

Abstract
Heterotrimeric G-proteins mainly relay the information from G-protein-coupled receptors (GPCRs) on the plasma membrane to the inside of cells to regulate various biochemical functions. Depending on the targeted cell types, tissues and organs, these signals modulate diverse physiological functions. The basic schemes of heterotrimeric G-proteins have been outlined. In this review we briefly summarize what is known about the regulation, signaling and physiological functions of G-proteins. We then focus on a few less explored areas such as regulation of G-proteins by non-GPCRs, and the physiological functions of G-proteins that can not be easily explained by the known G-protein signaling pathways. There are new signaling pathways and physiological functions for G-proteins to be discovered and further interrogated. With the advancements in structural and computational biological techniques, we are closer to having a better understanding of how G-proteins are regulated, and the specificity of G-protein interactions with their regulators.
Graphical abstract
1. Introduction
A structurally diverse repertoire of ligands, from photons to many hormones and neurotransmitters, activate G-protein-coupled receptors (GPCRs) to elicit their physiological functions [1, 2]. GPCRs comprise a large and diverse superfamily, and family members have been identified in organisms as evolutionarily distant as yeast and humans. Heterotrimeric guanine nucleotide-binding regulatory proteins (G-proteins) directly relay the signals from GPCRs [3-5]. These G-proteins are composed of α, β, and γ subunits. The β and γ subunits are tightly associated and can be regarded as one functional unit. G-proteins function as molecular binary switches with their biological activity determined by the bound nucleotide [3-5]. Upon agonist binding, GPCRs increase the exchange of GDP bound on the Gα subunit with GTP. This leads to the dissociation of Gα subunit from Gβγ dimer resulting in two functional subunits (Gα and Gβγ). Both Gα and Gβγ subunits signal to various cellular pathways.
G-proteins are identified by their Gα subunits. Based on the sequence and functional similarities, Gα proteins are grouped into four families: Gαs, Gαi, Gαq, and Gα12 (Figure 1). In the Gαs family, there are two members: Gαs and Gαolf. While Gαs (s stands for stimulation) is expressed in most types of cells, Gαolf (olf stands for olfaction) is specifically expressed in the olfactory sensory neurons. Gαi family is the largest and most diverse family, including Gαi1, Gαi2, Gαi3, Gαo, Gαt, Gαg and Gαz. Gαi proteins (i stands for inhibition) have been detected in most types of cells. Gαo is highly expressed in neurons and has two spliced variants: GαoA and GαoB. Gαt (t stands for transducin) has two isoforms. Gαt1 is expressed in the rod cells in the eye, while Gαt2 is in the cone cells of the eye. Gαg (g stands for gustducin) is found in taste receptor cells. Gαz is expressed in neuronal tissues and in platelets. In humans, the Gαq family consists of Gαq, Gα11, Gα14 and Gα16 (The mouse equivalent is Gα15). Gαq and Gα11 are ubiquitously expressed, while Gα14 and Gα15/16 expression is more restricted. Gα14 is mainly found in the kidney, lung and liver, and Gα15/16 is specifically expressed in hematopoietic cells. In the Gα12 family, there are Gα12 and Gα13, which are expressed in most types of cells.
Figure 1.
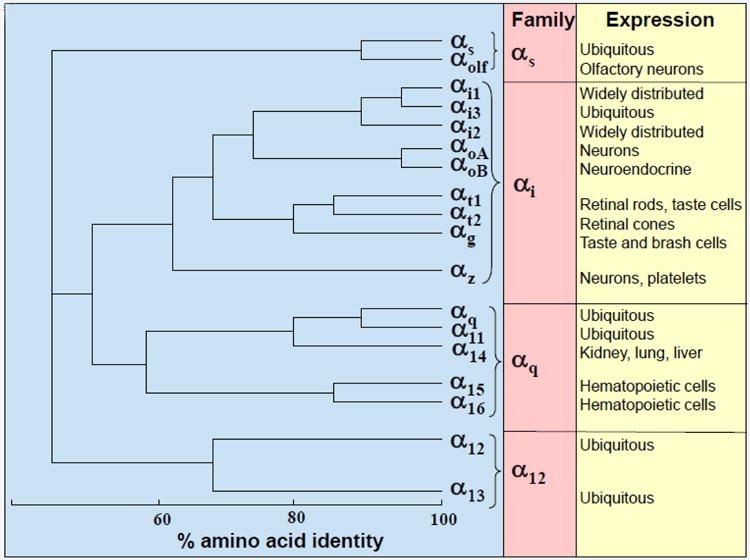
Phylogenetic relationship of human and mouse Gα subunits and their expression.
In addition to these Gα subunits, heterotrimeric G-proteins contain Gβγ subunits. There are 5 Gβ and 12 Gγ genes in the human and mouse genomes (Figures 2 and 3). Gβ1, Gβ2, Gβ3 and Gβ4 share high sequence similarities (between 80 to 90%), while Gβ5 is ∼50% similar to other Gβ subunits (Figure 2). While Gβ5 is mainly found in the brain, other Gβ subunits are widely distributed. Gγ subunits are more diverse and share sequence similarities ranging from 20% to 80% (Figure 3). Although purified Gβγ subunits with different Gβ and Gγ isoform combinations generally have similar biochemical activities in in vitro assays, gene-deletion experiments in mice show that at least some Gβ and Gγ genes have different physiological functions. These might reflect the different distributions and expression levels of Gβ and Gγ genes.
Figure 2.

Phylogenetic relationship of human Gβ subunits and their expression.
Figure 3.

Phylogenetic relationship of human Gγ subunits and their expression.
The crystal structures of several Gα, Gβγ, and Gαβγ have been solved (Figure 4). The structure of a Gα subunit consists of two domains: a Ras-like GTPase domain and an α-helical domain [6] (Figure 4a). These two domains are linked by Linker 1 and Linker 2 (Figure 4a). Between these two domains lies a deep cleft within which GDP or GTP is tightly bound (Figure 4a). The nucleotide is essentially occluded from the bulk solvent, leading to the proposal that the helical domain is the inhibitory barrier and provides the regulatory entry point by GPCRs or Gβγ subunits [7-10]. The structure of a Gβγ subunit shows that Gβ folds into a β-propeller with 7 blades (Figure 4b). Each blade consists of four-stranded β-sheets. The N-terminal α-helical segment of Gβ forms a tight coiled-coil interaction with the Gγ subunit (Figure 4b). The crystal structure of a Gαβγ heterotrimer illustrates that the two domains of Gα interact with different regions of Gβ (Figure 4 c and d). The Gα N-terminal α helix interacts with the side of the Gβ propeller. The Gα switch region II region interacts with the top of the Gβ propeller (Figure 4 c and d).
Figure 4.

Crystal structures of G-protein heterotrimer. Cartoon diagrams of Gαi1 (orange) with GDP in space filling representation (color by atom) (a), Gβ1γ2 dimer where blue is Gβ, and red is Gγ2 (b), Gαi1β1γ2 heterotrimer (c), and surface representation of Gαi1β1γ2 heterotrimer (d).
Great progress has been made in understanding the mechanisms by which heterotrimeric G-proteins regulate their downstream targets [6, 11]. Recently a series of crystal structures of GPCRs in the inactive and active states, bound with antagonists, inverse agonists or agonists, have elucidated the structural basis for the modulation and activation of GPCRs by ligands [1, 12-14]. A crystal structure of the complex of β2-adrenergic receptor (β2-AR) and Gs has revealed the structural changes in β2-AR and in Gs, as well as the interacting regions and residues between a GPCR and a G protein [15-17].
In this review, we summarize the activation of G-proteins by GPCRs, and regulation of G-proteins by non-GPCR proteins, such as Ric-8 protein, GPR-domain containing proteins, GBA-motif containing proteins, and RGS-domain containing proteins. Furthermore, we list some G-protein interacting proteins, in addition to the well-established G-protein effectors such as adenylyl cyclases and phospholipase C, although the physiological functions of many of these interactions are not clear. Finally, we describe two examples of non-canonical signaling events that deserve more attention: one is the role of G-protein signaling in cell division, and the other is the role of G-proteins in receptor tyrosine kinase signaling. This review does not intend to be complete, but rather illustrates that there is much more exciting work to be done in the G-protein signaling field.
2. Regulation of G-Proteins
a). General features of G-protein regulation
G-proteins biochemically function in a GTPase cycle (Figure 5). In the inactive form, GDP-bound Gα subunits bind tightly to the obligate heterodimer of Gβγ [1]. Upon agonist binding to a GPCR, downstream signaling is initiated. GPCRs act as guanine nucleotide exchange factors (GEFs), promoting the release of bound GDP from Gα [18, 19]. Nucleotide-free Gα then binds GTP, resulting in the dissociation of Gβγ. Both GTP-bound Gα and free Gβγ are capable of initiating signals by interacting with downstream effector proteins. Gα subunit signaling is terminated by the intrinsic GTPase activity of Gα which hydrolyzes the bound GTP to GDP. GTPase activating proteins (GAPs) (such as RGS proteins) bind with Gα to accelerate the intrinsically low GTPase activity of Gα subunit. The Gβγ signaling, on the other hand, is terminated by re-association with Gα·GDP. This process represents a G-protein cycle.
Figure 5.

G-protein cycle.
The nucleotide-binding pocket of G-proteins is highly conserved, and binding of GDP or GTP proceeds through a mechanism which is conserved. Generally described as a “switch” mechanism, guanine nucleotide loading and unloading take place in the Ras-like domain of G-proteins which serves as a platform for both guanine nucleotide binding as well as in GTP hydrolysis (Figure 4a) [20]. This Ras-like domain is a typical α/β nucleotide-binding fold, and consists of a six-stranded β-sheet with five helices located on both sides. Lining the binding pocket are four to five signature sequence elements attributable to G-proteins, two of which are the most important for guanine-base binding specificity. The first is a N/TKXD motif [where X is any amino acid] whose aspartic acid is thought to form a bifurcated H bond with the guanine base. The second is the highly conserved P-loop region which contains the sequence pattern GXXXXGKS/T, also known as a Walker A motif [21]. G-proteins possessing this motif belong to the “P-loop containing nucleotide triphosphate hydrolase superfamily”.
GEFs work to weaken the affinity for the nucleotide [22]. G-proteins have a high affinity for GDP (pmol in some cases) that causes the GDP disassociation process to be slow. To overcome this barrier, G-proteins employ the action of GEFs which reduce the affinity for bound GDP, and subsequently enable binding of the triphosphate moiety, due to its higher cellular concentration (Figure 5). While the interaction between G-proteins and either GDP or GTP is very high, affinity of the GEF for G-protein in either nucleotide-bound state is much lower. GEFs cause perturbations in the two switch regions of G-proteins. Bound GDP is encompassed by two loop regions called Switch I and Switch II, the latter of which is the location of the conserved N/TKXD motif. The switch regions together with the P-loop interact with the phosphates of the bound nucleotide as well as a coordinating Mg2+ ion. In the exchange reaction of a Ras-superfamily GTPase, a GEF interfaces with the GDP-bound GTPase in such a way that the nucleotide is partially competed from its binding site. More specifically, the Mg2+ ion is displaced by residues residing on the GTPase, causing disruption in the interactions between the P-loop region and the nucleotide phosphates. This produces a “push-pull” effect between the switch regions and the nucleotide. The affinity of the GTPase for the nucleotide is decreased as it is “loosened” from its binding site, and the nucleotide is eventually released. Finally a high affinity binary complex is formed between the GEF and GTPase, which is free to bind GTP. Not much structural information is available for the ternary complexes or the intermediate conformational states, and therefore it remains to be elucidated how the GEFs approach the G-protein, how the nucleotide is released from the binding site and how GTP is recruited.
b). Activation of G-proteins by GPCRs
The main physiological functions of G-proteins are to relay the signals from GPCRs which function as GEFs for G-proteins. Binding with exogenous or endogenous agonists induces GPCRs into an active conformational state which, in turn, influences intracellular binding of G-proteins or arrestin proteins [23, 24]. This superfamily of receptors shares seven transmembrane (TM) helical domains (TM1 to TM7) connected by alternating three intra- and three extra- cellular loops (ICL1, ECL1, ICL2, ECL2, ICL3, and ECL3) (Figure 6). GPCR targeted ligands are classified into agonists, inverse agonists, and antagonists. Agonists binding to GPCRs promote an active conformation, which in turn increases the signaling effect. Conversely inverse agonists inhibit spontaneous basal signaling activity by stabilizing an inactive conformation of GPCRs. Antagonists have no effect on the dynamic equilibrium between the active and inactive conformations of GPCRs, but prevent binding of both agonists and inverse agonists.
Figure 6.

Crystal structure of the complex of β2-adrenergic receptor and Gs. (a) Cartoon representation of β2-adrenergic receptor (green) and Gs (orange, blue and red), (b) Close view of the β2-adrenergic receptor and Gs interface, (c) Surface representation of the β2-adrenergic receptor and Gs. (d) Superposition of the α subunit of Gs from the apo form (cyan) and from its complex form with β2-adrenergic receptor (orange).
Various studies have suggested that ICL2, and the N- and C-terminal regions of ICL3 are G-protein interacting sites on GPCRs [25]. Additionally, the ICL1 and some C-terminal residues might contribute to the coupling of G-proteins [18]. Binding of a ligand at the extracellular region induces a structural rearrangement in the transmembrane core region which further leads to a conformational change in the intracellular region at the cytoplasmic side. This facilitates interaction with intracellular effectors such as G-proteins. Agonist binding alone may not be sufficient to stabilize a fully active conformation of GPCRs which might require the additional binding of an intracellular protein, such as a G-protein or arrestin, at the cytoplasmic side [15, 26].
For the interaction between GPCRs and G-proteins, it was originally proposed that GPCRs and G-proteins interact by collision. Both GPCRs and G-proteins diffuse freely within the plasma membrane, and that only activated (agonist-bound) GPCRs couple to and activate G-proteins [27]. However, some GPCRs and G-proteins were reported to form a preassembled (or precoupled) complex in the absence of any ligands [27, 28]. Agonist binding activates the GPCR which causing the conformational changes in the preassembled GPCR/G-protein complex, resulting in G-protein activation. The structure of the preassembled GPCR/G-protein complex is proposed to be different from that of the activated GPCR/G-protein complex. Collision coupling or preassembly can yield different kinetics of activation of G-proteins, and may underlie the different constitutive activities of GPCRs. These different GPCR/G-protein interaction modes depend on the specific GPCR/G-protein pair. Further investigations are needed to provide the structural basis for these different GPCR/G-protein interactions.
In the crystal structure of a complex of β2-AR and Gs, a large β2-AR-Gαs protein interface is formed by ICL2 and transmembrane helices TM5 and TM6 of the receptor, as well as by the α4- and α5-helices, the αN–β1 junction, and the β3-strand of Ras-like domain of Gαs [15] (Figure 6). Major interactions between β2-AR and Gαs protein occur along the carboxyl-terminal end of TM5 of the receptor. Here, residues Glu225, Gln229, and Lys232 belonging to TM5 of β2-AR are involved in strong electrostatic interactions with Asp381, Gln384, and Arg385 located at the carboxyl-terminal end of the α5-helix of Gαs (Figure 6b). Main chain oxygens of residues Thr136 and Ile135 at the carboxyl-terminus of TM3 are also involved in polar interactions with Arg380 and Gln384 of the same α5-helix of the Ras-domain of Gαs. When the structure of Gαs alone (cyan color in Figure 6d) is superimposed on top of its crystal structure in complex with β2–AR (in orange color), it shows that the α-helical domain of Gαs in the complex was rotated by about 127° towards the receptor relative to its apo-structure. The closed and open states of the cleft (the GDP/GTP binding site) are determined by the relative pivoting of the Ras-like domain and the α-helical domain at Linker 2. Given this pivot model, the two domains of Gα could be ‘clam-shell’ opening or ‘rolling-top’ expansion including a relative rotation of the Ras-like domain and the helical domain around an axis through the linkers [29]. In addition to the established role for α5-helix of Gα in connecting GPCRs to the guanine-nucleotide binding pocket, an extended Linker 2 connects GPCRs to the nucleotide binding pocket as well as the α-helical domain of Gα [29]. Many more structures in different states and with different GPCRs/G-proteins are needed to fully understand the biochemistry of the G-protein activation cycle.
c). GPCR dimers and oligomers
GPCRs can exist and function as dimers or oligomers [30-34]. Dimerization and oligomerization modulate various GPCR functions such as cell surface targeting, cooperativity, activation, G-protein coupling, signaling and internalization[33-35]. The requirement of dimers or tetramers of GPCRs for G-protein activation has not been firmly established. However, for the class-C family of GPCRs, the dimerization of two protomers (either heterodimers or homodimers) is essential for its biological function and G-protein activation.
In the crystal structure of β1-adrenergic receptor (β1-AR) oligomers in a membrane-like environment, there are two dimer interfaces: one involves TM1/TM2/H8 (H8: helix 8) and the other engages TM4/TM5/ICL2 [14] (Figure 7). These are the two major dimer interfaces observed in crystal structures of various GPCRs [36-39]. In the TM1/TM2/H8 dimer interface, interacting residues are mainly from TM1 (including Gln38, Gln39, Ala42, Leu46, Ala49, Leu50, Val52, Leu53, and Leu54) [14]. Residues from other parts of the receptor also contribute to this dimer interface, including residues from TM2 (Pro96, Ala99, Thr100 and Val103), the extracellular loop 1 (Thr106, Leu108, and Trp109), and the C-terminal H8 (Arg351, Lys354, Arg355 and Leu356) [14]. In addition to these hydrophobic and van der Waals interactions, Ser45 in one TM1 forms a hydrogen bond with Ser45 from another TM1. Glu41 in TM1 from one monomer forms a salt bridge with Arg104 in TM2 from the second monomer [14]. This dimer interface is similar to the one observed in the dimer of rhodopsin in the active state which uses TM1 and H8 as an interface [37, 38].
Figure 7.

Crystal structure of the oligomeric β1-adrenergic receptor in a membrane-like environment. (a) Cartoon representation of β1-AR as a tetramer in presence of molecular surface highlighting two distinct dimer interfaces. (b) Top view of β1-AR tetramer down the extracellular surface. (c) Interacting TM1/TM2/H8 and TM4/TM5/ICL2 segments in cartoon diagram in presence of the surface representation of β1-AR tetramer.
In the second TM4/TM5/ICL2 dimer interface, residues from both TM4 (including Leu171) and TM5 (including Arg205, Ala206, Ala210, Ile218 and Arg229) contribute to the hydrophobic interaction [14]. ICL2 also plays a significant role in this dimer interaction (including residues Tyr140, Leu141, Thr144, Ser145, Phe147, Arg148, Ser151, and Leu152) [14]. Two residues (Trp181 and Arg183) from extracellular loop 2 also participate in this interaction [14]. There is a variation of the TM4/TM5 dimer interface that uses TM5/TM6 as observed in the crystal structures of several GPCRs [40, 41]. ICL2 is critical for interacting with G-proteins based on the structural model of the β2-AR-Gs complex [15]. Participation of ICL2 in this dimer interface may prevent G-protein coupling to the dimer formed through TM4/TM5/ICL2 interface, or G-protein binding may disrupt this dimer interface. Therefore, if the signaling unit is a pentamer (two GPCRs and one trimeric G protein), the GPCR dimer interface in this signaling unit is likely TM1/TM2/H8. In this model, only one GPCR contacts with the G-protein trimer, and the other receptor is “spared” or could function through trans-protomer allosteric regulation.
A dimer interface involving TM1 was shown to be insensitive to ligand binding and the receptor activation state as shown for dopamine D2 receptors and serotonin 5HT2c receptors [42, 43]. The similarity of TM1/TM2/H8 dimer interface in the inactive β1-AR and in the active rhodopsin is consistent with the notion that this TM1/TM2/H8 dimer interface does not undergo significant conformational changes from inactive to active states of GPCRs. On the other hand, the TM4/TM5/ICL2 dimer interface makes structural rearrangement during the GPCR activation process, at least in the cases of dopamine D2 receptors and serotonin 5HT2c receptors [42, 44]. Based on the structures of active GPCRs, the intracellular end of TM5 moves away from the TM bundle core [15]. Therefore it is possible that, upon the agonist binding, the configuration change at the TM4/TM5/ICL2 dimer interface is part of the receptor activation process.
d). Regulation of G-proteins by non-GPCRs
In addition to GPCRs, other regulatory proteins exist for heterotrimeric G-proteins [45-49]. These regulators include Ric-8, GPR-domain containing proteins, GBA motif-containing proteins, and RGS-domain containing proteins. Although there might be other non-GPCR regulators, we will focus on a few as examples of G-protein regulation by non-GPCR proteins.
(i) Regulation of G-proteins by Ric-8
Ric-8 (synembryn) was originally identified in C. elegans through genetic analysis [50]. Ric-8 functions upstream of Gαq in regulating neurotransmitter secretion [50]. Ric-8 also acts upstream of Gαo and GPA16 (another Gα subunit in C. elegans) during asymmetric cell division of one-cell stage C. elegans embryos [51-53]. In Drosophila, Ric-8 is required for Gα-mediated spindle orientation and cell polarity during asymmetric cell division [54-56]. In Ric-8 mutants, Gαi failed to localize at the cell cortex [54-56]. Ric-8 has also been genetically shown to play a role in gastrulation and is involved in the fog-concertina pathway [55]. Concertina is the Drosophila homolog of Gα13 [57]. Fog (folded gastrulation) is an extracellular polypeptide growth factor [57]. Thus, Ric-8 has been genetically demonstrated to be involved in Gα13-mediated signaling in Drosophila.
There are two distinct mammalian Ric-8-like genes, Ric-8A and Ric-8B. Ric-8A was identified in yeast two-hybrid screens of a rat brain embryonic cDNA library as a protein that interacted with a Gαo bait [58]. The Ric-8A prey clone interacted with Gαo, Gαi1, Gαq and Gα13, but not Gαs baits in pair-wise two-hybrid interaction studies. In vitro biochemical studies have shown that Ric-8A is a GEF for Gαq, Gαi, Gαo, Gα12, Gα13 but not Gαs [58, 59]. On the other hand, Ric-8B interacts with Gαs and Gαq [58, 59]. Mechanistically, Ric-8A binds to GDP-bound Gα proteins, promotes rapid GDP release and forms a stable nucleotide free transition state complex with Gα that is disrupted upon GTP binding, thus leading to the formation of Gα-GTP. Ric-8 differs from GPCRs in that GPCRs work on the inactive Gα-GDP/Gβγ heterotrimer while Ric-8 acts only on the Gα-GDP monomer. Ric-8 could also work on Gα-GDP in complex with GPR proteins (see below). While Ric-8A mRNA is expressed in a variety of tissues, Ric-8B mRNA is mainly expressed in the olfactory epithelium [60]. Moreover, Ric-8A-/- mouse embryos died in the early stages of embryonic development [61].
Although the biochemical mechanism of activation of Gα by Ric-8 is not understood, it has been shown that Ric-8A induces structural changes in the nucleotide binding site of Gα which reduces its affinity for GDP, and subsequently releases the nucleotide and exchanges it for GTP [58] (Figure 8). A mechanistic study of the guanine nucleotide-exchange of GDP for GTP by Ric-8A using SDSL and DEER spectroscopy shed light on some of the structural features of this exchange [62]. Ric-8A binding induces structural displacements in Gαi, resulting in the separation of the α-helical and Ras-like domains. Thermostability studies of Gαi showed that not only do Gαi proteins exhibit characteristics of an inherently disordered protein without GDP bound, but that GDP-bound as well as Ric-8A-bound Gαi are thermodynamically more stable than the non-bound protein. Locally, Ric-8A is thought to instigate structural changes within the nucleotide binding site, producing a series of intermediate states which affect Switch I and Switch II regions, and decreases the affinity of Gαi for the bound GDP nucleotide. Ultimately these perturbations result in a solvent exposed, structurally heterogeneous binding site. These structural rearrangements lead to a viable entry and escape pathway for nucleotides. Though extensive functional studies have been carried out to study the interaction between Ric-8A and Gα, the lack of a crystal structure precludes any concrete determination of the exact mechanism of Ric-8A GEF activity toward Gα proteins.
Figure 8.

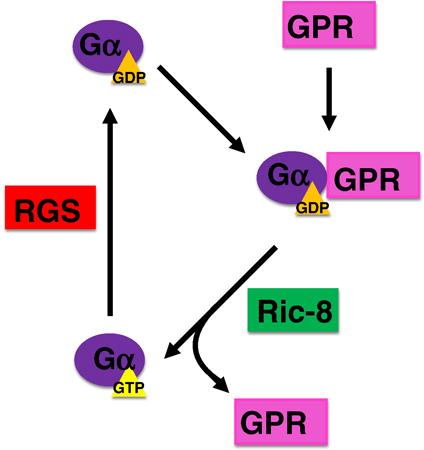
G-protein activation by Ric-8. A GDP-bound Gα subunit disassociates from the membrane and can interact with a GPR-domain containing protein. This complex can then interact with Ric-8, which facilitates the exchange of GTP for GDP. Once activated, Gα can go on to interact with downstream effectors. The signal is terminated when Gα-GTP interacts with a RGS protein, which promotes the hydrolysis of GTP to GDP.
In addition to its GEF activity, Ric-8 proteins have multiple other functions [58, 63]. They could act as a chaperone for the Gα subunit by regulating Gα folding and processing during its biosynthesis [64] [65]. They could regulate the membrane translocation of Gα subunits. In C. elegans, Ric-8 directly interacts with GPA-16, but has no GEF activity towards GPA-16 [66]. Instead, Ric-8 is required for the plasma membrane localization of GPA-16 [66]. In Drosophila, Ric-8 directly interacts with Gαi and is required for the plasma membrane localization of Gαi (in this case, Ric-8 also functions as a GEF for this Gαi)[54-56]. There are still many unanswered questions regarding functions and mechanisms of Ric-8 proteins.
(ii) Regulation of G-proteins by GRP proteins
The second group of non-GPCR regulators of G-proteins is the GPR-domain containing proteins. GPR (G-protein regulator) domains are ∼25 amino acid segments (also called the GoLoco domain) [67, 68]. GPR domains have been shown biochemically to bind the inactive GDP-bound Gαi/o. These GPR proteins inhibit nucleotide exchange in Gα, and compete with Gβγ for Gα binding [46, 48, 69]. Furthermore, Ric-8A can activate Gα by promoting exchange of GDP for GTP when Gα is bound to a GPR domain [70, 71], but not when Gα is bound to Gβγ [58] (Figure 8). Although in vitro biochemical studies implied that GPR domain-containing proteins could function as a GDI (guanine nucleotide dissociation inhibitor) and thus inhibit G-protein signaling, genetic studies in C. elegans showed that GPR domain-containing proteins promote G-protein signaling by either sequestering Gα from Gβγ so that free Gβγ can signal, and/or keeping Gα in a form that can be activated by Ric-8A to prolong the G-protein signaling (Figure 8).
Some AGS proteins (for activator of G protein signaling) are GPR-domain-containing proteins. In a yeast expression screen for GPCR-independent activators of Gβγ-dependent signaling, several AGS proteins were identified [46, 72]. One of these AGS proteins, AGS3, contains four GPR domains, and selectively binds to the GDP-bound form of the Gαi family, as well as acts as a guanine nucleotide dissociation inhibitor [46].
Additional GPR domain-containing proteins include the human proteins LGN, Pcp2, G181b, and Drosophila protein PINS (partner of inscuteable), as well as a related protein (F32A6.4) from C. elegans [46, 67, 73]. These proteins have been proposed to participate in GPCR-independent G-protein regulated events such as asymmetric cell division, mitotic spindle formation, and planar polarity of developing sensory organ precursor cells [46, 74, 75].
(iii) Regulation of G-proteins by GBA motif-containing proteins
The third group of non-GPCR activators of G-proteins is the GBA motif-containing proteins [76, 77]. GBA stands for Gα-binding and activating. GBA motif-containing proteins include Gα-Interacting-Vesicle-associated protein (GIV/Girdin), Daple, NUCB1, NUCB2 and GBAS-1. GIV was discovered as a Gα-interacting protein which could enhance Akt activation [76]. GIV could bind robustly to the Gαi family members and to a lesser extent to Gαs [78]. A series of in vitro studies with purified components demonstrated that GIV accelerates the rate of nucleotide exchange, leading to Gα subunit activation [77]. Further, GIV could not bind Gα subunits in the active GTP-bound conformation. This feature of GEFs ensures that GIV only engages with inactive Gα-GDP and thus promotes Gα signaling. It is not known whether GIV can directly activate a Gαi-βγ trimer in vitro. However, it was shown that GIV could displace Gβγ from a preformed Gαi-βγ trimer in vitro and enhance Gβγ-dependent signaling in cells [76].
GBA motif-containing proteins have been coined “molecular rheostats” due to their ability to fine-tune the duration and extent of signal transduction cascades, more specifically in cancer cell migration [79, 80]. Elevated levels of these proteins in circulating tumor cells and their role as G-protein activators suggest that hyperactivation of G-proteins, as opposed to gene mutations in G-proteins and GPCRs, might play a role in tumor metastatic progression.
e). Regulation of G-proteins by RGS proteins
A large family of RGS (regulators of G-protein signaling) proteins has been identified. After the active GTP-bound Gα exerts its effect on downstream effectors, this activation must be highly regulated to maintain the appropriate signal strength and duration. Although Gα subunits contain weak intrinsic GTP hydrolytic activity, the hydrolysis of GTP to GDP and the re-association of Gα with Gβγ subunits are steps in the G-protein deactivation cycle that can be modulated to achieve the desired cellular signal output, and these steps are regulated by RGS proteins [81]. RGS proteins act as GAPs for Gα subunits. Gα selectivity can be attributed to heterogeneity in the RGS domains of different RGS proteins which in turn result in different interactions with the α-helical domains and switch III regions of Gα subunits (Figure 9) [82, 83]. The interaction between the RGS domain and the switch and α-helical regions of Gα is thought to promote the transition state for GTP hydrolysis within the nucleotide binding domain and thus facilitate deactivation of Gα [83].
Figure 9.

Crystal structure of the complex of Gαi1 and RGS4. (a) Cartoon representation of Gαi1 (orange) in complex with RGS4 (magenta). (b) Surface representation of the complex. (c) Cartoon diagram of RGS4.
Crystal structures of the complexes of Gα and the RGS domains have highlighted the structural basis for the interaction between RGS proteins and Gα [84]. While these structures locally and globally describe the interacting domains in the context of both proteins, the exact mechanism for how RGS proteins approach and regulate G-protein deactivation is a subject of speculation. RGS proteins have been proposed as attractive targets for drug development [85].
3. Signaling by G-Proteins
a). Well-defined G-protein signaling pathways
Accumulated data have established several well-defined signaling effectors for G-proteins (Table 1). Both Gαs and Gαi families of G-proteins could regulate adenylyl cyclases. Gαs stimulates adenylyl cyclase to convert ATP into cyclic AMP (cAMP) (Figure 10a) [86]. Elevated cAMP results in the activation of cAMP-regulated proteins, such as protein kinase A, cyclic nucleotide-gated channels, and EPAC (a GEF for Rap small GTP-binding proteins) [86]. Gαi proteins, on the other hand, can inhibit certain isotypes of adenylyl cyclases, leading to reduced intracellular cAMP levels. Gαq family G-proteins activate the β-isoforms of phospholipase C (PLC-β1-4), which cleaves phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] into inositol trisphosphate (IP3) and membrane-bound diacylglycerol (DAG) [87] (Figure 10b). IP3 then opens the calcium channel IP3 receptor on the ER membrane, and DAG activates protein kinase C. Within the Gα12/13 family, Gα13 (but not Gα12) is able to directly increase the activity of p115RhoGEF and related RhoGEF proteins [PDZ-RhoGEF and leukemia-associated RhoGEF (LARG)] by membrane recruitment and direct interaction [88]. However, one should be aware that other G-proteins, such as Gαq, could also increase the activity of other RhoGEFs, such as p63RhoGEF, linked to RhoA activation [89, 90] (Figure 10c). Activation of RhoA does not imply a Gα13-dependent signaling pathway. In endothelial cells, RhoA activation is normal in Gα13-knockout cells, but is impaired in Gαq-knockout cells [91]. For Gα12, several proteins were reported to interact with Gα12. These proteins include Btk-family tyrosine kinases, Gap1, rasGAP, cadherins, α-SNAP, and p120-caterin [92-95]. Similarly, several proteins (such as Btk-family tyrosine kinase, cadherins, radixin, Hax-1, and Integrin αiiibβ3) were reported to interact with Gα13 [93, 96-100] (Table 1).
Table 1. Gα subunit interacting proteins.
| Gα subunit | Well-defined G-protein effectors | Other G-protein interacting proteins |
|---|---|---|
| Gαs, Gαolf | Adenylate cyclase (+) | Tubulin, Calnuc, Src tyrosine kinase, axin |
| Gαo, Gαi1–3, Gαt1,2, Gαg, Gαz | Adenylate cyclase (-), cGMP phosphodiesterase (+) | Rap1Gapll, Calnuc, Src tyrosine kinase, nucleobindin 2 (NUCB2), Tubulin, Pins, Pcp1, LGN, GRIN1, Eya2, Pcp2 |
| Gαq, Gα11, Gα14, Gα15/16 | Phospholipase C-β (+), p63RhoGEF | GRK2, actin, tubulin, PI3K, TPR1, Btk tyrosine kinase, Phospholipase C-ε, TRPM8 |
| Gα12, Gα13 | p115RhoGEF, LARG and PDZ-RhoGEF | Gap1, rasGap, Btk tyrosine kinase, Radixin, Hax-1, Cadherins, α-SNAP, p120caterin, Integrin αlllbβ3 |
(+) indicates stimulation. (-) represents inhibition.
Figure 10.

Crystal structures of Gα in complex with different downstream effectors. (a) Cartoon representation of Gαs (orange) with adenylyl cyclase (AC) C1A (magenta) and C2A domains (green). (b) Representation of Gαq (orange) with phospholipase Cβ3(blue). (c) Representation of Gαq (orange) with p63RhoGEF (blue) and RhoA (green).
In addition to Gα proteins, Gβγ can also signal to downstream effectors. Gβγ can regulate adenylyl cyclases, phospholipase Cβ, inwardly rectifying K+ channel, and voltage-gated Ca2+ channels [101] (Table 2). Due to the relatively higher amounts of Gi families of G-proteins in cells, Gi activation is thought to be the primary source for Gβγ-mediated signaling processes.
Table 2. Gβγ subunit effectors.
| Well-defined Gβγ effectors | Other Gβγ interacting proteins |
|---|---|
| Adenylate cyclase (+), Phospholipase C-β (+), Phosphoinositide 3 Kinases, G protein-coupled receptor kinases, K+ and Ca2+ channels | Btk-family tyrosine kinase, IP3 receptors, Raf kinase, Protein kinase D, Histone deacetylase 5 (HDAC5), Tubulin, F-actin, Vinculin, ElmoE, Rab11, mitofusinl, Radil, activator protein 1, TFE3, TRPM1 |
b) Non-canonical G-protein signaling pathways
Other than the well-established downstream effectors, such as adenylyl cyclase and phospholipase Cβ, there are many proteins reported to interact with G-proteins (Tables 1 and 2). Although the physiological significances of these interactions are currently not well established, these interactions might underlie cell type-specific G-protein functions, and need to be further explored.
4. Physiological Functions of G-Proteins
a). Physiological functions of G-proteins as observed in gene-knockout mice
There are many physiological functions described for various G-protein signaling pathways. Here we focus on the physiological studies in G-protein gene knockout mice.
Gs and Gi pathways contribute to cardiac functions such as contractility [102]. Studies from null mutation of the Gαs maternal and paternal alleles suggest that Gαs gene is imprinted [103-105] (Table 3). Further studies showed that Gαs gene is imprinted in a tissue-specific matter, being primarily expressed from the maternal allele in renal proximal tubules, thyroid, pituitary and ovary.
Table 3. Physiological and Pathological functions of Gαs family.
| Physiological functions which could be explained by the well-studied cellular signaling pathways | Functions without well-established G-protein signaling pathways |
|---|---|
| Myocardial hypertrophy, increase in hydrolysis of triglyceride, decrease in amino acid uptake, increase in the conversion of glycogen to glucose, inhibition of synthesis of glycogen, increase in synthesis of estrogen and progesterone, increase in synthesis of aldosterone and Cortisol, increase in excitation/contraction and sympathetic cardiac activation and hypertrophy, iodide organification, secretion of thyroxine, thyroid cell mitogenesis, increase in reabsorption of calcium from bone, fluid secretion, inhibition of platelet aggregation and secretion | Mg2+ uptake, hematopoietic stem cell engraftment in bone marrow |
Each gene encoding for individual Gαi-proteins has been knocked out in mice, and these studies revealed that each knockout has a specific set of deficiencies and abnormalities [106] (Table 4). Heart tissues analyzed from Gαi-deficient mice suggest that deletion of one Gαi isoform might be accompanied by an increased protein expression for the non-targeted Gαi subunit in the heart [107]. Gαi2 and Gαi3 show isoform-specific roles in the regulation of macrophage migration and hepatic autophagy but exhibit redundant functions in other processes such as macrophage activation [108].
Table 4. Physiological and Pathological functions of Gαi family.
| Physiological functions which could be explained by the well-studied cellular signaling pathways | Functions without well-established G-protein signaling pathways |
|---|---|
| Vision, taste, vomeronasal function, Cardiac activation (contractility), regulation of cardiac L-type Ca2+ channels, leukaryote activation and migration, hepatic authophagy, developmental processes, lipid metabolism, regulation of immune cells, renal function, platelet activation, chemokine-induced lymphocyte migration | Transformation of fibroblast, spindle positioning during cell division, regulation of diacylglycerol kinase (DGK), neurotransmitter release in synapses, cell migration |
Mice lacking Gαq and Gα11 genes have multiple defects (Table 5). These include impaired motor coordination associated with cerebellar development, defective platelet activation, cardiac malformation and craniofacial defects associated with embryonic cardiomyocyte proliferation and craniofacial development, and hyperparathyroidism [86]. Overall, the Gq family of G-proteins collectively regulate a wide range of cell- and tissue-specific responses via coupling to PLC β-isoforms. Furthermore, Gαq family members can induce Rho-mediated responses including activation of RhoA in smooth muscle cells via p63RhoGEF, and acetylcholine vesicle release at neuromuscular junctions in C elegans as well as regulation of pharynx pumping, speed of locomotion and egg laying via TrioC (a Rho GEF) [109, 110].
Table 5. Physiological and Pathological functions of Gαq family.
| Physiological functions which could be explained by the well-studied cellular signaling pathways | Functions without well-established G-protein signaling pathways |
|---|---|
| Myocardial hypertrophy, smooth muscle tone, platelet activation, hormone release in anterior pituitary, synaptic transmission at Purkinje cell synapse | Insulin secretion by β-cells, leukocyte migration and activation, embryonic myocardial growth, neural crest development, transformation of fibroblast |
While Gα12 gene-deleted mice were normal, Gα13-/- mouse embryos died at E9.5 [111, 112] (Table 6). Gα13 is essential for blood vessel formation, and highly expressed in endothelial cells [113]. The Gα13-/- mouse embryos have defective vascular systems that show no blood vessels [111]. Therefore, deletion of Gα13 gene impaired the ability of endothelial cells to develop into an organized vascular system, resulting in intrauterine death. Endothelial cell-specific Gα13 knockout embryos also died at ∼E9.5, similar to the above mentioned global Gα13-/- mice [113]. Although it is clear that Gα13 is essential for blood vessel formation, the biochemical signaling mechanisms of Gα13 in endothelial cells are not completely understood. Furthermore, ablation of the Gα12 and Gα13 genes in the nervous system results in neuronal ectopia of the cerebral and cerebellar cortices, indicating that both G-proteins are required for proper positioning of migrating cortical plate neurons and Purkinje cells during development [114].
Table 6. Physiological and Pathological functions of Gα12 family.
| Physiological functions which could be explained by the well-studied cellular signaling pathways | Functions without well-established G-protein signaling pathways |
|---|---|
| Platelet activation, smooth muscle contraction, leukocyte migration, and neuronal axon guidance | Leukocyte activation and proliferation, lymphocyte development, embryonic development of blood vessels and angiogenesis, transformation of fibroblast, cancer cell invasion and metastasis |
b). Physiological functions without well-defined signaling pathways
There are many reports of physiological and pathological functions of G-proteins that can not be easily explained by the currently well-defined G-protein signaling effectors [115]. Here we will focus on two examples, one of which is the role of G-proteins in cell division, and the other is the role of G-proteins in mediating signals from receptor tyrosine kinases (RTKs).
(i) Role of G-proteins in cell division
Genetic studies in C. elegans and Drosophila demonstrate that G-protein subunits function in asymmetric cell division [75, 116-120]. In one-cell C. elegans embryo, the Par3/Par6/aPKC complex localizes to the anterior cortex, and a complex of Gα and GPR domain-containing protein localizes to the posterior cortex before the first cleavage, thus setting up a series of events that result in the generation of an anterior daughter cell that is larger than its posterior sister (Figure 11a). The difference in sibling size is the result of a shift in the cleavage plane toward the posterior pole, caused by pulling of the posterior spindle pole toward that direction. Depletion of two Gαi family members in C. elegans, GOA-1 and GPA-16, produces a loss of asymmetry characterized by equal but greatly decreased pulling forces on both anterior and posterior spindle poles [53, 75]. Gβγ signaling is also necessary for proper asymmetric cell division. Loss of GPB-1, one of the two C. elegans Gβ subunits, or GPC-2, one of the two C. elegans Gγ subunits, results in incorrect positioning of the mitotic spindle axes. This improper orientation of cell cleavage results in developmental defects in the embryo [75, 116]. However, the biochemical mechanisms of how G-proteins are regulated during this process are not clearly defined. Nevertheless, numerous studies provide a possible model for how G-proteins function to regulate microtubule pulling forces in cell division (Figure 11b). The key element that comprises this non-canonical pathway is that the GDP bound form of Gα, rather than the GTP bound form, is the important form in the force generation mechanism. Gαi- GDP binds directly to GPR-1/2 through the GRP-domain [118, 119]. At the same time, GPR-1/2 binds to the coiled-coiled protein LIN-5. Deletion of either GPR-1/2 or LIN-5 in C. elegans embryos leads to defects in asymmetry cell division (Figure 11b) [118]. Furthermore, the key microtubule motor protein dynein forms a complex with GPR-1/2 and LIN-5. A decrease or loss of function of dynein resulted in decreased pulling forces during asymmetric cell division [121]. Thus, the current model for how G-proteins function in asymmetric cell division in C. elegans involves plasma membrane Gα-GDP forming a complex with GPR1/2 and LIN-5 at the cell cortex which recruits dynein that generates the force necessary to control spindle pole positioning (Figure 11b).
Figure 11.

An example of the non-canonical roles of G-protein signaling. In the control of embryonic spindle positioning in C. eiegans fertilized eggs, the Par3/Par6/aPKC complex is localized at the anterior while the Gα and GPR complex is in the posterior (a). The Gα/GPR complex is linked to the spindle through LIN-5 and dynein proteins (b).
This G-protein signaling does not require GPCR involvement, but the nucleotide bound states of Gα are shown to be regulated by other regulator proteins such as RGS-7 and Ric-8. Loss of RGS-7 resulted in increased force at the anterior spindle pole [52], while loss of Ric-8 leads to decreased pulling forces on both anterior and posterior spindle poles [53-56, 66, 121-123]. Hence, Gα needs to cycle between GDP/GTP bound states to carry out its function in asymmetric cell division.
(ii) Roles of G-proteins in mediating signals from receptor tyrosine kinases (RTKs)
Another example of non-canonical functions for G-proteins is their roles in RTK signaling. There are many reports that G-proteins could function downstream of RTKs [77, 124-136]. However, the mechanisms are not well defined. It is not clear whether GPCRs are involved in all these pathways. In some cases, trans-activation of GPCRs by RTKs has been proposed. These areas deserve more biochemical and genetic investigations. Here we will use the role of Gα13 in RTK-induced actin cytoskeletal reorganization as one example.
The earliest ultra-structural changes of cells treated with growth factors are the intensive bursts of ruffling of the dorsal surface plasma membranes as seen under the phase-contrast microscope [137-139]. The physiological functions of dorsal ruffles, including macropinocytosis, cell migration and invasion, are continually expanding [140-143]. It has been suggested that one major function of dorsal ruffles is to reorganize the actin cytoskeleton to prepare a static cell for motility [144]. In serum-starved fibroblasts, platelet-derived growth factor (PDGF) induces at least two types of membrane ruffles: peripheral membrane ruffles (or lamellipodia) and dorsal ruffles [145]. In addition to fibroblast cells, dorsal ruffle formation in response to growth factors has been reported in glial cells, endothelial cells, hippocampal neurons, kidney epithelial cells and tumor cells [137, 138, 144, 146, 147]. Dorsal ruffles are dynamic structures, and they form and disassemble rapidly [148, 149]. Intensive investigations have revealed several critical regulators of the dorsal ruffle formation in response to growth factors such as Rac, PAK1 and WAVE1 [145].
Gα13 is essential for RTK-induced cell migration and actin cytoskeletal reorganization in fibroblasts and endothelial cells [150]. Gα13 has been shown to control actin cytoskeletal reorganization by regulating the disassembly of dorsal ruffles [151]. In the absence of Gα13, the activity of Rac stays much longer than in normal cells, indicating Gα13 controls the duration of Rac activation [151]. Re-expression of Gα13 shortens the duration of Rac activity in cells [151]. Furthermore, Ric-8A is critical in relaying RTK signals to Gα13 [152]. Down-regulation of Ric-8A in cells impaired PDGF-induced dorsal ruffle turnover and decreased PDGF-initiated cell migration. Deficiency of Ric-8A impaired the translocation of Gα13 to the cell cortex. Therefore, Ric-8A is involved in the PDGFR-Gα13 pathway and possibly functions as the GEF for GPCR-independent activation of Gα13 in this PDGF-initiated pathway. Furthermore, atypical protein kinase C (aPKC) could phosphorylate Ric-8A after PDGF stimulation [153]. aPKC exists in cells as a complex of PAR3/PAR6/aPKC [154]. This complex regulates cell polarity, cell division and cell migration. The main mechanism by which this PAR3/PAR6/aPKC complex regulates cell function is to modulate the subcellular localization of downstream proteins. Unphosphorylated Ric-8A is observed mainly in the nucleus whereas phosphorylated Ric-8A is observed mainly in the cytoplasm [153]. Hence, RTKs likely signal through aPKC to regulate the subcellular localization of Ric-8A, which in turn controls the subcellular translocation of Gα13. Further investigations of the molecular mechanisms by which G-proteins are regulated by RTKs will help us understand the physiological functions of G-proteins in general.
5. Perspectives
In this review, we have briefly summarized the regulation, signaling and physiological functions of heterotrimeric G-proteins. We reviewed the regulation of G-proteins by its main activator (the GPCRs) and by some non-GPCR activators (such as GPR proteins and Ric-8A) as well as its negative regulation by RGS proteins. We reviewed the well-studied cellular signaling pathways controlled by G-proteins. In addition, we mentioned additional G-protein interacting proteins although their physiological importance is not clear or well established at the present time. We have also described some well-known physiological functions of G-proteins, and pointed out that there are many more physiological and pathological functions of G-proteins that we don't yet understand.
We still have a long way to go to fully understand how GPCRs and non-GPCR activators regulate G-proteins. In addition to more crystal structures of different GPCRs with various G-proteins, and of non-GPCR activators and G-proteins, to gain insights on the specific coupling between the activators and G-proteins, we also need to investigate the dynamic process of these activation processes since the crystal structures represent snapshots of particular states. Recent uses of cryo-EM structural studies of different states of receptor ion channels illustrate one example of the types of studies that could be utilized to investigate the activation processes of G-proteins, in addition to computational simulation studies.
There are many hints that G-proteins might have other signaling pathways (in addition to the well-known targets and pathways) which could expand the physiological functions of G-proteins. These additional signaling pathways and physiological functions need to be rigorously investigated with open minds.
Highlights.
Heterotrimeric G-proteins can be regulated by G-protein-coupled receptors (GPCRs), and by non-receptor proteins such as Ric-8, GRP and RGS proteins.
Many proteins interacting with G-proteins have been identified.
G-proteins regulate various physiological functions.
Combinations of structural and computational biological tools can be used to understand the mechanisms of activation of G-proteins by GPCRs and non-GPCR regulators. Multidisciplinary studies are needed to systematically investigate the biochemical mechanisms, cellular effects and physiological functions in processes involving apparently non-canonical G-protein signaling.
Acknowledgments
Work in the Huang lab is supported by grants R01CA193815 and R01HL091525 from the NIH, and NPRP 7-709-3-195 from the Qatar National Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–63. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 4.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–32. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 5.Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802–8. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 6.Sprang SR, Chen Z, Du X. Structural basis of effector regulation and signal termination in heterotrimeric Galpha proteins. Adv Protein Chem. 2007;74:1–65. doi: 10.1016/S0065-3233(07)74001-9. [DOI] [PubMed] [Google Scholar]
- 7.Noel JP, Hamm HE, Sigler PB. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 1993;366:654–63. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 8.Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science. 1994;265:1405–12. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 9.Iiri T, Farfel Z, Bourne HR. G-protein diseases furnish a model for the turn-on switch. Nature. 1998;394:35–8. doi: 10.1038/27831. [DOI] [PubMed] [Google Scholar]
- 10.Cherfils J, Chabre M. Activation of G-protein Galpha subunits by receptors through Galpha-Gbeta and Galpha-Ggamma interactions. Trends Biochem Sci. 2003;28:13–7. doi: 10.1016/s0968-0004(02)00006-3. [DOI] [PubMed] [Google Scholar]
- 11.Tesmer JJ. The quest to understand heterotrimeric G protein signaling. Nat Struct Mol Biol. 2010;17:650–2. doi: 10.1038/nsmb0610-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe HW, Park JH, Kim YJ, Ernst OP. Transmembrane signaling by GPCRs: insight from rhodopsin and opsin structures. Neuropharmacology. 2011;60:52–7. doi: 10.1016/j.neuropharm.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–56. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Chen S, Zhang JJ, Huang XY. Crystal structure of oligomeric beta1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol. 2013;20:419–25. doi: 10.1038/nsmb.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–55. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramachandran S, Cerione RA. How GPCRs hit the switch. Nat Struct Mol Biol. 2006;13:756–7. doi: 10.1038/nsmb0906-756. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz TW, Sakmar TP. Structural biology: snapshot of a signalling complex. Nature. 2011;477:540–1. doi: 10.1038/477540a. [DOI] [PubMed] [Google Scholar]
- 18.Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 19.Lappano R, Maggiolini M. GPCRs and cancer. Acta Pharmacol Sin. 2012;33:351–62. doi: 10.1038/aps.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem. 1997;66:639–78. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- 21.Wolf YI, Brenner SE, Bash PA, Koonin EV. Distribution of protein folds in the three superkingdoms of life. Genome Res. 1999;9:17–26. [PubMed] [Google Scholar]
- 22.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 23.Rajagopal K, Lefkowitz RJ, Rockman HA. When 7 transmembrane receptors are not G protein-coupled receptors. The Journal of clinical investigation. 2005;115:2971–4. doi: 10.1172/JCI26950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szczepek M, Beyriere F, Hofmann KP, Elgeti M, Kazmin R, Rose A, et al. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wess J. G-protein-coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J. 1997;11:346–54. [PubMed] [Google Scholar]
- 26.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–94. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 27.Strange PG. Signaling mechanisms of GPCR ligands. Curr Opin Drug Discov Devel. 2008;11:196–202. [PubMed] [Google Scholar]
- 28.Neubig RR, Gantzos RD, Thomsen WJ. Mechanism of agonist and antagonist binding to alpha 2 adrenergic receptors: evidence for a precoupled receptor-guanine nucleotide protein complex. Biochemistry. 1988;27:2374–84. doi: 10.1021/bi00407a019. [DOI] [PubMed] [Google Scholar]
- 29.Huang J, Sun Y, Zhang JJ, Huang XY. Pivotal role of extended linker 2 in the activation of Galpha by G protein-coupled receptor. J Biol Chem. 2015;290:272–83. doi: 10.1074/jbc.M114.608661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–86. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- 31.Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–35. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 32.Filizola M, Weinstein H. The study of G-protein coupled receptor oligomerization with computational modeling and bioinformatics. FEBS J. 2005;272:2926–38. doi: 10.1111/j.1742-4658.2005.04730.x. [DOI] [PubMed] [Google Scholar]
- 33.Milligan G. The role of dimerisation in the cellular trafficking of G-protein-coupled receptors. Curr Opin Pharmacol. 2010;10:23–9. doi: 10.1016/j.coph.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Palczewski K. Oligomeric forms of G protein-coupled receptors (GPCRs) Trends Biochem Sci. 2010;35:595–600. doi: 10.1016/j.tibs.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lohse MJ. Dimerization in GPCR mobility and signaling. Curr Opin Pharmacol. 2010;10:53–8. doi: 10.1016/j.coph.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, et al. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103:16123–8. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–7. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 38.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 39.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–32. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–6. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–71. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mancia F, Assur Z, Herman AG, Siegel R, Hendrickson WA. Ligand sensitivity in dimeric associations of the serotonin 5HT2c receptor. EMBO Rep. 2008;9:363–9. doi: 10.1038/embor.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, et al. Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 2008;27:2293–304. doi: 10.1038/emboj.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo W, Shi L, Filizola M, Weinstein H, Javitch JA. Crosstalk in G protein-coupled receptors: changes at the transmembrane homodimer interface determine activation. Proc Natl Acad Sci U S A. 2005;102:17495–500. doi: 10.1073/pnas.0508950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato M, Ribas C, Hildebrandt JD, Lanier SM. Characterization of a G-protein activator in the neuroblastoma-glioma cell hybrid NG108-15. J Biol Chem. 1996;271:30052–60. doi: 10.1074/jbc.271.47.30052. [DOI] [PubMed] [Google Scholar]
- 46.Takesono A, Cismowski MJ, Ribas C, Bernard M, Chung P, Hazard S, 3rd, et al. Receptor-independent activators of heterotrimeric G-protein signaling pathways. J Biol Chem. 1999;274:33202–5. doi: 10.1074/jbc.274.47.33202. [DOI] [PubMed] [Google Scholar]
- 47.Cismowski MJ, Ma C, Ribas C, Xie X, Spruyt M, Lizano JS, et al. Activation of heterotrimeric G-protein signaling by a ras-related protein. Implications for signal integration. J Biol Chem. 2000;275:23421–4. doi: 10.1074/jbc.C000322200. [DOI] [PubMed] [Google Scholar]
- 48.Bernard ML, Peterson YK, Chung P, Jourdan J, Lanier SM. Selective interaction of AGS3 with G-proteins and the influence of AGS3 on the activation state of G-proteins. J Biol Chem. 2001;276:1585–93. doi: 10.1074/jbc.M005291200. [DOI] [PubMed] [Google Scholar]
- 49.Kroslak T, Koch T, Kahl E, Hollt V. Human phosphatidylethanolamine-binding protein facilitates heterotrimeric G protein-dependent signaling. J Biol Chem. 2001;276:39772–8. doi: 10.1074/jbc.M106991200. [DOI] [PubMed] [Google Scholar]
- 50.Miller KG, Emerson MD, McManus JR, Rand JB. RIC-8 (Synembryn): a novel conserved protein that is required for G(q)alpha signaling in the C. elegans nervous system. Neuron. 2000;27:289–99. doi: 10.1016/s0896-6273(00)00037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller KG, Rand JB. A role for RIC-8 (Synembryn) and GOA-1 (G(o)alpha) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics. 2000;156:1649–60. doi: 10.1093/genetics/156.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hess HA, Roper JC, Grill SW, Koelle MR. RGS-7 completes a receptor-independent heterotrimeric G protein cycle to asymmetrically regulate mitotic spindle positioning in C. elegans. Cell. 2004;119:209–18. doi: 10.1016/j.cell.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 53.Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, et al. RIC-8 is required for GPR-1/2-dependent Galpha function during asymmetric division of C. elegans embryos. Cell. 2004;119:219–30. doi: 10.1016/j.cell.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Ng KH, Qian H, Siderovski DP, Chia W, Yu F. Ric-8 controls Drosophila neural progenitor asymmetric division by regulating heterotrimeric G proteins. Nat Cell Biol. 2005;7:1091–8. doi: 10.1038/ncb1317. [DOI] [PubMed] [Google Scholar]
- 55.Hampoelz B, Hoeller O, Bowman SK, Dunican D, Knoblich JA. Drosophila Ric-8 is essential for plasma-membrane localization of heterotrimeric G proteins. Nat Cell Biol. 2005;7:1099–105. doi: 10.1038/ncb1318. [DOI] [PubMed] [Google Scholar]
- 56.David NB, Martin CA, Segalen M, Rosenfeld F, Schweisguth F, Bellaiche Y. Drosophila Ric-8 regulates Galphai cortical localization to promote Galphai-dependent planar orientation of the mitotic spindle during asymmetric cell division. Nat Cell Biol. 2005;7:1083–90. doi: 10.1038/ncb1319. [DOI] [PubMed] [Google Scholar]
- 57.Parks S, Wieschaus E. The Drosophila gastrulation gene concertina encodes a G alpha-like protein. Cell. 1991;64:447–58. doi: 10.1016/0092-8674(91)90652-f. [DOI] [PubMed] [Google Scholar]
- 58.Tall GG, Krumins AM, Gilman AG. Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. J Biol Chem. 2003;278:8356–62. doi: 10.1074/jbc.M211862200. [DOI] [PubMed] [Google Scholar]
- 59.Tall GG, Gilman AG. Purification and functional analysis of Ric-8A: a guanine nucleotide exchange factor for G-protein alpha subunits. Methods in enzymology. 2004;390:377–88. doi: 10.1016/S0076-6879(04)90023-7. [DOI] [PubMed] [Google Scholar]
- 60.Von Dannecker LE, Mercadante AF, Malnic B. Ric-8B, an olfactory putative GTP exchange factor, amplifies signal transduction through the olfactory-specific G-protein Galphaolf. J Neurosci. 2005;25:3793–800. doi: 10.1523/JNEUROSCI.4595-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tonissoo T, Koks S, Meier R, Raud S, Plaas M, Vasar E, et al. Heterozygous mice with Ric-8 mutation exhibit impaired spatial memory and decreased anxiety. Behav Brain Res. 2006;167:42–8. doi: 10.1016/j.bbr.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 62.Van Eps N, Thomas CJ, Hubbell WL, Sprang SR. The guanine nucleotide exchange factor Ric-8A induces domain separation and Ras domain plasticity in Galphai1. Proc Natl Acad Sci U S A. 2015;112:1404–9. doi: 10.1073/pnas.1423878112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan P, Gabay M, Wright FA, Tall GG. Ric-8B is a GTP-dependent G protein alphas guanine nucleotide exchange factor. J Biol Chem. 2011;286:19932–42. doi: 10.1074/jbc.M110.163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gabay M, Pinter ME, Wright FA, Chan P, Murphy AJ, Valenzuela DM, et al. Ric-8 proteins are molecular chaperones that direct nascent G protein alpha subunit membrane association. Sci Signal. 2011;4:ra79. doi: 10.1126/scisignal.2002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan P, Thomas CJ, Sprang SR, Tall GG. Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein alpha subunits. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1220943110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Afshar K, Willard FS, Colombo K, Siderovski DP, Gonczy P. Cortical localization of the Galpha protein GPA-16 requires RIC-8 function during C. elegans asymmetric cell division. Development. 2005;132:4449–59. doi: 10.1242/dev.02039. [DOI] [PubMed] [Google Scholar]
- 67.Mochizuki N, Cho G, Wen B, Insel PA. Identification and cDNA cloning of a novel human mosaic protein, LGN, based on interaction with G alpha i2. Gene. 1996;181:39–43. doi: 10.1016/s0378-1119(96)00456-8. [DOI] [PubMed] [Google Scholar]
- 68.Kimple RJ, Kimple ME, Betts L, Sondek J, Siderovski DP. Structural determinants for GoLoco-induced inhibition of nucleotide release by Galpha subunits. Nature. 2002;416:878–81. doi: 10.1038/416878a. [DOI] [PubMed] [Google Scholar]
- 69.Natochin M, Lester B, Peterson YK, Bernard ML, Lanier SM, Artemyev NO. AGS3 inhibits GDP dissociation from galpha subunits of the Gi family and rhodopsin-dependent activation of transducin. J Biol Chem. 2000;275:40981–5. doi: 10.1074/jbc.M006478200. [DOI] [PubMed] [Google Scholar]
- 70.Tall GG, Gilman AG. Resistance to inhibitors of cholinesterase 8A catalyzes release of Galphai-GTP and nuclear mitotic apparatus protein (NuMA) from NuMA/LGN/Galphai-GDP complexes. Proc Natl Acad Sci U S A. 2005;102:16584–9. doi: 10.1073/pnas.0508306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas CJ, Tall GG, Adhikari A, Sprang SR. Ric-8A catalyzes guanine nucleotide exchange on G alphai1 bound to the GPR/GoLoco exchange inhibitor AGS3. J Biol Chem. 2008;283:23150–60. doi: 10.1074/jbc.M802422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cismowski MJ, Takesono A, Ma C, Lizano JS, Xie X, Fuernkranz H, et al. Genetic screens in yeast to identify mammalian nonreceptor modulators of G-protein signaling. Nat Biotechnol. 1999;17:878–83. doi: 10.1038/12867. [DOI] [PubMed] [Google Scholar]
- 73.Yu F, Morin X, Cai Y, Yang X, Chia W. Analysis of partner of inscuteable, a novel player of Drosophila asymmetric divisions, reveals two distinct steps in inscuteable apical localization. Cell. 2000;100:399–409. doi: 10.1016/s0092-8674(00)80676-5. [DOI] [PubMed] [Google Scholar]
- 74.Schaefer M, Petronczki M, Dorner D, Forte M, Knoblich JA. Heterotrimeric G proteins direct two modes of asymmetric cell division in the Drosophila nervous system. Cell. 2001;107:183–94. doi: 10.1016/s0092-8674(01)00521-9. [DOI] [PubMed] [Google Scholar]
- 75.Gotta M, Ahringer J. Distinct roles for Galpha and Gbetagamma in regulating spindle position and orientation in Caenorhabditis elegans embryos. Nat Cell Biol. 2001;3:297–300. doi: 10.1038/35060092. [DOI] [PubMed] [Google Scholar]
- 76.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc Natl Acad Sci U S A. 2009;106:3178–83. doi: 10.1073/pnas.0900294106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV/Girdin transmits signals from multiple receptors by triggering trimeric G protein activation. J Biol Chem. 2015;290:6697–704. doi: 10.1074/jbc.R114.613414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le-Niculescu H, Niesman I, Fischer T, DeVries L, Farquhar MG. Identification and characterization of GIV, a novel Galpha i/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J Biol Chem. 2005;280:22012–20. doi: 10.1074/jbc.M501833200. [DOI] [PubMed] [Google Scholar]
- 79.Ghosh P, Garcia-Marcos M, Farquhar MG. GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh Migr. 2011;5:237–48. doi: 10.4161/cam.5.3.15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barbazan J, Dunkel Y, Li H, Nitsche U, Janssen KP, Messer K, et al. Prognostic Impact of Modulators of G proteins in Circulating Tumor Cells from Patients with Metastatic Colorectal Cancer. Sci Rep. 2016;6:22112. doi: 10.1038/srep22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature. 1996;383:172–5. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 82.Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, et al. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc Natl Acad Sci U S A. 2008;105:6457–62. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–61. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 84.Baltoumas FA, Theodoropoulou MC, Hamodrakas SJ. Interactions of the alpha-subunits of heterotrimeric G-proteins with GPCRs, effectors and RGS proteins: a critical review and analysis of interacting surfaces, conformational shifts, structural diversity and electrostatic potentials. J Struct Biol. 2013;182:209–18. doi: 10.1016/j.jsb.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Sjogren B. Regulator of G protein signaling proteins as drug targets: current state and future possibilities. Adv Pharmacol. 2011;62:315–47. doi: 10.1016/B978-0-12-385952-5.00002-6. [DOI] [PubMed] [Google Scholar]
- 86.Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 87.Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272:15045–8. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- 88.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–11. doi: 10.1126/science.280.5372.2109. see comments. [DOI] [PubMed] [Google Scholar]
- 89.Lowry WE, Huang J, Ma YC, Ali S, Wang D, Williams DM, et al. Csk, a critical link of g protein signals to actin cytoskeletal reorganization. Developmental cell. 2002;2:733–44. doi: 10.1016/s1534-5807(02)00175-2. [DOI] [PubMed] [Google Scholar]
- 90.Lutz S, Freichel-Blomquist A, Yang Y, Rumenapp U, Jakobs KH, Schmidt M, et al. The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem. 2005;280:11134–9. doi: 10.1074/jbc.M411322200. [DOI] [PubMed] [Google Scholar]
- 91.Worzfeld T, Wettschureck N, Offermanns S. G(12)/G(13)-mediated signalling in mammalian physiology and disease. Trends in pharmacological sciences. 2008;29:582–9. doi: 10.1016/j.tips.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 92.Jiang Y, Ma W, Wan Y, Kozasa T, Hattori S, Huang XY. The G protein G alpha12 stimulates Bruton's tyrosine kinase and a rasGAP through a conserved PH/BM domain. Nature. 1998;395:808–13. doi: 10.1038/27454. [DOI] [PubMed] [Google Scholar]
- 93.Meigs TE, Fields TA, McKee DD, Casey PJ. Interaction of Galpha 12 and Galpha 13 with the cytoplasmic domain of cadherin provides a mechanism for beta -catenin release. Proc Natl Acad Sci U S A. 2001;98:519–24. doi: 10.1073/pnas.021350998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dhanasekaran N, Dermott JM. Signaling by the G12 class of G proteins. Cell Signal. 1996;8:235–45. doi: 10.1016/0898-6568(96)00048-4. [DOI] [PubMed] [Google Scholar]
- 95.Suzuki N, Nakamura S, Mano H, Kozasa T. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci U S A. 2003;100:733–8. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cvejic S, Jiang Y, Huang X. Signaling of G(alpha)(12) family of G proteins through a tyrosine kinase and a Ras-GAP. Trends in cardiovascular medicine. 2000;10:160–5. doi: 10.1016/s1050-1738(00)00054-2. [DOI] [PubMed] [Google Scholar]
- 97.Huang W, Morales JL, Gazivoda VP, Lai J, Qi Q, August A. The zinc-binding region of IL-2 inducible T cell kinase (Itk) is required for interaction with Galpha13 and activation of serum response factor. The international journal of biochemistry & cell biology. 2013;45:1074–82. doi: 10.1016/j.biocel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vaiskunaite R, Adarichev V, Furthmayr H, Kozasa T, Gudkov A, Voyno-Yasenetskaya TA. Conformational activation of radixin by G13 protein alpha subunit. J Biol Chem. 2000;275:26206–12. doi: 10.1074/jbc.M001863200. [DOI] [PubMed] [Google Scholar]
- 99.Radhika V, Onesime D, Ha JH, Dhanasekaran N. Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem. 2004;279:49406–13. doi: 10.1074/jbc.M408836200. [DOI] [PubMed] [Google Scholar]
- 100.Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, et al. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327:340–3. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure JP, Labbe JC, et al. The expanding roles of Gbetagamma subunits in G protein-coupled receptor signaling and drug action. Pharmacol Rev. 2013;65:545–77. doi: 10.1124/pr.111.005603. [DOI] [PubMed] [Google Scholar]
- 102.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 103.Yu S, Gavrilova O, Chen H, Lee R, Liu J, Pacak K, et al. Paternal versus maternal transmission of a stimulatory G-protein alpha subunit knockout produces opposite effects on energy metabolism. The Journal of clinical investigation. 2000;105:615–23. doi: 10.1172/JCI8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu S, Yu D, Lee E, Eckhaus M, Lee R, Corria Z, et al. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein alpha-subunit (Gsalpha) knockout mice is due to tissue-specific imprinting of the gsalpha gene. Proc Natl Acad Sci U S A. 1998;95:8715–20. doi: 10.1073/pnas.95.15.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu S, Castle A, Chen M, Lee R, Takeda K, Weinstein LS. Increased insulin sensitivity in Gsalpha knockout mice. J Biol Chem. 2001;276:19994–8. doi: 10.1074/jbc.M010313200. [DOI] [PubMed] [Google Scholar]
- 106.Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L. Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci U S A. 2001;98:3577–82. doi: 10.1073/pnas.051632598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rudolph U, Spicher K, Birnbaumer L. Adenylyl cyclase inhibition and altered G protein subunit expression and ADP-ribosylation patterns in tissues and cells from Gi2 alpha-/- mice. Proc Natl Acad Sci U S A. 1996;93:3209–14. doi: 10.1073/pnas.93.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gohla A, Klement K, Piekorz RP, Pexa K, vom Dahl S, Spicher K, et al. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci U S A. 2007;104:3003–8. doi: 10.1073/pnas.0611434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Williams SL, Lutz S, Charlie NK, Vettel C, Ailion M, Coco C, et al. Trio's Rho-specific GEF domain is the missing Galpha q effector in C. elegans. Genes & development. 2007;21:2731–46. doi: 10.1101/gad.1592007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Steven R, Zhang L, Culotti J, Pawson T. The UNC-73/Trio RhoGEF-2 domain is required in separate isoforms for the regulation of pharynx pumping and normal neurotransmission in C. elegans. Genes & development. 2005;19:2016–29. doi: 10.1101/gad.1319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Offermanns S, Mancino V, Revel JP, Simon MI. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science. 1997;275:533–6. doi: 10.1126/science.275.5299.533. [DOI] [PubMed] [Google Scholar]
- 112.Gu JL, Muller S, Mancino V, Offermanns S, Simon MI. Interaction of G alpha(12) with G alpha(13) and G alpha(q) signaling pathways. Proc Natl Acad Sci U S A. 2002;99:9352–7. doi: 10.1073/pnas.102291599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ruppel KM, Willison D, Kataoka H, Wang A, Zheng YW, Cornelissen I, et al. Essential role for Galpha13 in endothelial cells during embryonic development. Proc Natl Acad Sci U S A. 2005;102:8281–6. doi: 10.1073/pnas.0503326102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moers A, Nieswandt B, Massberg S, Wettschureck N, Gruner S, Konrad I, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nature medicine. 2003;9:1418–22. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- 115.Manning DR. Evidence mounts for receptor-independent activation of heterotrimeric G proteins normally in vivo: positioning of the mitotic spindle in C. elegans. Sci STKE. 2003;2003:pe35. doi: 10.1126/stke.2003.196.pe35. [DOI] [PubMed] [Google Scholar]
- 116.Zwaal RR, Ahringer J, van Luenen HG, Rushforth A, Anderson P, Plasterk RH. G proteins are required for spatial orientation of early cell cleavages in C. elegans embryos. Cell. 1996;86:619–29. doi: 10.1016/s0092-8674(00)80135-x. [DOI] [PubMed] [Google Scholar]
- 117.Srinivasan DG, Fisk RM, Xu H, van den Heuvel S. A complex of LIN-5 and GPR proteins regulates G protein signaling and spindle function in C elegans. Genes & development. 2003;17:1225–39. doi: 10.1101/gad.1081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Colombo K, Grill SW, Kimple RJ, Willard FS, Siderovski DP, Gonczy P. Translation of polarity cues into asymmetric spindle positioning in Caenorhabditis elegans embryos. Science. 2003;300:1957–61. doi: 10.1126/science.1084146. [DOI] [PubMed] [Google Scholar]
- 119.Gotta M, Dong Y, Peterson YK, Lanier SM, Ahringer J. Asymmetrically distributed C. elegans homologs of AGS3/PINS control spindle position in the early embryo. Curr Biol. 2003;13:1029–37. doi: 10.1016/s0960-9822(03)00371-3. [DOI] [PubMed] [Google Scholar]
- 120.Siller KH, Doe CQ. Spindle orientation during asymmetric cell division. Nat Cell Biol. 2009;11:365–74. doi: 10.1038/ncb0409-365. [DOI] [PubMed] [Google Scholar]
- 121.Couwenbergs C, Labbe JC, Goulding M, Marty T, Bowerman B, Gotta M. Heterotrimeric G protein signaling functions with dynein to promote spindle positioning in C. elegans. J Cell Biol. 2007;179:15–22. doi: 10.1083/jcb.200707085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Couwenbergs C, Spilker AC, Gotta M. Control of embryonic spindle positioning and Galpha activity by C. elegans RIC-8. Curr Biol. 2004;14:1871–6. doi: 10.1016/j.cub.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 123.Woodard GE, Huang NN, Cho H, Miki T, Tall GG, Kehrl JH. Ric-8A and Gi alpha recruit LGN, NuMA, and dynein to the cell cortex to help orient the mitotic spindle. Mol Cell Biol. 2010;30:3519–30. doi: 10.1128/MCB.00394-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Waters C, Sambi B, Kong KC, Thompson D, Pitson SM, Pyne S, et al. Sphingosine 1-phosphate and platelet-derived growth factor (PDGF) act via PDGF beta receptor-sphingosine 1-phosphate receptor complexes in airway smooth muscle cells. J Biol Chem. 2003;278:6282–90. doi: 10.1074/jbc.M208560200. [DOI] [PubMed] [Google Scholar]
- 125.Waters C, Pyne S, Pyne NJ. The role of G-protein coupled receptors and associated proteins in receptor tyrosine kinase signal transduction. Semin Cell Dev Biol. 2004;15:309–23. doi: 10.1016/j.semcdb.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 126.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–60. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 127.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–8. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 128.Moxham CM, Malbon CC. Insulin action impaired by deficiency of the G-protein subunit G ialpha2. Nature. 1996;379:840–4. doi: 10.1038/379840a0. [DOI] [PubMed] [Google Scholar]
- 129.Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, et al. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science. 2001;291:1800–3. doi: 10.1126/science.1057559. [DOI] [PubMed] [Google Scholar]
- 130.Kluk MJ, Colmont C, Wu MT, Hla T. Platelet-derived growth factor (PDGF)-induced chemotaxis does not require the G protein-coupled receptor S1P1 in murine embryonic fibroblasts and vascular smooth muscle cells. FEBS Lett. 2003;533:25–8. doi: 10.1016/s0014-5793(02)03742-0. [DOI] [PubMed] [Google Scholar]
- 131.Alderton F, Rakhit S, Kong KC, Palmer T, Sambi B, Pyne S, et al. Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J Biol Chem. 2001;276:28578–85. doi: 10.1074/jbc.M102771200. [DOI] [PubMed] [Google Scholar]
- 132.Sun H, Chen Z, Poppleton H, Scholich K, Mullenix J, Weipz GJ, et al. The juxtamembrane, cytosolic region of the epidermal growth factor receptor is involved in association with alpha-subunit of Gs. J Biol Chem. 1997;272:5413–20. [PubMed] [Google Scholar]
- 133.Pyne NJ, Pyne S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: out of the shadow? Trends in pharmacological sciences. 2011;32:443–50. doi: 10.1016/j.tips.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 134.Alberelli MA, De Candia E. Functional role of protease activated receptors in vascular biology. Vascular pharmacology. 2014;62:72–81. doi: 10.1016/j.vph.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 135.Marty C, Ye RD. Heterotrimeric G protein signaling outside the realm of seven transmembrane domain receptors. Mol Pharmacol. 2010;78:12–8. doi: 10.1124/mol.110.063453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aznar N, Kalogriopoulos N, Midde KK, Ghosh P. Heterotrimeric G protein signaling via GIV/Girdin: Breaking the rules of engagement, space, and time. Bioessays. 2016;38:379–93. doi: 10.1002/bies.201500133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mellstroom K, Hoglund AS, Nister M, Heldin CH, Westermark B, Lindberg U. The effect of platelet-derived growth factor on morphology and motility of human glial cells. J Muscle Res Cell Motil. 1983;4:589–609. doi: 10.1007/BF00712117. [DOI] [PubMed] [Google Scholar]
- 138.Schliwa M, Nakamura T, Porter KR, Euteneuer U. A tumor promoter induces rapid and coordinated reorganization of actin and vinculin in cultured cells. J Cell Biol. 1984;99:1045–59. doi: 10.1083/jcb.99.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mellstrom K, Heldin CH, Westermark B. Induction of circular membrane ruffling on human fibroblasts by platelet-derived growth factor. Exp Cell Res. 1988;177:347–59. doi: 10.1016/0014-4827(88)90468-5. [DOI] [PubMed] [Google Scholar]
- 140.Warn R, Brown D, Dowrick P, Prescott A, Warn A. Cytoskeletal changes associated with cell motility. Symp Soc Exp Biol. 1993;47:325–38. [PubMed] [Google Scholar]
- 141.Dowrick P, Kenworthy P, McCann B, Warn R. Circular ruffle formation and closure lead to macropinocytosis in hepatocyte growth factor/scatter factor-treated cells. European journal of cell biology. 1993;61:44–53. [PubMed] [Google Scholar]
- 142.Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Developmental cell. 2003;5:595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- 143.Bretscher MS. Getting membrane flow and the cytoskeleton to cooperate in moving cells. Cell. 1996;87:601–6. doi: 10.1016/s0092-8674(00)81380-x. [DOI] [PubMed] [Google Scholar]
- 144.Krueger EW, Orth JD, Cao H, McNiven MA. A dynamin-cortactin-Arp2/3 complex mediates actin reorganization in growth factor-stimulated cells. Mol Biol Cell. 2003;14:1085–96. doi: 10.1091/mbc.E02-08-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–57. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 146.Blume-Jensen P, Claesson-Welsh L, Siegbahn A, Zsebo KM, Westermark B, Heldin CH. Activation of the human c-kit product by ligand-induced dimerization mediates circular actin reorganization and chemotaxis. Embo J. 1991;10:4121–8. doi: 10.1002/j.1460-2075.1991.tb04989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ruthel G, Banker G. Role of moving growth cone-like “wave” structures in the outgrowth of cultured hippocampal axons and dendrites. J Neurobiol. 1999;39:97–106. doi: 10.1002/(sici)1097-4695(199904)39:1<97::aid-neu8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 148.Hedberg KM, Bengtsson T, Safiejko-Mroczka B, Bell PB, Lindroth M. PDGF and neomycin induce similar changes in the actin cytoskeleton in human fibroblasts. Cell Motil Cytoskeleton. 1993;24:139–49. doi: 10.1002/cm.970240207. [DOI] [PubMed] [Google Scholar]
- 149.Dharmawardhane S, Sanders LC, Martin SS, Daniels RH, Bokoch GM. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J Cell Biol. 1997;138:1265–78. doi: 10.1083/jcb.138.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shan D, Chen L, Wang D, Tan YC, Gu JL, Huang XY. The g protein galpha(13) is required for growth factor-induced cell migration. Developmental cell. 2006;10:707–18. doi: 10.1016/j.devcel.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 151.Wang D, Tan YC, Kreitzer GE, Nakai Y, Shan D, Zheng Y, et al. G proteins G12 and G13 control the dynamic turnover of growth factor-induced dorsal ruffles. J Biol Chem. 2006;281:32660–7. doi: 10.1074/jbc.M604588200. [DOI] [PubMed] [Google Scholar]
- 152.Wang L, Guo D, Xing B, Zhang JJ, Shu HB, Guo L, et al. Resistance to inhibitors of cholinesterase-8A (Ric-8A) is critical for growth factor receptor-induced actin cytoskeletal reorganization. J Biol Chem. 2011;286:31055–61. doi: 10.1074/jbc.M111.253427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xing B, Wang L, Guo D, Huang J, Espenel C, Kreitzer G, et al. Atypical protein kinase Clambda is critical for growth factor receptor-induced dorsal ruffle turnover and cell migration. J Biol Chem. 2013;288:32827–36. doi: 10.1074/jbc.M113.489427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–87. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]