

Abstract
Mutations in the SLC13A5 gene that codes for the Na+/citrate cotransporter, NaCT, are associated with early onset epilepsy, developmental delay and tooth dysplasia in children. In this study, we identify additional SLC13A5 mutations in nine epilepsy patients from six families. To better characterize the syndrome, families with affected children answered questions about the scope of illness and the treatment strategies. Currently, there are no effective treatments, but some antiepileptic drugs targeting the γ-aminobutyric acid system reduce seizure frequency. Acetazolamide, a carbonic anhydrase inhibitor and atypical antiseizure medication, decreases seizures in four patients. In contrast to previous reports, the ketogenic diet and fasting resulted in worsening of symptoms. The effects of the mutations on NaCT transport function and protein expression were examined by transient transfections of COS-7 cells. There was no transport activity from any of the mutant transporters, although some of the mutant transporter proteins were present on the plasma membrane. The structural model of NaCT suggests that these mutations can affect helix packing or substrate binding. We tested various treatments, including chemical chaperones and low temperatures, but none improved transport function in the NaCT mutants. Interestingly, coexpression of NaCT and the mutants results in decreased protein expression and activity of the wild-type transporter, indicating functional interaction. In conclusion, this study has identified additional SLC13A5 mutations in patients with chronic epilepsy starting in the neonatal period, with the mutations producing inactive Na+/citrate transporters.
INTRODUCTION
The onset of seizures in the first weeks of life has been associated with a large number of metabolic and genetic causes (1) including recently identified heterozygous mutations in the SLC13A5 gene that codes for the Na+/citrate transporter, NaCT (2,3). Relatively few children with SLC13A5 disorder have been reported in the literature, and the full extent of their neurologic and epileptic phenotypes is only beginning to emerge. It appears that most children present with epilepsy very early within the first few weeks of life and then have lifelong seizures. This disorder is also characterized by limited and slow motor progress, with children described as unable to sit or walk independently. Reports of tone are also variable, and patients exhibit a range of symptoms: increased tone, decreased tone and poor motor coordination to choreoathetoid movements (2,3). Language is limited with a few patients speaking in single words and others being nonverbal. These findings suggest heterogeneity within the disorder, though to date no clear phenotype–genotype correlate has been found. No effective treatments have been identified yet.
The mechanisms by which mutations in SLC13A5 result in epilepsy are not understood. The inability to meet neuronal energy demand is believed to cause epilepsy and diminished intracellular citrate might result in neuronal energy failure (4). The NaCT transporter encoded by SLC13A5 is found on the plasma membrane in liver and brain (5,6). NaCT is found in neurons in rats (7) and both astrocytes and neurons in mice (8). NaCT cotransports sodium and citrate from the extracellular fluid into the cells (5). Citrate is an important precursor of lipid and cholesterol biosynthesis, and intracellular citrate and its metabolites are key signaling molecules in the regulation of energy expenditure (9). In human hepatocytes, NaCT expression is correlated with lipid accumulation and triglyceride synthesis (10,11). Furthermore, inhibition of NaCT-mediated citrate transport by the liver is emerging as a therapeutic approach for the treatment of metabolic disorders, such as diabetes (11,12). Treatment of animals with small molecule NaCT inhibitors (that do not enter the central nervous system (CNS)) increased citrate excretion in urine, decreased hepatic lipid production and reduced plasma glucose (11). It is clear that NaCT in the liver plays an important metabolic role, but there is very little information on the role of NaCT in brain.
The goal of this study was to identify additional mutations in epileptic patients and to examine potential therapy. We report new mutations in the SLC13A5 gene in nine patients from six families. We examine the mutated NaCT transporters in transiently transfected cell cultures to determine whether the mutations affect citrate transport activity or protein targeting. We also examine potential treatments with chemical chaperones and temperature changes. Furthermore, we use a homology model of NaCT to rationalize changes in function as a result of the mutations. Understanding the pathogenesis of epilepsy as a result of mutations in SLC13A5 will provide critical insights for developing effective therapy.
MATERIALS AND METHODS
Clinical and Genetic Information about the Patients
This study includes nine patients with SLC13A5 mutations and epileptic encephalopathy from six families. The parents of patients contacted the TESS research foundation (tessresearch.org) and voluntarily completed a Stanford REDCap questionnaire (Supplementary Document S1). The families provided SLC13A5 genotype, identified by whole exome sequencing. The questionnaire was designed to understand how patients with SLC13A5 mutations present to medical care, their long-term physical, mental and neurologic outcomes. We focused on antiseizure treatment to identify harmful and helpful treatment strategies. In addition, we attempted to identify phenotype–genotype correlations. The institutional review board of Stanford Medical School approved the study protocol and parents signed an informed consent document.
Expression of NaCT Mutants in COS-7 Cells
Site-directed mutagenesis of human NaCT (hNaCT) in pcDNA3.1 vector was done using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), as described (13). A total of six NaCT mutants were prepared: four point mutations identified in this study, Y82C, G219R, T227M and L492P (Table 1 and Figure 1), and one mutation, L488P, identified previously (2). In addition, we prepared the DelG mutant of hNaCT, with a nucleotide deletion of g511 (relative to the start codon), which produces a frameshift after Leu170 and a premature stop codon (Table 1). The total length of the DelG truncated protein was 187 amino acids and included the first four transmembrane domains and the hairpin loop (HPin) (Figure 1). The recombinant plasmids were expressed in mammalian cells, COS-7 or HEK-293, as described previously (14,15). COS-7 cells (ATCC CRL-1651) were cultured in Dulbecco’s modified Eagle medium containing glutamax and 25 mmol/L HEPES (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum, 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C in 5% CO2. HEK-293 cell medium also contained 1 mmol/L sodium pyruvate and 0.1 mmol/L nonessential amino acids. Cells were plated on collagen-coated (COS-7) or poly-D-lysine-coated (HEK-293) 24-well plates at 0.6 × 105 cells per well and transfected with NaCT plasmids using FuGene 6 (Roche) at a 9:3 ratio (1.8 μL FuGene 6 and 0.6 μg plasmid DNA) (13). Treatments were applied 6 h after transfection and continued for 42 h.
Table 1.
SLC13A5 mutations associated with epilepsy.
| Family | cDNA change | Protein change | Zygosity | Number of patients | Reference |
|---|---|---|---|---|---|
| This study: | |||||
| A | c. 655 G > A | p. G219R | Heterozygous | 2 | |
| c. 1475 T > C | p. L492P | ||||
| B | c. 680 C > T | p. T227M | Homozygous | 1 | |
| C | c. 245 A > G | p. Y82C | Heterozygous | 2 | |
| c. 655 G > A | p. G219R | ||||
| D | c. 511delG | p. E171SfsX16 | Homozygous | 1 | |
| (DelG) | |||||
| E | c. 1276-1G > A | Affect splice site | Heterozygous | 2 | |
| c. 103-1G > A | & add intron** | ||||
| F | c. 655 G > A | p. G219R | Homozygous | 1 | |
| Previous studies: | |||||
| A | c. 1280 C > T | p. S427L | Homozygous | (3) | |
| B | c. 1022 G > A | p. T341* | Heterozygous | (3) | |
| c. 1207_1217dupl 1 | p. P407Rfs*12 | ||||
| C | c. 425 C > T | p. T142M | Heterozygous | (3) | |
| c. 655 G > A | p. G219R | ||||
| D | c. 680 C > T | p. T227M | Heterozygous | (3) | |
| c. 1570 G > C | p. D524H | ||||
| 1 | c. 655 G > A | p. G219R | Heterozygous | (2) | |
| c. 680 C > T | p. T227M | ||||
| 2 | c. 1463T > C | p. L488P | Homozygous | (2) | |
| 3 | c. 655G > A | p. G219R | Heterozygous | (2) | |
| c. 680C > T | p. T227M | ||||
**Describes mRNA changes as a result of cDNA change.
Figure 1.

Secondary structure model of NaCT showing location of mutations in this study. The 11 transmembrane helices are shown as numbered rectangles and the opposing hairpin loops are labeled HP. The outside of the cell is at the top of the image.
Transport Assays
Transport assays were carried out 48 h after transfections, also as described (13,16). The sodium buffer contained 140 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2 and 10 HEPES (all in mmol/L), pH adjusted to 7.4 with 1 M Tris. Choline buffers contained equimolar choline Cl in place of NaCl. For the assays, each well was washed twice with choline buffer, then incubated with 0.25 mL sodium buffer containing [1,5–14C]-citrate (112 mCi/mmol; Moravek) and nonradioactive citrate (100 μmol/L) for 30 min at 37°C. The uptake assays were stopped and surface radioactivity was removed with 4 × 1 mL washes of choline buffer. Cells were dissolved in 1% SDS, transferred to scintillation vials and counted. For all experiments, uptake rates in vector-transfected cells were subtracted from those measured in NaCT plasmid-transfected cells to correct for background counts.
Western Blots
Cells were grown in six-well plates and lysed with RIPA buffer (BioWorld) supplemented with protease inhibitors (10 μg/mL pepstatin, 10 μg/mL leupeptin and 0.5 mmol/L phenylmethanesulfonyl fluoride). Protein concentrations were measured with a DC Protein Assay Kit (Bio-Rad). Samples were separated by Tricine SDS-PAGE, 7.5% (w/v) as described previously (17). Equal amounts of protein were loaded per well, approximately 6 μg, made to a total volume of 20 μL. Cell surface proteins were identified using cell-surface biotinylation with sulfo-NHS-LC-biotin (Thermo Scientific) as described (16,18) and three wells of a six-well plate were combined for each group. Chemiluminescent size standards were MagicMark XP (Invitrogen). The anti-NaCT antibodies were a gift from Pfizer. For Western blots, the primary antibody, anti-NaCT, was applied at 1:750 dilution followed by secondary antibody at 1:7,500 dilution, peroxidase-conjugated anti-rabbit IgG (Jackson Laboratories). Protein loading was verified using duplicate blots probed with anti-GAPDH antibodies (1:20,000 dilution) (Ambion AM4300), followed by secondary antibody at 1:5,000 dilution, peroxidase-conjugated anti-mouse IgG (Jackson Laboratories). Detection was done with Supersignal West Pico Chemiluminescent Reagent (Thermo Scientific) and Image Station 4000R (Carestream Scientific).
Statistics
Duplicate or quadruplicate measurements were made for each data point. The experiments were repeated with two or three different batches of transfected cells from different passage numbers. Significant differences between groups were identified by Student’s t test or ANOVA with p < 0.05. Data are reported as means ± SEM.
Homology Modeling
We modeled the NaCT dimer based on the atomic structure of vcINDY, which was determined as a dimer (PDB ID: 4F35) (19). Models were constructed with MODELLER-9v14 (20) using a previously published NaCT-vcINDY alignment and modeling protocol (14,21). In brief, we built 100 models, which were assessed and ranked by the statistical potential Z-DOPE (22). The Z-DOPE score of the top model was –0.35, suggesting that approximately 55% of their Cα atoms are within 3.5 Å of their correct positions (23). The models were constructed with atoms of nonprotein elements including a sodium ion and the ligand citrate based on their coordinates in the vcINDY template structure. All models were visualized with PyMOL (Schrodinger, LLC (2010) The PyMOL Molecular Graphics System, Version 1.3r1).
All supplementary materials are available online at www.molmed.org.
RESULTS
SLC13A5 Mutations in Pediatric Epilepsy Patients
This study identifies new autosomal recessive mutations in SLC13A5 in children with early onset epilepsy from six families (Table 1). The families are from the United States, the Netherlands and Brazil (Supplementary Table S1), and the children age range from 2 to 18.7 years (Table 2). Three of the families had homozygous mutations: T227M and G219R, both reported previously in compound heterozygous form (2,3), and a new deletion mutation c511DelG (abbreviated DelG in this paper). The c511DelG mutation produces a frameshift leading to a premature stop codon, with a resulting predicted truncated protein of only 187 amino acids rather than the full 568 amino acids (5). Two families had compound heterozygous mutations, some of which have not been reported previously (Y82C and L492P), and one family had compound heterozygous mutations affecting RNA splicing (Table 1).
Table 2.
Background and basic neurodevelopmental information on children with SLC13A5 mutations in this study.
| Subject | Family | Age (years) | Sex | Motor delay | Language delay | Regression | Muscle tone | Movement disorder | MRI brain | Dental issues |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | 11.8 | F | Y | Y | Y | Low | Y-dystonia | Normal | Teeth hypoplasia |
| 2 | A | 2 | M | Y | Y | N | Low | N | Normal | Teeth hypoplasia |
| 3 | B | 2.9 | F | Y | Y | Y | NR | NR | Normal | Teeth hypoplasia + Gingival hyperplasia |
| 4 | C | 5.7 | F | Y | Y | N | Low | N | Focal frontal lobe thickening | Gingival hyperplasia |
| 5 | C | 8.7 | M | Y | Y | N | Low | N | Normal | Gingival hyperplasia |
| 6 | D | 9.6 | F | Y | Y | NR | NR | NR | Normal | NR |
| 7 | E | 18.7 | F | Y | Y | N | Normal | N | Hyperintense foci in parietal white matter | Teeth hypoplasia + amelogenesis imperfecta |
| 8 | E | 15.5 | M | Y | Y | N | Normal | N | Normal | Amelogenesis imperfecta |
| 9 | F | 4 | M | Y | Y | Y | Increased | Y-dystonia | Focal loss of gray matter | N |
F, female; M, male; Y, yes; N, no; NR, not reported.
Clinical Features of Patients
The pregnancy and birth history were unremarkable in seven of the nine patients, with two being born premature (Supplementary Table S1). None of the patients were small for gestational age. MRI was normal in six of the nine patients, and evidence of possible perinatal injury was present in one child. All of the patients in this study presented with epilepsy early, with eight of the nine patients presenting in the first week of life, and all had motor and language delays (Table 2). The seizure semiology varied with most having focal seizures, some with secondary generalization and myoclonic seizures. The seizure frequency also varied greatly, as high as >100 myoclonic seizures per day to rare focal seizures. Frequency of seizures and severity of developmental delay varied even between siblings with the same mutations, but all patients had severe neurologic phenotypes without clear genotype–phenotype correlations. All patients reported episodes of prolonged seizures (Supplementary Table S2).
The one frequent physical/morphologic abnormality that seven of the nine patients reported, in agreement with previous reports (3), was dental abnormalities consisting of teeth hypoplasia and gingival hyperplasia (Table 2). Two patients had constipation but, in general, they seemed to be in otherwise good health outside of their neurologic disorder. Parents described them as social and happy to engage, though all had limited communication, with only three of the nine being described as verbal (Supplementary Table S3). All patients reported delayed motor development with only two walking independently; one subject, who was a sibling of one of the ambulatory patients, had learned to crawl, and another patient was described as standing independently and using a walker to ambulate.
Epilepsy Treatments
All of the patients had been tried on numerous antiseizure medications, ranging between 4 and 13 per patient (Supplementary Table S2), in an effort to control their seizures without complete seizure control. No patients were reported as successfully being weaned off their seizure medications and a resective surgery was tried in one patient without success. The majority of patients showed improvement in seizures by antiepileptic drugs that work on the γ-aminobutyric acid (GABA) system (24,25), including diazepam, lorazepam, phenobarbital, clonazepam, clobazam and midazolam (Table 3). In addition, several patients reported improvement with phenytoin and lamotrigine, drugs that target sodium channels (26). In one patient, however, phenytoin was mentioned as worsening myoclonus. Acetazolamide, the carbonic anhydrase inhibitor, has been used for treating epilepsy (27). In the four children who tried acetazolamide, parents reported improved seizure control. It is not known if this correlated with a change in urinary citrate excretion or serum level. Two siblings from family A while treated with acetazolamide reported elevated plasma citrate levels of 235 μmol/L (Patient 2) and 110 μmol/L (Patient 1), where the upper range of normal is 100 μmol/L. Neither patient has developed an acidosis while on the acetazolamide. Acetazolamide typically decreases urinary citrate excretion by inducing metabolic acidosis (28,29), which should increase citrate reabsorption by the renal NaDC1 transporter (SLC13A2) (30,31).
Table 3.
Information on seizures in children with SLC13A5 mutations in this study.
| Subject | Seizure onset | Seizure types | Current seizure frequency | Most effective medications | Ketogenic diet effects | Seizures worse with fasting |
|---|---|---|---|---|---|---|
| 1 | First week of life | Complex partial GTC Myoclonic | >100/day | Acetazolamide Carbamazepine Diazepam Felbamate Phenobarbital Phenytoin* | Seizures worse Dystonia worse | Y |
| 2 | First week of life | Atonic hemiplegia | 0–5/month | Acetazolamide Phenobarbital | NT | N |
| 3 | First week of life | Complex partial Focal motor Myoclonic | 100 s/day with 3–5 wks seizure-free periods | Diazepam Lamotrigine Phenobarbital Phenytoin | NT | Y |
| 4 | First week of life | Complex partial Focal motor Myoclonic Tonic | >50/day | Clobazam Clonazepam Lorazepam Midazolam | Seizures worse | Y |
| 5 | First week of life | Focal motor Myoclonic | 1/month | Clobazam Clonazepam Lamotrigine | NT | N |
| 6 | First week of life | Complex partial GTC | 1/month | Acetazolamide Phenytoin | NT | N |
| 7 | <1 year old | Absence Complex partial Focal motor GTC Myoclonic | Rare (no specific frequency reported) | Acetazolamide Clonazepam Lamotrigine Valproic acid | No effect reported | N |
| 8 | First week of life | Focal motor GTC Myoclonic | 1/month to 3/year | Acetazolamide Clonazepam Lamotrigine Valproic acid | NT | N |
| 9 | First week of life | Absence Atonic Complex partial Focal motor GTC Infantile spasms Myoclonic Tonic | 1/wk | Lamotrigine Oxcarbazepine Topiramate | Seizures worse | N |
GTC, generalized tonic clonic; NT, has not been treated with ketogenic diet; Y, yes; N, no. *Phenytoin improved seizures, but caused myoclonus.
The use of the ketogenic diet, a high-fat, low-carbohydrate diet, is successful in many children with refractory epilepsy and was reported to be beneficial for seizure control or level of alertness and development in three patients with SLC13A5 mutations (3). However, in this study, the ketogenic diet was reported as causing longer or more frequent seizures in three children and had no effect in another (Table 3). A limited carbohydrate diet (Atkins) was also noted to worsen seizures in one patient (Supplementary Table S2). Three patients had worsening of seizures with fasting though six others did not notice any worsening. Three described increased seizures with fever and illness.
Function and Expression of NaCT Mutants
To determine the effects of the NaCT mutations on function and protein expression, we prepared the mutants and expressed them in COS-7 cells, a cell line derived from monkey kidney that has very low background transport of citrate and succinate. As shown in Figure 2, most of the mutants had no citrate transport activity, whereas the DelG and the G219R mutants had a small amount of citrate transport activity above background. We next examined NaCT protein expression in cell lysates transfected with plasmids coding for the NaCT mutants (Figure 3A). The anti-NaCT antibodies are specific for human NaCT and do not recognize NaDC1 or NaDC3 (13). The antibodies recognize multiple bands at ~45 and 75 kDa in cells transfected with NaCT, which correspond to differently glycosylated forms of the transporter (unpublished results), similar to Western blots of other SLC13 family members (13). There was no detectable NaCT protein in control cells transfected with vector plasmid, pcDNA3.1, as well as in cells transfected with DelG deletion mutant, T227M and L488P mutants (Figure 3A). The DelG mutant has an early stop codon and would be undetectable using our antibodies because the early stop codon is before the antigenic site. We also examined cell surface expression of the mutants by biotinylation with a membrane-impermeant reagent, sulfo-NHS-LC-biotin. Figure 3B shows that the Y82C, G219R and L492P mutants are found on the plasma membrane.
Figure 2.
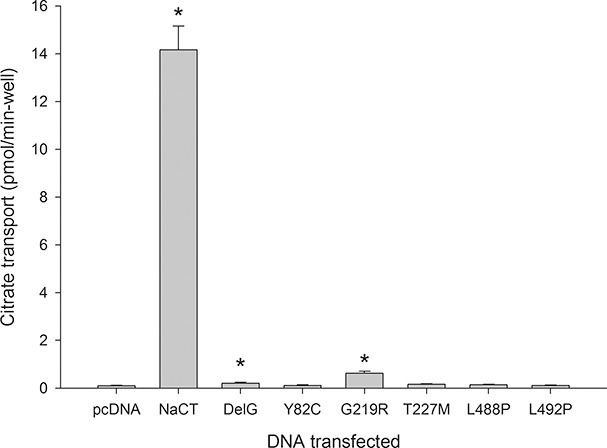
Transport activity of hNaCT wild-type and mutant transporters expressed in COS-7 cells. 14C-citrate transport (100 μmol/L) was measured at 37°C for 30 min. Data are means ± SEM, n = 8 (pcDNA and G219R), n = 7 (wild-type NaCT), n = 5 (DelG) and n = 3 (all others). * denotes p < 0.05 relative to pcDNA (empty vector plasmid) group.
Figure 3.

Western blots. (A) Total cell protein. Protein lysates from COS-7 cells transfected with empty vector, pcDNA, wild-type NaCT (WT) and mutants. Duplicate blots were probed with anti-NaCT (top) and GAPDH antibodies (bottom). Size standards (kDa) are shown at left. (B) Cell surface biotinylated proteins from HEK-293 cells treated with membrane-impermeant sulfo-NHS-LC-biotin followed by streptavidin beads. The HEK-293 cells were used for biotinylations because the signal was stronger than with COS-7 cells.
Characterization of the DelG and G219R Mutant Functions
The functional characteristics of the DelG and G219R mutants were tested to determine whether the measured transport activity was due to activation of an endogenous protein or intrinsic to the mutant transporter. We compared citrate, succinate and glutarate transport activity in the presence and absence of sodium. The transport activity in cells transfected with the DelG mutant was very low (Figure 4A). There was some sodium-dependent citrate transport, but the succinate and glutarate transport were sodium independent. In contrast, the cells expressing the G219R mutant had sodium-dependent transport of all three substrates: citrate, succinate and glutarate. There was also some succinate and glutarate transport in the absence of sodium in G219R. This transport specificity profile is different from the wild-type SLC13 transporters (Figure 4C). These transporters are sodium dependent, with no transport activity reported in the absence of sodium (6). Wild-type human NaCT transported both citrate and succinate, but there was no measurable transport of glutarate, in agreement with previous observations (5,32). Human NaDC1 and NaDC3 transported all three substrates, and the highest activity was with succinate (Figure 4C) (30,33,34). NaDC3 had high transport activity with glutarate. Therefore, cells expressing DelG and G219R express transport activities that are different from any of the human SLC13 transporters. This activity was seen in transfected COS-7 cells but not in HEK-293 cells; in two experiments, there was no transport in HEK-293 cells transfected with G219R or DelG (not shown). COS-7 cells transfected with G219R cultured at cool temperature (28°C) did not have transport activity (Supplementary Table S4). Finally, a truncated NaCT construct that is four amino acids longer than DelG induced sodium-dependent citrate, succinate and glutarate transport activity with a similar profile but higher activity than the G219R mutant (results not shown).
Figure 4.
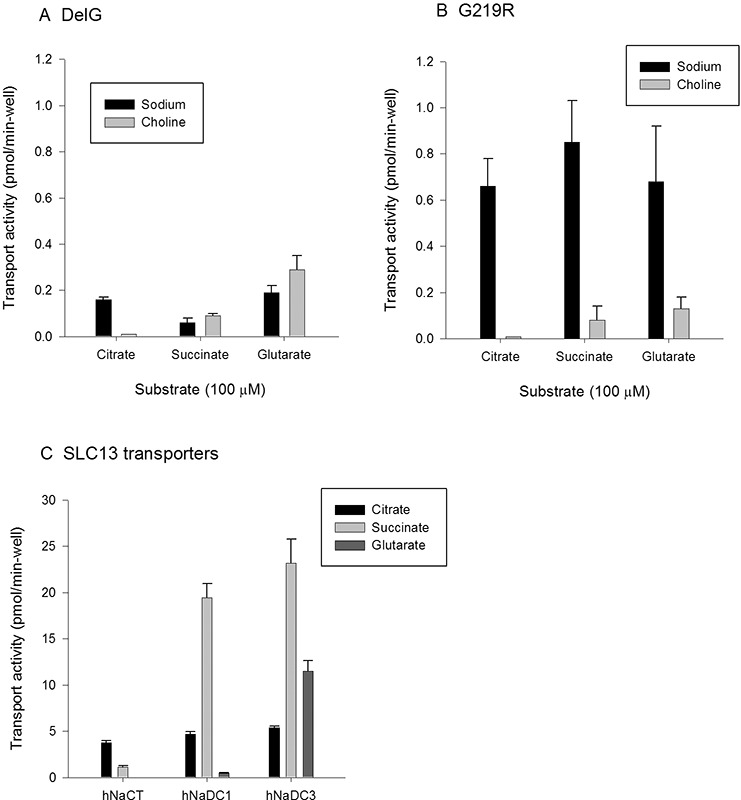
Characterization of substrate transport by mutant and wild-type SLC13 transporters. Transport of 14C-citrate, 14C-succinate and 14C-glutarate (100 μmol/L each) in sodium or choline buffers. (A) DelG mutant. Bars show mean and range, n = 2 experiments. (B) G219R mutant. Bars show means ± SEM, n = 3–4; range is shown for n = 2 (succinate, glutarate with choline). (C) Comparison of citrate, succinate and glutarate transport in wild-type NaCT, NaDC1 and NaDC3 transporters. Transport was measured in sodium buffer. Bars show means ± SEM, n = 3 replicates from single experiment. In all experiments, backgrounds were corrected by subtracting counts in pcDNA-transfected cells. Note the 25-fold difference in scale between (C) and (A), (B).
We examined various treatments in an effort to increase the expression and activity of the mutant transporter proteins in transiently transfected COS-7 cells. None of the treatments affected the NaCT mutants, although several affected the citrate transport activity of the wild-type transporter (Supplementary Table S4). We tested treatments that have been shown to increase expression of CFTR or aquaporin mutants in cell culture, including culture at 28°C (35), or the addition of 4-phenylbutyric acid (36), but they had no effect on function of the NaCT mutants. The chemical chaperone glycerol, which increases the expression of NaDC1 mutants (37), had no effect on the NaCT mutants. Glycerol decreased the activity of the wild-type NaCT, similar to our previous findings with NaDC1 (37). We tested treatments that were shown to affect other transporters: 4-aminopyrimidine and NBCe1 (38), heat shock and SGLT1 (39), but these treatments had no effect on wild-type or mutant NaCT. Sebacic acid was tested because of a report that some mice treated with this compound had higher expression of SLC13A5 (40), but it had no effect in this study. We also examined whether some of the compounds used for therapy (acetazolamide and heptanoic acid) would have a direct effect on the transporters and they did not. Finally, we tested the addition of methotrexate to the transport solutions, which we have found in previous studies to increase activity and protein at the plasma membrane in NaCT and NaDC1 (results not shown). However, methotrexate did not have consistent effects on the NaCT mutants.
Because many of the patients are heterozygous with different NaCT mutations, we examined whether the proteins might interact by coexpressing a mutant transporter with the wild-type NaCT transporter. Citrate transport activity was decreased when HEK-293 cells were cotransfected with wild-type NaCT and any of the missense mutations (Figure 5). In particular, the coexpression of NaCT and T227M led to very low citrate transport activity, less than 5% of the control. The DelG truncated protein did not affect citrate transport by NaCT (Figure 5A). The protein expression in cell lysates paralleled the transport activity (Figure 5B) indicating that the low transport activity is due to low protein abundance in the cells. The control group was transfected with plasmid for wild-type NaCT combined with empty vector plasmid, pcDNA3.1. There was a strong protein band at around 55 kDa representing NaCT. This protein signal was very similar in cells transfected with plasmids for wild-type NaCT and DelG, indicating that these proteins do not interact. However, when wild-type NaCT was combined with plasmids for the point mutants, the protein signal was greatly decreased, indicating that the mutant proteins interact with the wild type and reduce the amount of protein in the cells (Figure 5B). The amount of total cell protein loaded in each lane was similar, as shown from the GAPDH signal in the lower panel of Figure 5B.
Figure 5.

Effects of cotransfection of wild-type NaCT with vector plasmid (pcDNA) or mutant transporters. (A) Citrate transport activity. All groups were transfected with the same amount of wild-type NaCT DNA and a second plasmid. Activity is expressed as a percentage of the NaCT + pcDNA control group. All counts were corrected for background counts determined from cells transfected only with pcDNA plasmid. Bars are means ± SEM, n = 3–5, or range, n = 2 (DelG). * denotes p < 0.05 relative to the NaCT + pcDNA group. (B) Western blots. Duplicate blots were probed with anti-NaCT (top) and anti-GAPDH (bottom) antibodies. Groups were the same as for panel (A). The samples in the lane next to the size standards (labeled none) were transfected only with pcDNA, the other samples had two plasmid DNAs (NaCT and a second plasmid). All cells were transfected with the same total amount of DNA.
NaCT Homology Model
We modeled NaCT based on the dimeric structure of the homolog protein vcINDY from Vibrio cholerae (29% sequence identity), using similar parameters to those used in our previously published NaCT monomer model (20). The location of the five mutations on the structural models of NaCT provides a potential explanation for their effects on the transport function (Figure 6). In particular, Y82 is located in TM4a at the subunit interface (Figures 6 and 7) and may be involved in stabilizing the dimer. The substitution of a cysteine residue at this location may induce the formation of a disulfide bridge with a nearby cysteine at position 79 (Figure 6A). G219 is located in TM5a and is involved in helix–helix packing by hydrophobic effect with residues in HPin. Therefore, the G219R mutation is likely to disrupt the packing of the helices, which would affect the function of this loop that is very important for substrate binding (Figure 6B). Additionally, G219 is in proximity to the sodium ion (7 Å). A mutation of this residue to arginine, a large, positively charged residue, changes the chemical properties of the binding site, including its shape and charge, which might disrupt the interaction with the ion. T227 is located in the binding site formed between TM5b and the two hairpin loops, HPin and HPout. T227 is involved in substrate binding and ion coordination (Figure 6C). This residue is highly conserved in the SLC13 family, and our previous study showed that mutation of T253, the equivalent residue in NaDC3 (SLC13A3), affects substrate transport and selectivity as well as cation binding (14). Substitution of threonine by methionine in the NaCT T227M mutant should change the chemical properties of the binding site. Finally, L488 and L492 are located in TM10a and are part of a leucine zipper motif (Figure 6D). The substitution by proline residues should break the helix, which would affect the overall folding of the protein.
Figure 6.

Homology model of hNaCT showing the location of the point mutations. (A) Y82 is located in TM4, (B) G219 (TM5a), (C) T227 (TM5b) and (D) L488 and L492 (both located in TM10). In all panels, the transmembrane helices are shown as cyan cartoons; key amino acids represented by sticks with the mutated residues colored in orange. The purple sphere in panels (B) and (C) represents the sodium ion that binds the Na1 binding site. Citrate is shown in green sticks in panel (C).
DISCUSSION
Mutations in the SLC13A5 gene that codes for the NaCT have been reported to cause epilepsy, developmental delay and tooth dysplasia in children (2,3). In this study, we identified six families with new SLC13A5 mutations. We surveyed the families of the patients to identify potential treatments and diagnostic features and also examined the functional consequences of the mutations on the NaCT transporter in vitro. Our results show that the new mutations in NaCT are also associated with severe epilepsy, developmental delays and dental abnormalities. To date, there is no treatment that fully abolishes the epilepsy, although some antiepileptic drugs reduce the symptoms. The severity of the disease is in agreement with the functional findings: none of the NaCT mutants had functional activity although some were found on the plasma membrane. None of the treatments tested in vitro could rescue the function of the mutated transporters. Furthermore, the mutated transporters appear to interact with one another.
Several key features may aid in the identification of SLC13A5 disorder in a clinical setting. The onset of seizures in the first week of life and the presence of dental abnormalities agree with previous reports (3) and might be clues to help clinicians. The diagnosis to date requires sequencing of the SLC13A5 gene, available via gene panel and exome sequencing. Most of the children have ongoing seizures of varied types (myoclonic, convulsions and focal seizures), although the frequency varied greatly (many per day to rare, less than once a year). Because of the small study size and variety of mutations reported, we were unable to identify any clear genotype–phenotype correlations either within our study population or when comparing our data with prior studies.
Treatments that seemed to improve seizure control in several patients were primarily drugs that affect the GABA neurotransmitter system (phenobarbital, clobazam, diazepam and lorazepam). Several parents also noted that sodium channel blocking agents, phenytoin and lamotrigine, were helpful though may have exacerbated myoclonus in one patient. Several families have felt that acetazolamide has been helpful. This is interesting in light of its ability to alter urinary citrate excretion, possibly as a consequence of changes in urinary or renal cell pH (28,29). However, it is also possible that the effects of acetazolamide are related to changes in pH or metabolism in the CNS (27). We do not yet know whether elevated plasma citrate contributes to the pathogenesis of this disorder. In contrast to previous reports (3), we found that the ketogenic diet or low caloric intake seemed to exacerbate seizures, including episodes of status epilepticus in two patients. Unlike some other disorders of CNS energy depletion that respond favorably to the ketogenic diet, such as glucose transporter deficiency or pyruvate dehydrogenase deficiency, lack of citrate transport cannot be rescued by a high-fat diet (4,41).
The mechanism by which mutations in SLC13A5 result in epilepsy is not yet known. To address this, we examined the functional properties of NaCT mutants in cultured cells. The wild-type NaCT transporter had high citrate transport activity above background, but none of the mutants found in epilepsy patients had activity. The structural models suggest that the mutations alter substrate binding or helix packing, which would be expected to have severe effects on transport function. Two of the mutants, DelG and G219R, appeared to induce citrate, succinate and glutarate activity in COS-7 cells, which may indicate that these mutants could potentially induce other transporters in vivo, although this remains to be tested. An alternate explanation for the function seen in cells expressing G219R is that G219R has intrinsic activity with altered substrate specificity. However, transport activity with the same substrate profile was seen with a deletion mutant of NaCT, which is unlikely to have activity because it is missing most of the substrate binding site. Three of the mutants tested in this study, Y32C, G219R and L492P, were located on the plasma membrane showing that these mutations have severe effects on function. We were unable to detect T227M and L488P proteins in whole cell lysates. A previous study examined the cellular distribution of T227M and G219R (3) using constructs that added an epitope tag at the N-terminus, which may alter tissue distribution of membrane proteins. Our cell surface biotinylation results with untagged G219R using specific anti-NaCT antibodies agree with the previous study showing that tagged G219R is found on the plasma membrane (3), but unlike that study, our antibodies identified fully glycosylated G219R rather than just one lower mass band. Furthermore, we did not find protein expression of T227M, whereas the tagged T227M was found on the plasma membrane in the previous study (3).
One surprising finding in this study was the potential interaction between wild-type and mutant NaCT transporters in cotransfection experiments. The DelG mutant did not affect NaCT protein expression or activity, suggesting that DelG does not interact with NaCT. The truncated DelG protein may be missing key interaction sites. However, the point mutants all decreased protein expression, with subsequent decreased activity, of the wild-type transporter. In particular, coexpression of T227M and wild-type NaCT had very low protein expression and transport activity. This result might suggest that heterozygotes between wild type and mutants could have reduced NaCT expression and activity, although the health consequences of reduced activity are not known. There are no reports of seizures or other neurologic problems from a heterozygous SLC13A5 state of the parents or siblings, suggesting that reduced activity is not deleterious, in contrast to the absent NaCT transport activity in the patients. Interestingly, heterozygotes may have some benefit relating to reduced liver citrate transport. A current focus in diabetes drug development targets the liver NaCT with small molecule inhibitors that do not penetrate the CNS (11,12). There is some evidence from other members of the SLC13 family that dimer formation can alter function, both homodimers (19) and heterodimers with other proteins (42). For example, the transport activity of the Na+/dicarboxylate cotransporter (NaDC1 and SLC13A2) is regulated by heterodimer formation with the organic anion exchanger SLC26A6 (42).
Potential therapeutic approaches for SLC13A5-related epilepsy involve three general areas. The first approach would be to improve expression or function of the mutant transporters. For example, one of the approaches to treating cystic fibrosis patients with the F508del mutation involves small molecule correctors and stabilizers to target misfolded proteins to the plasma membrane (43). However, at present, our studies with transfected cells show that none of the chaperone treatments resulted in increased transport activity of NaCT mutants, possibly because the affected proteins have no function. The second therapeutic approach would involve using a second citrate transporter in the brain to bypass the inactive NaCT. The related transporter NaDC3 (SLC13A3) carries citrate and it is expressed in brain (7,8), but it is not known whether there is overlap in distribution between NaDC3 and NaCT in humans. Another possible citrate transporter, SLC25A1, is normally located in the mitochondrion, but a splice variant localizes to the plasma membrane in prostate, although it has not been studied in the CNS (44). Finally, a third approach is to identify the physiological role of intracellular citrate in the brain, and then to compensate for the lack of plasma membrane citrate transport in the mutants, possibly with other metabolites. Recent studies have suggested that epilepsy is related to alterations in energy metabolism (45), and citrate is a key metabolite. Related to this, it would be important to determine whether elevated plasma citrate contributes to development of seizures, so that altering plasma citrate might be a therapeutic approach. A recent study shows that in humans the liver accounts for most of the clearance of citrate from the plasma, through the activity of NaCT, consistent with the mild elevation in plasma citrate in two of the children (46).
There are currently no models for epilepsy associated with SLC13A5 mutations. The functional properties of human NaCT are different from the rat and mouse transporters. The human NaCT has a Km for citrate around 600 μmol/L and functions as a citrate transporter (5). In contrast, the rat and mouse NaCT have Km values for citrate 20–50 μmol/L and broad substrate selectivity for citrate, succinate, fumarate and malate (47–49). Deletion of the mouse Slc13a5 gene (mNaCT and mIndy) results in beneficial metabolic changes including decreased lipid accumulation, resistance to a high-fat diet and increased insulin sensitivity (49). The Slc13a5–/– mice do not appear to have seizures (50). It is possible that the Slc13a5–/– mice are protected from seizures because mice express two citrate transporters, NaCT and NaDC3, on neurons and astrocytes (8), and the citrate might be transported via NaDC3 when NaCT is absent. Rats express NaDC3 on astrocytes and NaCT in neurons (7), and thus may be more likely to develop seizures after inhibition or deletion of NaCT. It is clear that further study is needed to establish the distribution and function of NaCT in human brain.
CONCLUSION
We have identified nine new patients from six families with new compound heterozygous or homozygous mutations in SLC13A5 that codes for NaCT. All of the patients developed epilepsy early in life, most within the first week, and these patients show teeth hypoplasia, both of which appear to be diagnostic characteristics of the disorder. The mutated NaCT transporters had no functional activity although some were found on the plasma membrane. We also observed an interaction between mutant and wild-type NaCT transporters, indicating that these transporters may interact with other subunits or other proteins. Future studies are needed to identify the role of citrate in the brain and to identify potential treatments for this disorder.
Supplementary Material
ACKNOWLEDGMENTS
Thanks to Kim Nye of the TESS foundation for many discussions throughout the course of this study. This research was funded by a gift from the TESS Foundation and a UCSD Academic Senate Bridging grant (AMP), the Child Neurology Chief Fund (BP), the National Institute of Health (GM108911 to AS and CC) and the Department of Defense (W81XWH-15-1-0539 to AS and CC).
Footnotes
Online address: http://www.molmed.org
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Klotz J, Porter BE, Colas C, Schlessinger A, Pajor AM. (2016) Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 22:310–21.
REFERENCES
- Gursoy S, Ercal D. Diagnostic approach to genetic causes of early-onset epileptic encephalopathy. J Child Neurol. 2016;31:523–32. doi: 10.1177/0883073815599262. [DOI] [PubMed] [Google Scholar]
- Thevenon J, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. 2014;95:113–20. doi: 10.1016/j.ajhg.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies K, et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. 2015;138:3238–50. doi: 10.1093/brain/awv263. [DOI] [PubMed] [Google Scholar]
- Zsurka G, Kunz WS. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015;14:956–66. doi: 10.1016/S1474-4422(15)00148-9. [DOI] [PubMed] [Google Scholar]
- Inoue K, Zhuang L, Ganapathy V. Human Na+-coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem Biophys Res Commun. 2002;299:465–71. doi: 10.1016/s0006-291x(02)02669-4. [DOI] [PubMed] [Google Scholar]
- Pajor AM. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. 2014;466:119–30. doi: 10.1007/s00424-013-1369-y. [DOI] [PubMed] [Google Scholar]
- Yodoya E, et al. Functional and molecular identification of sodium-coupled dicarboxylate transporters in rat primary cultured cerebrocortical astrocytes and neurons. J Neurochem. 2006;97:162–73. doi: 10.1111/j.1471-4159.2006.03720.x. [DOI] [PubMed] [Google Scholar]
- Lamp J, et al. Glutaric aciduria type 1 metabolites impair the succinate transport from astrocytic to neuronal cells. J Biol Chem. 2011;286:17777–84. doi: 10.1074/jbc.M111.232744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol. 1999;276:E1–18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- Li L, et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol Pharmacol. 2015;87:674–82. doi: 10.1124/mol.114.097287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard K, et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5) Sci Rep. 2015;5:17391. doi: 10.1038/srep17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard K, et al. Optimization of a dicarboxylic series for in vivo inhibition of citrate transport by the solute carrier 13 (SLC13) family. J Med Chem. 2016;59:1165–75. doi: 10.1021/acs.jmedchem.5b01752. [DOI] [PubMed] [Google Scholar]
- Pajor AM, Sun NN. Single nucleotide polymorphisms in the human Na+-dicarboxylate cotransporter affect transport activity and protein expression. Am J Physiol Renal Physiol. 2010;299:F704–11. doi: 10.1152/ajprenal.00213.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger A, Sun NN, Colas C, Pajor AM. Determinants of substrate and cation transport in the human Na+/dicarboxylate cotransporter, NaDC3. J Biol Chem. 2014;289:16998–17008. doi: 10.1074/jbc.M114.554790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor AM, Sun NN. Non-steroidal anti-inflammatory drugs and other anthranilic acids inhibit the Na+/dicarboxylate symporter from Staphylococcus aureus. Biochemistry. 2013;52:2924–32. doi: 10.1021/bi301611u. [DOI] [PubMed] [Google Scholar]
- Pajor AM, Randolph KM. Conformationally sensitive residues in extracellular loop 5 of the Na+/dicarboxylate co-transporter. J Biol Chem. 2005;280:18728–35. doi: 10.1074/jbc.M501265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor AM, Sun N. Characterization of the rabbit renal Na+/dicarboxylate cotransporter using anti-fusion protein antibodies. Am J Physiol. 1996;271:C1808–16. doi: 10.1152/ajpcell.1996.271.6.C1808. [DOI] [PubMed] [Google Scholar]
- Weerachayaphorn J, Pajor AM. Sodium-dependent extracellular accessibility of Lys-84 in the sodium/dicarboxylate cotransporter. J Biol Chem. 2007;282:20213–20. doi: 10.1074/jbc.M701113200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancusso R, Gregorio GG, Liu Q, Wang DN. Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature. 2012;491:622–7. doi: 10.1038/nature11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Colas C, Pajor AM, Schlessinger A. Structure-based identification of inhibitors for the SLC13 family of Na(+)/dicarboxylate cotransporters. Biochemistry. 2015;54:4900–8. doi: 10.1021/acs.biochem.5b00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–24. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramian D, Eswar N, Shen MY, Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17:1881–93. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne TR, Penry JK. Benzodiazepines in the treatment of epilepsy. A review. Epilepsia. 1973;14:277–310. doi: 10.1111/j.1528-1157.1973.tb03965.x. [DOI] [PubMed] [Google Scholar]
- Robertson MM. Current status of the 1,4- and 1,5-benzodiazepines in the treatment of epilepsy: The place of clobazam. Epilepsia. 1986;27(Suppl 1):S27–41. doi: 10.1111/j.1528-1157.1986.tb05730.x. [DOI] [PubMed] [Google Scholar]
- Wuttke T V, Lerche H. Novel anticonvulsant drugs targeting voltage-dependent ion channels. Expert Opin Investig Drugs. 2006;15:1167–77. doi: 10.1517/13543784.15.10.1167. [DOI] [PubMed] [Google Scholar]
- Aggarwal M, Kondeti B, McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: A patent review. Expert Opin Ther Pat. 2013;23:717–24. doi: 10.1517/13543776.2013.782394. [DOI] [PubMed] [Google Scholar]
- Hamm LL. Renal handling of citrate. Kidney Int. 1990;38:728–35. doi: 10.1038/ki.1990.265. [DOI] [PubMed] [Google Scholar]
- Unwin RJ, Capasso G, Shirley DG. An overview of divalent cation and citrate handling by the kidney. Nephron Physiol. 2004;98:15–20. doi: 10.1159/000080259. [DOI] [PubMed] [Google Scholar]
- Pajor AM, Sun N. Functional differences between rabbit and human Na+-dicarboxylate cotransporters, NaDC-1 and hNaDC-1. Am J Physiol. 1996;271:F1093–9. doi: 10.1152/ajprenal.1996.271.5.F1093. [DOI] [PubMed] [Google Scholar]
- Aruga S, et al. Chronic metabolic acidosis increases NaDC-1 mRNA and protein abundance in rat kidney. Kidney Int. 2000;58:206–15. doi: 10.1046/j.1523-1755.2000.00155.x. [DOI] [PubMed] [Google Scholar]
- Brauburger K, Burckhardt G, Burckhardt BC. The sodium-dependent di- and tricarboxylate transporter, NaCT, is not responsible for the uptake of D-, L-2-hydroxyglutarate and 3-hydroxyglutarate into neurons. J Inherit Metab Dis. 2011;34:477–82. doi: 10.1007/s10545-010-9268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekuda R, et al. Primary structure and functional characteristics of a mammalian sodium-coupled high affinity dicarboxylate transporter. J Biol Chem. 1999;274:3422–9. doi: 10.1074/jbc.274.6.3422. [DOI] [PubMed] [Google Scholar]
- Kaufhold M, et al. Differential interaction of dicarboxylates with human sodium-dicarboxylate cotransporter 3 and organic anion transporters 1 and 3. Am J Physiol Renal Physiol. 2011;301:F1026–34. doi: 10.1152/ajprenal.00169.2011. [DOI] [PubMed] [Google Scholar]
- Denning GM, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–4. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Suaud L, et al. 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J Biol Chem. 2011;286:45083–92. doi: 10.1074/jbc.M111.293282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AD, Pajor AM. Role of conserved prolines in the structure and function of the Na+/dicarboxylate cotransporter 1, NaDC1. Biochemistry. 2006;45:4231–9. doi: 10.1021/bi052064y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodl-Haussel M, Theparambil SM, Deitmer JW, Roussa E. Regulation of functional expression of the electrogenic sodium bicarbonate cotransporter 1, NBCe1 (SLC4A4), in mouse astrocytes. Glia. 2015;63:1226–39. doi: 10.1002/glia.22814. [DOI] [PubMed] [Google Scholar]
- Ikari A, Nakano M, Kawano K, Suketa Y. Up-regulation of sodium-dependent glucose transporter by interaction with heat shock protein 70. J Biol Chem. 2002;277:33338–43. doi: 10.1074/jbc.M200310200. [DOI] [PubMed] [Google Scholar]
- Iaconelli A, et al. Effect of oral sebacic acid on postprandial glycemia, insulinemia, and glucose rate of appearance in type 2 diabetes. Diabetes Care. 2010;33:2327–32. doi: 10.2337/dc10-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass HR, et al. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure. 2016;35:83–87. doi: 10.1016/j.seizure.2016.01.011. [DOI] [PubMed] [Google Scholar]
- Ohana E, Shcheynikov N, Moe OW, Muallem S. SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J Am Soc Nephrol. 2013;24:1617–26. doi: 10.1681/ASN.2013010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby EF, Heard AS, Wang XR. Enhancing the potency of F508del correction: a multi-layer combinational approach to drug discovery for cystic fibrosis. J Pharmacol Clin Toxicol. 2013;1:1007. [PMC free article] [PubMed] [Google Scholar]
- Mazurek MP, et al. Molecular origin of plasma membrane citrate transporter in human prostate epithelial cells. EMBO Rep. 2010;11:431–7. doi: 10.1038/embor.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sada N, Lee S, Katsu T, Otsuki T, et al. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science. 2015;347:1362–7. doi: 10.1126/science.aaa1299. [DOI] [PubMed] [Google Scholar]
- Li Z, Erion DM, Maurer TS. Model-based assessment of plasma citrate flux into the liver: Implications for NaCT as a therapeutic target. CPT Pharmacometrics Syst Pharmacol. 2016;5:132–9. doi: 10.1002/psp4.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Zhuang L, Maddox DM, Smith SB, et al. Structure, function and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J Biol Chem. 2002;277:39469–76. doi: 10.1074/jbc.M207072200. [DOI] [PubMed] [Google Scholar]
- Inoue K, Fei YJ, Zhuang L, Gopal E, et al. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem J. 2004;378:949–57. doi: 10.1042/BJ20031261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011;14:184–95. doi: 10.1016/j.cmet.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.