown in Supplemental Figure 10. Asterisk denotes statistical significance (p<0.05) for post-hoc comparison (Tukey's HSD) to control within each time point. N=5. Bars±SEM.
4. Discussion
Here, we have investigated how reduced mtDNA content affects sensitivity of the in vivo model organism C. elegans to secondary mitochondrial toxicant exposure, and report that reduced mtDNA content causes mild sensitization to certain environmental toxicants. Collectively, our results provide the first line of evidence that reduced mtDNA content may sensitize an organism to certain mitochondrial toxicants, while also demonstrating that this is not likely to be a universal outcome for all mitotoxicants.
4.1 Use of EtBr to reduce mtDNA genome copy number in germline-deficient nematodes
Because development of the nematode germline around the L3/L4 transition causes large increases in whole-body mtDNA content that would complicate the interpretation of results [38], we used germline-deficient glp-4 nematodes for our studies, which allows for the investigation of somatic cell effects of mtDNA knockdown. To reduce mtDNA, we used the DNA intercalating agent, EtBr, which has been used to generate rho0 (mtDNA-deficient) cells for decades by preferentially inhibiting mtDNA replication over nucDNA replication [39–41]. EtBr has previously been used to reduce mtDNA copy number in C. elegans; however, the timing and dose of EtBr exposure is crucial. High larval doses of EtBr (≥5.0μg/ml) induce larval growth arrest [38, 46], while the lower doses of EtBr (1.0μg/ml) used in the present study caused no growth delay (S. Figure 2). Interestingly, high doses of EtBr (≥5.0μg/ml) have also been shown extend nematode lifespan through induction of the mitochondrial unfolded protein response (UPRmt) [38, 45]. Because induction of the UPRmt represent a protective response that could protect nematodes from secondary toxicant exposure, we utilized lower doses of EtBr to reduce mtDNA. Low dose EtBr (1.0μg/ml) mildly induced the expression of several genes (hsp-6, hsp60b, but not hsp-60a) known to play a role in the UPRmt (Figure 2). However, chronic exposure to low dose EtBr did not extend nematode lifespan (S. Figure 1) suggesting only a mild induction of the UPRmt.
Although chronic exposure to low dose EtBr reduced mtDNA 35-55% over the first 8 days of the nematode lifespan, the knockdown was lost by day 12 (Figure 1). The loss in knockdown may be explained by the nematode cuticle (a protective collagenous barrier), which has been shown to limit toxicant uptake, and is impermeable to ionic compounds [42–44]. However, nematode feeding also decreases with age [57, 58], which is expected to reduce intestinal uptake of EtBr, and contribute to the loss of mtDNA knockdown later in life. Interestingly, nematodes exposed to EtBr had increased maximal OCR after 8 days of exposure (Figure 3A). As compensatory changes in mtDNA and function have been reported following cessation of NRTI-induced mtDNA knockdown [47], we hypothesized that increased maximal OCR may be due to compensatory changes in mtDNA replication or transcription. To test this, we measured the expression of the mitochondrial DNA polymerase (polg-1), and the nematode TFAM homolog (hmg-5), which is required for mtDNA replication and transcription. However, chronic exposure to EtBr did not significantly affect polg-1 expression, and only mildly induced hmg-5 expression (Figure 4). It is possible that the small induction of hmg-5 expression on day 8, which coincided with the increase in maximal OCR, facilitates a compensatory increase in mtDNA transcription. Additional experiments would be needed to test this possibility. Based on these findings, all secondary mitochondrial toxicant exposures were initiated in young adult (2 day old) nematodes, when the largest reduction of mtDNA was observed (55%), and all experiments were completed on or before day 8, when increased maximal OCR was observed.
4.2 C. elegans as a model for studying mtDNA depletion
As mtDNA encodes 13 critical ETC subunits [1], depletion of mtDNA due to mutations in nuclear encoded genes that control mitochondrial mtDNA replication (POLG1, C10orf) or the mitochondrial dNTP pool (TK2, DGUOK, RRM2B, TYMP, SUCLA2, SUCLG1) cause severe human respiratory chain disease. In agreement with the pathogenic threshold effect frequently described in other species, a 35-55% loss of mtDNA did not cause severe mitochondrial dysfunction in C. elegans. This is further supported by the fact that the NRTI, zidovudine, which reduced mtDNA content by greater than 80%, also reduced mitochondrial respiration in nematodes [59], while mutations in the sole mitochondrial polymerase, polg-1, resulted in a greater than 90% loss of mtDNA and altered mitochondrial morphology in C. elegans [60].
Although EtBr can be used to reduce mtDNA, and major mitochondrial dysfunction is not observed following a 35-55% reduction in mtDNA, there are limitations associated with the use of C. elegans to study mtDNA depletion. In humans, mtDNA depletion due to nuclear mutations typically only affects certain, energetically demanding tissues such as the brain, liver, kidney, or muscle [6, 7], and exposure to NRTIs has also been shown to deplete mtDNA in energetically demanding tissues (e.g. brain, heart, liver) in humans and primates [12, 61]. Although nematodes lack many of the well-defined organs found in humans, they do have many well-defined tissues such as the cuticle, excretory system, gonad, hypodermis, intestine, muscles, neurons, and a pharynx [62]. Some of these tissues, including the neurons, pharynx, and gonad, are more energetically demanding, and highly sensitive to certain toxicant exposures. For example, the nematode gonad is one of the most sensitive tissues to doxycycline- and chloramphenicol-induced mitochondrial toxicity [63]. Thus, it is possible that EtBr is preferentially depleting mtDNA in the more energetically demanding tissues of C. elegans; however, due to the small size of nematodes (adults are 1.0mm in length), it is difficult to isolate biochemically relevant amounts of purified tissue, making this hypothesis difficult to test.
4.2 Secondary toxicant exposures
Given that high mtDNA copy number provides a cell with genetic redundancy and buffers against DNA damaging events, we hypothesized that reducing mtDNA copy number would sensitize nematodes to secondary mitochondrial toxicant exposure. Surprisingly, secondary toxicant exposure only mildly exacerbated mitochondrial dysfunction when initiated in the context of a 55% reduction in mtDNA. Several factors could explain this. First, many of the toxicants used in the present study have been shown to have tissue-specific effects in human and other mammalian models. For example, AfB1 targets the liver [64, 65], while rotenone and paraquat target the central nervous system [66, 67], and arsenite targets many organs, such as the bladder, kidney, liver, lungs, and skin [68, 69]. Given that more energetically demanding tissues are typically more sensitive to mtDNA depletion [6, 7], it is likely that the toxicants used in the present study are having tissue-specific effects or even exacerbating mitochondrial dysfunction in the context of reduced mtDNA copy number in a tissue-specific manner; however, it is also likely that these effects are being diluted at the organismal level. This is supported by the fact that UVC, which indiscriminately damages mtDNA and nucDNA (irreparable in mtDNA) in all tissues of the transparent nematode [52, 53], reduced ATP levels in the context of reduced mtDNA, while AfB1 and paraquat, both of which preferentially damage mtDNA [15–17], did not exacerbate mitochondrial dysfunction. Alternatively, the nematode cuticle may limit the uptake and thus toxicity of AfB1, arsenite, paraquat, and rotenone, while not limiting UVC-induced damage. However, this seems unlikely as we have previously shown that the concentrations of AfB1, paraquat and UVC used in the current study cause similar amounts of mtDNA damage [15, 53]. Finally, EtBr can act synergistically with UV light (365nm) [70]; however, we have previously shown that EtBr at this level does not exacerbate UVC (100–280nm)-induced mtDNA damage [35].
As previously mentioned, high concentrations of EtBr (≥5 μg/ml) have been shown to induce the expression of numerous xenobiotic metabolizing genes, including myriad CYPs [35]; however, little is known about how low-dose EtBr exposure will effect gene expression. Since steady-state ATP levels recovered faster in nematodes pretreated with EtBr than in nematodes not treated with EtBr following rotenone exposure, and rotenone is mainly metabolized via CYPs [54], low dose EtBr may be inducing the expression of CYPs, allowing EtBr-treated nematodes to metabolize and recover from rotenone exposure faster than nematodes not treated with EtBr. In agreement with this, CYP35B2 expression was induced by low doses of EtBr (Figure 6). Although the precise CYPs that metabolize rotenone in mammals and nematodes remain unknown, both CYP35B2 and CYP33C6 have previously been shown to be inducible by high doses (5μg/ml) of EtBr [35], and are known to play a role in xenobiotic metabolism is nematodes [55, 56], providing a plausible explanation for why EtBr treated nematodes recovered more rapidly from rotenone exposure than control nematodes. However, it is important to note that induction of CYPs is not expected to be protective for all toxicant exposures, as paraquat is, in general, poorly metabolized [71], while CYPs metabolically activate AfB1 to its DNA damaging epoxide (although it is unclear if the necessary CYPs are being induced (CYP2A6, 2B6, 3A4); [72, 73]). Finally, nematodes lack arsenite methyl transferase [74], which plays a major role in arsenite metabolism and excretion in humans [75].
Interestingly, a trend in increased steady-state ATP levels was observed in nematodes exposed to arsenite, while reducing mtDNA copy number prevented this trend (Figure 7A). Although growing evidence suggests that the mitochondrion is an important target of arsenic toxicity [36, 48–50], low dose arsenic has also been linked to hormesis in which the induction of protective mechanisms is associated with therapeutic effects [76, 77]. The reported hormetic effects of arsenic include: reduced risk for non-melanoma skin cancer in Denmark [78], increased growth advantage of cells in culture [79, 80], and lifespan extension in C. elegans [79]. Furthermore, arsenite-induced lifespan extension in nematodes is accompanied by increased mitochondrial respiration, and increased mitochondrial protein content [79]. Although the concentrations of arsenite used in the present study are much higher (50–100μM vs. 100nM), the exposure duration is brief (48h vs. lifelong), which could result in increased mitochondrial function and steady-state ATP levels, while reduced mtDNA copy number may prevent this effect by limiting the number of mtDNA transcripts available for transcription and translation of ETC subunits. Alternatively, chronic arsenic exposure can induce metabolic shifts from OXPHOS to aerobic glycolysis (Warburg effect) in vitro, and in C. elegans [36, 50]. Therefore, we hypothesized that asenite-induced glycolysis persists after arsenite exposure, resulting in increased steady-state ATP levels. However, this does not appear to be the case, as sensitivity to ATP depletion following inhibition of glycolysis with 2-deoxy-D-glucose was not persistent in arsenite or EtBr and arsenite treated nematodes (Figure 7B).
Finally, given the long half-life of mtDNA in C. elegans [81], it is not surprising that we did not see reduced steady-state ATP levels in EtBr-treated nematodes exposed to UVC until 48h after UVC exposure. Although UVC induces irreparable photodimers in mtDNA, it is not surprising that steady-state ATP levels completely recovered by 96h-post UVC exposure, as we have previously shown that the processes of autophagy, and mitochondrial fission, fusion, and mitophagy play a role in the slow removal of irreparable mtDNA damage through the recycling of damaged genomes [46]. It is also possible that these protective mechanisms limit the adverse effects of the other mitochondrial toxicants tested.
5. Conclusions
Overall, our data suggests that individuals with reduced mtDNA content may be more susceptible to certain environmental toxicants. However, interpretation of our data is complicated by the fact that energetically demanding tissues tend to be more susceptible to mtDNA depletion, while many toxicants target specific tissues. It is likely, and an important area for future research, that adverse interactions between reduced mtDNA content and environmental toxicants will occur in a tissue specific manner.
Supplementary Material
Highlights.
Chronic, low dose EtBr exposure reduces mtDNA content 35-55% in C. elegans
This level of reduced mtDNA content doesn't induce major mitochondrial dysfunction
This doesn't sensitize C. elegans to afB1, paraquat, or rotenone
This sensitizes C. elegans to secondary arsenite and UVC exposures
Acknowledgments
Funding This work was supported by the National Institute of Environmental Health Sciences and National Institute of Health (R01-ES017540-01A2 and F31ES026859). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson S, et al. Sequence and organization of the human mitochondrial genome. 1981 doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.Naviaux RK. Mitochondrial DNA disorders. European journal of pediatrics. 2000;159(3):S219–S226. doi: 10.1007/pl00014407. [DOI] [PubMed] [Google Scholar]
- 3.Wallace DC. A mitochondrial bioenergetic etiology of disease. The Journal of clinical investigation. 2013;123(4):1405–1412. doi: 10.1172/JCI61398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.García-Rodríguez LJ. Appendix 1. Basic properties of mitochondria. Methods in cell biology. 2006;80:809–812. doi: 10.1016/S0091-679X(06)80040-3. [DOI] [PubMed] [Google Scholar]
- 5.Sarzi E, et al. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. The Journal of pediatrics. 2007;150(5):531–534. e6. doi: 10.1016/j.jpeds.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 6.Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes–many genes, common mechanisms. Neuromuscular Disorders. 2010;20(7):429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 7.El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10(2):186–198. doi: 10.1007/s13311-013-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renaldo F, et al. MFN2, a new gene responsible for mitochondrial DNA depletion. Brain. 2012;135(8):e223–e223. doi: 10.1093/brain/aws111. [DOI] [PubMed] [Google Scholar]
- 9.Rouzier C, et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy `plus' phenotype. Brain. 2011:awr323. doi: 10.1093/brain/awr323. [DOI] [PubMed] [Google Scholar]
- 10.Poirier MC, et al. Long-term mitochondrial toxicity in HIV-uninfected infants born to HIV-infected mothers. Journal of acquired immune deficiency syndromes (1999) 2003;33(2):175–183. doi: 10.1097/00126334-200306010-00010. [DOI] [PubMed] [Google Scholar]
- 11.Divi RL, et al. Transplacentally exposed human and monkey newborn infants show similar evidence of nucleoside reverse transcriptase inhibitor-induced mitochondrial toxicity. Environmental and molecular mutagenesis. 2007;48(3–4):201–209. doi: 10.1002/em.20201. [DOI] [PubMed] [Google Scholar]
- 12.Divi RL, et al. Progressive mitochondrial compromise in brains and livers of primates exposed in utero to nucleoside reverse transcriptase inhibitors (NRTIs) Toxicological Sciences. 2010:kfq235. doi: 10.1093/toxsci/kfq235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer JN, et al. Mitochondria as a target of environmental toxicants. toxicological sciences. 2013;134(1):1–17. doi: 10.1093/toxsci/kft102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought? Environmental and molecular mutagenesis. 2010;51(5):417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 15.González-Hunt CP, et al. Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. PloS one. 2014;9(12):e114459. doi: 10.1371/journal.pone.0114459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niranjan B, Bhat N, Avadhani N. Preferential attack of mitochondrial DNA by aflatoxin B1 during hepatocarcinogenesis. Science. 1982;215(4528):73–75. doi: 10.1126/science.6797067. [DOI] [PubMed] [Google Scholar]
- 17.Niranjan BG, et al. Protection of mitochondrial genetic system against aflatoxin B1 binding in animals resistant to aflatoxicosis. Cancer research. 1986;46(7):3637–3641. [PubMed] [Google Scholar]
- 18.Backer JM, Weinstein IB. Interaction of Benzo (a) pyrene and Its Dihydrodiol-Epoxide Derivative with Nuclear and Mitochondrial DNA in C3H10T½ Cell Cultures. Cancer Research. 1982;42(7):2764–2769. [PubMed] [Google Scholar]
- 19.Tsang WY, Lemire BD. The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2003;1638(2):91–105. doi: 10.1016/s0925-4439(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 20.Braeckman BP, Houthoofd K, Vanfleteren JR. Intermediary metabolism. In: T.C.e.R. Community, editor. WormBook. WormBook; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okimoto R, et al. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992;130(3):471–498. doi: 10.1093/genetics/130.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagido C, McLaggan D, Glover LA. A Screenable In Vivo Assay for Mitochondrial Modulators Using Transgenic Bioluminescent Caenorhabditis elegans. JoVE (Journal of Visualized Experiments) 2015;(104):e53083–e53083. doi: 10.3791/53083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagido C, et al. Bridging the phenotypic gap: real-time assessment of mitochondrial function and metabolism of the nematode Caenorhabditis elegans. BMC physiology. 2008;8(1):7. doi: 10.1186/1472-6793-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beanan MJ, Strome S. Characterization of a germ-line proliferation mutation in C. elegans. Development. 1992;116(3):755–766. doi: 10.1242/dev.116.3.755. [DOI] [PubMed] [Google Scholar]
- 25.Boyd WA, et al. Application of a mathematical model to describe the effects of chlorpyrifos on Caenorhabditis elegans development. PLoS One. 2009;4(9):e7024. doi: 10.1371/journal.pone.0007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- 27.Stiernagle T. Maintenance of C. elegans. C. elegans. 1999;2:51–67. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng S-Q, et al. Drug absorption efficiency in Caenorhbditis elegans delivered by different methods. PloS one. 2013;8(2):e56877. doi: 10.1371/journal.pone.0056877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croteau DL, et al. Cooperative damage recognition by UvrA and UvrB: identification of UvrA residues that mediate DNA binding. DNA repair. 2008;7(3):392–404. doi: 10.1016/j.dnarep.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rooney JP, et al. PCR based determination of mitochondrial DNA copy number in multiple species. Mitochondrial Regulation: Methods and Protocols. 2015:23–38. doi: 10.1007/978-1-4939-1875-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Hunt CP, et al. PCR-Based Analysis of Mitochondrial DNA Copy Number, Mitochondrial DNA Damage, and Nuclear DNA Damage. Current Protocols in Toxicology. 2016:20.11. 1–20.11. 25. doi: 10.1002/0471140856.tx2011s67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luz AL, et al. Seahorse Xfe24 Extracellular Flux Analyzer-Based Analysis of Cellular Respiration in Caenorhabditis elegans. Current Protocols in Toxicology. 2015:25.7. 1–25.7. 15. doi: 10.1002/0471140856.tx2507s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luz AL, et al. Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PloS one. 2015;10(6):e0130940. doi: 10.1371/journal.pone.0130940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoogewijs D, et al. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC molecular biology. 2008;9(1):1. doi: 10.1186/1471-2199-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung MC, et al. Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacology and Toxicology. 2013;14(1):9. doi: 10.1186/2050-6511-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luz AL, et al. Arsenite uncouples mitochondrial respiration and induces a Warburg-like effect in Caenorhabditis elegans. Toxicol Sci. 2016 doi: 10.1093/toxsci/kfw185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luz A, et al. In Vivo determination of mitochondrial function using luciferase-expressing Caenorhabditis elegans: Contribution of oxidative phosphorylation, glycolysis, and fatty acid oxidation to toxicant-induced dysfunction. Current Protocols in Toxicology. 2016;69:25.8.1–25.8.22. doi: 10.1002/cptx.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsang WY, Lemire BD. Mitochondrial genome content is regulated during nematode development. Biochemical and biophysical research communications. 2002;291(1):8–16. doi: 10.1006/bbrc.2002.6394. [DOI] [PubMed] [Google Scholar]
- 39.Gaines G, Attardi G. Intercalating drugs and low temperatures inhibit synthesis and processing of ribosomal RNA in isolated human mitochondria. Journal of molecular biology. 1984;172(4):451–466. doi: 10.1016/s0022-2836(84)80017-0. [DOI] [PubMed] [Google Scholar]
- 40.Zylbee E, Vesco C, Penman S. Selective inhibition of the synthesis of mitochondria-associated RNA by ethidium bromide. Journal of molecular biology. 1969;44(1):195–204. doi: 10.1016/0022-2836(69)90414-8. [DOI] [PubMed] [Google Scholar]
- 41.Nass MM. Differential effects of ethidium bromide on mitochondrial and nuclear DNA synthesis in vivo in cultured mammalian cells. Experimental cell research. 1972;72(1):211–222. doi: 10.1016/0014-4827(72)90583-6. [DOI] [PubMed] [Google Scholar]
- 42.Au C, et al. SMF-1, SMF-2 and SMF-3 DMT1 orthologues regulate and are regulated differentially by manganese levels in C. elegans. PloS one. 2009;4(11):e7792. doi: 10.1371/journal.pone.0007792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Partridge FA, et al. The C. elegans glycosyltransferase BUS-8 has two distinct and essential roles in epidermal morphogenesis. Developmental biology. 2008;317(2):549–559. doi: 10.1016/j.ydbio.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe M, et al. A mutation in a cuticle collagen causes hypersensitivity to the endocrine disrupting chemical, bisphenol A, in Caenorhabditis elegans. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;570(1):71–80. doi: 10.1016/j.mrfmmm.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497(7450):451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bess AS, et al. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic acids research. 2012:gks532. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poirier MC, et al. Fetal consequences of maternal antiretroviral nucleoside reverse transcriptase inhibitor use in human and nonhuman primate pregnancy. Current opinion in pediatrics. 2015;27(2):233–239. doi: 10.1097/MOP.0000000000000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen S, et al. Arsenic binding to proteins. Chemical reviews. 2013;113(10):7769–7792. doi: 10.1021/cr300015c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naranmandura H, et al. Mitochondria are the main target organelle for trivalent monomethylarsonous acid (MMAIII)-induced cytotoxicity. Chemical research in toxicology. 2011;24(7):1094–1103. doi: 10.1021/tx200156k. [DOI] [PubMed] [Google Scholar]
- 50.Zhao F, et al. Arsenic exposure induces the Warburg effect in cultured human cells. Toxicology and applied pharmacology. 2013;271(1):72–77. doi: 10.1016/j.taap.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanner CM, et al. Rotenone, paraquat, and Parkinson's disease. Environmental health perspectives. 2011;119(6):866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasiviswanathan R, et al. Human mitochondrial DNA polymerase γ exhibits potential for bypass and mutagenesis at UV-induced cyclobutane thymine dimers. Journal of Biological Chemistry. 2012;287(12):9222–9229. doi: 10.1074/jbc.M111.306852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer JN, et al. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol. 2007;8(5):R70. doi: 10.1186/gb-2007-8-5-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukami J.-i., Yamamoto I, Casida JE. Metabolism of rotenone in vitro by tissue homogenates from mammals and insects. Science. 1967;155(3763):713–716. doi: 10.1126/science.155.3763.713. [DOI] [PubMed] [Google Scholar]
- 55.Menzel R, Bogaert T, Achazi R. A systematic gene expression screen of Caenorhabditis elegans cytochrome P450 genes reveals CYP35 as strongly xenobiotic inducible. Archives of Biochemistry and Biophysics. 2001;395(2):158–168. doi: 10.1006/abbi.2001.2568. [DOI] [PubMed] [Google Scholar]
- 56.Menzel R, et al. CYP35: xenobiotically induced gene expression in the nematode Caenorhabditis elegans. Archives of biochemistry and biophysics. 2005;438(1):93–102. doi: 10.1016/j.abb.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 57.Wolkow CA. Identifying factors that promote functional aging in Caenorhabditis elegans. Experimental gerontology. 2006;41(10):1001–1006. doi: 10.1016/j.exger.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 58.Croll NA, Smith JM, Zuckerman BM. The aging process of the nematode Caenorhabditis elegans in bacterial and axenic culture. Experimental aging research. 1977;3(3):175–189. doi: 10.1080/03610737708257101. [DOI] [PubMed] [Google Scholar]
- 59.de Boer R, et al. Caenorhabditis elegans as a Model System for Studying Drug Induced Mitochondrial Toxicity. PloS one. 2015;10(5) doi: 10.1371/journal.pone.0126220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bratic I, et al. Mitochondrial DNA level, but not active replicase, is essential for Caenorhabditis elegans development. Nucleic acids research. 2009:gkp018. doi: 10.1093/nar/gkp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Divi RL, et al. Cardiac mitochondrial compromise in 1-yr-old Erythrocebus patas monkeys perinatally-exposed to nucleoside reverse transcriptase inhibitors. Cardiovascular toxicology. 2005;5(3):333–346. doi: 10.1385/ct:5:3:333. [DOI] [PubMed] [Google Scholar]
- 62.Altun ZF, Hall DH. Handbook of C. elegans Anatomy. WormAtlas. 2005 http://www.wormatlas.org/ver1/handbook/contents.htm.
- 63.Tsang WY, et al. Mitochondrial Respiratory Chain Deficiency inCaenorhabditis elegans Results in Developmental Arrest and Increased Life Span. Journal of Biological Chemistry. 2001;276(34):32240–32246. doi: 10.1074/jbc.M103999200. [DOI] [PubMed] [Google Scholar]
- 64.Croy R, et al. Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver. Proceedings of the National Academy of Sciences. 1978;75(4):1745–1749. doi: 10.1073/pnas.75.4.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quezada T, et al. Effects of aflatoxin B 1 on the liver and kidney of broiler chickens during development. Comparative Biochemistry and Physiology Part C: Pharmacology, Toxicology and Endocrinology. 2000;125(3):265–272. doi: 10.1016/s0742-8413(99)00107-3. [DOI] [PubMed] [Google Scholar]
- 66.Pan-Montojo F, et al. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. PloS one. 2010;5(1):e8762. doi: 10.1371/journal.pone.0008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ossowska K, et al. Degeneration of dopaminergic mesocortical neurons and activation of compensatory processes induced by a long-term paraquat administration in rats: implications for Parkinson's disease. Neuroscience. 2006;141(4):2155–2165. doi: 10.1016/j.neuroscience.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 68.IARC . Some drinking-water disinfectants and contaminants, including arsenic. Vol. 84. IARC; 2004. [PMC free article] [PubMed] [Google Scholar]
- 69.ATSDR, U. Toxicological profile for arsenic. Agency for Toxic Substances and Disease Registry, Division of Toxicology; Atlanta, GA: 2007. [PubMed] [Google Scholar]
- 70.Juliani M, Hixon S, Moustacchi E. Mitochondrial genetic damage induced in yeast by a photoactivated furocoumarin in combination with ethidium bromide or ultraviolet light. Molecular and General Genetics MGG. 1976;145(3):249–254. doi: 10.1007/BF00325820. [DOI] [PubMed] [Google Scholar]
- 71.Fuke C, et al. In vitro studies of the metabolism of paraquat and diquat using rat liver homogenates--isolation and identification of the metabolites of paraquat and diquat. Nihon hoigaku zasshi= The Japanese journal of legal medicine. 1993;47(1):33–45. [PubMed] [Google Scholar]
- 72.Egner PA, et al. Identification of aflatoxin M1-N7-guanine in liver and urine of tree shrews and rats following administration of aflatoxin B1. Chemical research in toxicology. 2003;16(9):1174–1180. doi: 10.1021/tx034106u. [DOI] [PubMed] [Google Scholar]
- 73.Mace K, et al. Aflatoxin B1-induced DNA adduct formation and p53 mutations in CYP450-expressing human liver cell lines. Carcinogenesis. 1997;18(7):1291–1297. doi: 10.1093/carcin/18.7.1291. [DOI] [PubMed] [Google Scholar]
- 74.Thomas DJ, et al. Arsenic (+ 3 oxidation state) methyltransferase and the methylation of arsenicals. Experimental Biology and Medicine. 2007;232(1):3–13. [PMC free article] [PubMed] [Google Scholar]
- 75.Engström K, et al. Polymorphisms in arsenic (+ III oxidation state) methyltransferase (AS3MT) predict gene expression of AS3MT as well as arsenic metabolism. Environmental health perspectives. 2011;119(2):182. doi: 10.1289/ehp.1002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Calabrese EJ, Baldwin LA. Inorganics and hormesis. Critical reviews in toxicology. 2003;33(3–4):215–304. doi: 10.1080/713611040. [DOI] [PubMed] [Google Scholar]
- 77.Snow ET, et al. Arsenic, mode of action at biologically plausible low doses: what are the implications for low dose cancer risk? Toxicology and applied pharmacology. 2005;207(2):557–564. doi: 10.1016/j.taap.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 78.Baastrup R, et al. Arsenic in drinking-water and risk for cancer in Denmark. Environmental Health Perspectives. 2008;116(2):231. doi: 10.1289/ehp.10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmeisser S, et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging cell. 2013;12(3):508–517. doi: 10.1111/acel.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schmidt CM, et al. Hormesis effect of trace metals on cultured normal and immortal human mammary cells. Toxicology and industrial health. 2004;20(1–5):57–68. doi: 10.1191/0748233704th192oa. [DOI] [PubMed] [Google Scholar]
- 81.Rooney J, et al. Effects of 5′-fluoro-2-deoxyuridine on mitochondrial biology in Caenorhabditis elegans. Experimental gerontology. 2014;56:69–76. doi: 10.1016/j.exger.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.