

Abstract
Idiopathic trigeminal neuralgia (TN) is a debilitating pain disorder characterized by episodic unilateral facial pain along the territory of branches of the trigeminal nerve. Human pain disorders, but not TN, have been linked to gain-of-function mutations in peripheral voltage-gated sodium channels (NaV1.7, NaV1.8 and NaV1.9). Gain-of-function mutations in NaV1.6, which is expressed in myelinated and unmyelinated central nervous system (CNS) and peripheral nervous system neurons and supports neuronal high-frequency firing, have been linked to epilepsy but not to pain. Here, we describe an individual who presented with evoked and spontaneous paroxysmal unilateral facial pain and carried a diagnosis of TN. Magnetic resonance imaging showed unilateral neurovascular compression, consistent with pain in areas innervated by the second branch of the trigeminal nerve. Genetic analysis as part of a phase 2 clinical study in patients with TN conducted by Convergence Pharmaceuticals Ltd revealed a previously undescribed de novo missense mutation in NaV1.6 (c.A406G; p.Met136Val). Whole-cell voltage-clamp recordings show that the Met136Val mutation significantly increases peak current density (1.5-fold) and resurgent current (1.6-fold) without altering gating properties. Current-clamp studies in trigeminal ganglia (TRG) neurons showed that Met136Val increased the fraction of high-firing neurons, lowered the current threshold and increased the frequency of evoked action potentials in response to graded stimuli. Our results demonstrate a novel NaV1.6 mutation in TN, and show that this mutation potentiates transient and resurgent sodium currents and leads to increased excitability in TRG neurons. We suggest that this gain-of-function NaV1.6 mutation may exacerbate the pathophysiology of vascular compression and contribute to TN.
INTRODUCTION
Trigeminal neuralgia (TN) is characterized by recurring episodes of unilateral and lancinating facial pain along one or more branches of the trigeminal nerve (1). Neurovascular compression (NVC) of the trigeminal nerve is prevalent in TN with no other known comorbidities, and has been classically regarded as causing TN (1). Ultrastructural studies on biopsied trigeminal roots showed axonal loss, demyelination and dysmyelination consistent with NVC of the trigeminal nerve (2). However, recent studies have suggested that only severe neurovascular contact, causing displacement or atrophy of the nerve, is associated with pain in classical TN (3,4), and some studies have questioned whether NVC is necessary and sufficient for TN, since pain can occur and recur in the absence of nerve compression (5,6). A recent study reported that TN without NVC is more prevalent in younger individuals (6). These findings suggest contributions of genetic and epigenetic factors to the pathophysiology of classical TN, especially in patients without severe NVC. Rare cases of familial idiopathic trigeminal pain with an autosomal dominant inheritance pattern (7–10) support a genetic component for TN.
The molecular basis for TN has thus far remained elusive. Its pathophysiology has been modeled by the “ignition theory” (11,12), which posits that injured trigeminal neurons fire spontaneously and have a lower threshold for evoked after-discharges leading to paroxysmal pain. Intraoperative microneurographic recordings in trigeminal nerve fibers after stimulation of the known trigger zone in a TN patient demonstrated afterdischarges that were absent when unaffected facial areas were stimulated (13), and consistent with the predictions of ignition theory, the period of intense afterdischarge was followed by a prolonged period refractory to additional stimulation. Voltage-gated sodium channel blockers carbamazepine and oxcarbazepine are the first-line pharmacotherapy for classical TN (1); thus, it is reasonable to suggest an involvement of voltage-gated sodium channels in its pathophysiology.
Rare and common painful disorders, but not TN, have been genetically and functionally linked to gain-of-function mutations in peripheral sodium channels NaV1.7, NaV1.8 and NaV1.9 (14–16). Although gain-of-function mutations of NaV1.6 have been shown to cause infantile epileptic encephalopathy (17–19), there has been no genetic evidence thus far for a role for this channel in human pain disorders. We report here a novel NaV1.6 mutation in an individual with TN. Functional testing shows that the substitution confers gain-of-function attributes on the channel and increases the excitability of trigeminal ganglion neurons that express the mutant channel. This data support a role of NaV1.6 in the pathophysiology of TN, adding to the spectrum of excitability disorders linked to this channel.
MATERIAL AND METHODS
Molecular Genetics
The study was approved by an institutional review board. Genomic analysis was performed as part of a phase 2 clinical study in patients with trigeminal neuralgia conducted by Convergence Pharmaceuticals Ltd (NCT01540630; Tate et al., 2014). Next-generation sequencing utilizing a customized Agilent Haloplex Enrichment Panel and the Illumina MiSeq instrument was done to sequence the exonic regions of the targeted genes encoding voltage-gated sodium channels (NaV1.1, 1.2, 1.3, 1.6 and 1.7) and calcium channels (CaV2.1 and 2.2). Genomic DNA was isolated from whole-blood samples using a Qiagen DNA column-based purification kit. Quality and quantification checks were performed prior to PCR to ensure high-quality DNA. Bioinformatics analysis of the sequencing data and detection of mutations in the target channels were performed using GATK (Broad Institute of Massachusetts Institute of Technology, https://www.broadinstitute.org/gatk/). The incidence of mutations was evaluated in a matching population using the 1000 Genome database.
Plasmid and Animals
A plasmid carrying human NaV1.6 (hNaV1.6), purchased from Genionics, carries optimized codons for expression in mammalian cells. The channel was rendered resistant to tetrodotoxin (TTX-R) by a Tyr371Ser substitution so that it could be studied in voltage-clamp in isolation in ND7/23 cells (a rat DRG/mouse neuroblastoma hybrid cell line) and trigeminal ganglion (TRG) neurons from NaV1.8-null mice (20). The Tyr371Ser and Met136Val substitutions were introduced into the channel using Quick Change XL site-directed mutagenesis kit (Stratagene). The identity of the insert was confirmed by Sanger sequencing at the Keck sequencing facility at Yale University.
Animal care followed a protocol approved by the Institutional Animal Care and Use Committee at the Veterans Administration Connecticut Healthcare System, West Haven. TRG from adult NaV1.8-null mice were isolated and used for recording the resurgent current, while TRGs were isolated from male Sprague-Dawley albino rats (4–6 wks old, Harlan) and used for current-clamp studies.
Transient transfection of ND7/23 cells
hNaV1.6R (WT) and hNaV1.6-M136V (called Met136Val hereafter) were cotransfected with enhanced green fluorescent protein (EGFP) constructs into ND7/23 cells as previously described (21–23). Briefly, cells were plated at low density on 10 mm coverslips in 24-well plates and incubated at 37°C for 24 h before transfection. Cells were cotransfected with WT or Met136Val (0.5 ug/well) and EGFP (0.1 ug/well) (pEGFP, Clontech). Lipofectamine 2000 reagent (1.5 μL) and DNA (0.6 μg total) were each mixed with 50 μL Opti-MEM (serum- and antibiotic-free) and incubated at room temperature (RT, 20° ± 1°C) for 5 min. The two solutions were combined and incubated at RT for 20 min. The combined mixture (100 μL/well) was added to the ND7/23 cell culture medium (0.5 mL Dulbecco’s modified Eagle medium [DMEM] plus 10% fetal bovine serum [FBS] per well) and incubated at 37°C for 3 h. Culture medium was replaced and cells were incubated at 37°C for 48 h before electrophysiological recordings.
Voltage-clamp in ND7/23 cells
ND7/23 cells with robust green fluorescence and WT or Met136Val were recorded on the same day under similar conditions (21–23). Whole-cell voltage-clamp recordings were performed using an EPC10-double amplifier and Patchmaster software (HEKA Electronik) at RT (20° ± 1°C). The extra-cellular solution contained (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), 5 CsCl, 20 TEA-Cl, 0.1 CdCl2, pH 7.3 with NaOH (osmolarity adjusted to 320 mOsm with sucrose). Tetrodotoxin (TTX) (500 nM) was added to the bath to block endogenous TTX-sensitive sodium currents in ND7/23 cells (21–23). Patch pipettes were fabricated from borosilicate glass (1.65/1.1, OD/ID; World Precision Instruments) using a Sutter Instruments P-97 puller and had a resistance of 0.8-1.5 MΩ. Transfected ND7/23 cells were held at –120 mV for all parameters examined. To minimize voltage errors, 70–90% series resistance compensation was applied. Currents were acquired 5 min after establishing whole-cell configuration. Recordings were acquired at 10 kHz with low-pass Bessel filter and digitized at 50 kHz. Linear leak and residual capacitance artifacts were removed using P/6 subtraction.
To measure current-voltage (I-V) relationships, a range of potentials from –80 mV to +40 mV in 5 mV increments were applied from the holding potential (–120 mV) for 100 ms at 5 s intervals. Current density was calculated by normalizing maximal peak currents with cell capacitance. Peak inward currents obtained from activation protocols were converted to conductance values using the equation G = I/(Vm – ENa), where G is the conductance, I is the peak inward current, Vm is the membrane potential used to elicit the response and ENa is the reversal potential for sodium. Conductance data were normalized by the maximum conductance value and fit with a Boltzmann equation, G = Gmin + (Gmax-Gmin)/(1 + exp[Vm-V1/2]/k), where V1/2 is the midpoint of activation and k is the slope factor. Ramp currents were elicited with ramp depolarizations from –120 to +20 mV at a rate of 0.2 mV/ms. The ramp current was normalized to the maximal peak inward current recorded during the activation protocol for each cell.
Steady-state fast inactivation was assessed with a series of 100 ms prepulses (–140 mV to 0 mV in 10 mV increments) followed by a 40 ms step depolarization to –10 mV to activate the remaining noninactivated channels. Peak inward currents from steady-state fast inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation, I = Imin + 1/(1 + exp[Vm-V1/2]/k), where I is the current amplitude measured during the test depolarization, V1/2 is the midpoint of fast-inactivation and k is the slope factor.
Steady-state slow inactivation was assessed with 30 s prepulses at a range of potentials from –120 to +20 mV in 10 mV increments, followed by a 20 ms pulse to –120 mV to allow recovery from fast inactivation, and then a 20 ms depolarizing step to –10 mV to elicit a test response, which reflects the remaining channels available for activation. Peak inward currents from steady-state slow inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation, I = Imin + 1/(1 + exp[Vm-V1/2]/k), where I is the current amplitude measured during the test depolarization, V1/2 is the midpoint of slow inactivation and k is the slope factor
Isolation and transfection of NaV1.8-null TRG neurons
TRG neurons from 4-to-8-wk-old NaV1.8-null mice were harvested and dissociated as previously described (24). Briefly, dissected trigeminal ganglia were placed in ice-cold oxygenated complete saline solution (CSS) (in mM: 137 NaCl, 5.3 KCl, 1 MgCl2, 25 sorbitol, 3 CaCl2, 10 HEPES; pH 7.2). After removal of roots and desheathing, ganglia were minced into small pieces and incubated with gentle agitation at 37°C for 20 min in oxygenated CSS containing 1.5 mg/mL Collagenase A (Roche Applied Science) and 0.6 mM ethylenediaminetetraacetic acid (EDTA), followed by a 17-min incubation with gentle agitation at 37°C in oxygenated CSS containing 1.5 mg/mL Collagenase D (Roche Applied Science), 0.6 mM EDTA and 30 U/mL papain (Worthington Biochemical). The solution was then aspirated and the ganglia carefully triturated in TRG media (DMEM/Fl2 [1:1, Invitrogen] with 100 U/mL penicillin, 0.1 mg/mL streptomycin [Invitrogen], 2 mM L-glutamine [Invitrogen] and 10% FBS [Hyclone]) containing 1.5 mg/mL bovine serum albumin (Sigma-Aldrich) and 1.5 mg/mL trypsin inhibitor (Sigma-Aldrich). After filtration through a 70-μm nylon mesh (BD Biosciences), the cell suspension was centrifuged at 900 × rpm for 3 min and cell pellet was ready for transfection. TRG neurons were cotransfected with EGFP and WT or Met136Val NaV1.6R constructs using Nucleofector IIS (Lonza) and Basic Neuron SCN Nucleofector Kit (VSPI-1003; Amaxa). Recordings were conducted 40–48 h after transfection.
Resurgent current recordings in TRG neurons
Transfected small TRG neurons (20–25 μm) with robust green fluorescence and no apparent neurites were selected for whole-cell patch-clamp recordings of the resurgent current within 40–48 h after transfection, using an EPC10-double amplifier and Patchmaster software (HEKA Electronik) at room temperature (20° ± 1°C). The composition of the extracellular and pipette solutions and physical characteristics of the patch pipettes were the same as described for transfected ND7/23 cells above. TRG neurons were held at –100 mV and linear leak currents were subtracted using the P/6 method. To minimize voltage errors, we applied 80–90% series resistance compensation. Recordings were acquired 8 min after establishing whole-cell configuration to allow steady-state dialysis between the cytoplasm and pipette solution. Recordings were acquired at 10 kHz with low-pass Bessel filter and digitized at 50 kHz.
Resurgent currents were recorded with a two-step protocol where the membrane was depolarized to +30 mV for 20 ms from a holding potential of –100 mV, followed by repolarization pulses to voltages from –10 to –80 mV in 5 mV increments for 100 ms, before returning to the holding potential. Only cells that showed slow rise and slow decay upon repolarization, typical of resurgent current (25), were included in our analysis. Resurgent current density was calculated by normalizing maximal peak resurgent current with cell capacitance.
Isolation and transfection of TRG neurons from adult rat
TRG from adult male Sprague-Dawley albino rats (4–6 wks old, Harlan) were isolated and cultured as described previously (24). WT and Met136Val NaV1.6R channels were transiently transfected into TRG neurons, along with EGFP, by electroporation with AmaxaTM Basic Neuron SCN Nucleofector Kit (Lonza) using program SCN-BNP6 on Nucleofector IIS (Lonza), as described previously (26). The ratio of sodium channel to GFP constructs was 10:1 (2 μg of WT or Met136Val plus 0.2 μg of GFP). Transfected neurons were allowed to recover for 5 min at 37°C in 0.1 mL of calcium-free DMEM (Invitrogen), then diluted in TRG media containing 1.5 mg/mL bovine serum albumin and 1.5 mg/mL trypsin inhibitor. The cell suspension (100 μl) was plated on each of 12 mm circular poly-D-lysine/laminin-coated coverslips (BD Biosciences) and incubated at 37°C in 5% CO2 for 45 min for neurons to attach to the coverslips. TRG media (final volume of 1 mL/well), supplemented with 50 ng/mL (final concentration) each of mNGF (Alomone Labs) and GDNF (Peprotec), was then added and the cells maintained at 37°C in a humidified 5% CO2 incubator.
Current-clamp recordings in rat TRG neurons
Whole-cell current-clamp recordings were performed in transfected TRG neurons. The electrophysiologist was blinded to the sodium channel expressed in the TRG neurons until data were pooled into WT and Met136Val groups. Recordings were acquired at 10 kHz with a low-pass Bessel filter and digitized at 50 kHz using a HEKA EPC10-double amplifier. The extracellular solution contained (in mM): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, 10 dextrose; pH 7.3 with NaOH (osmolarity adjusted to 320 mOsm w/sucrose). Patch pipettes (0.9–1.5 MΩ) were fabricated from borosilicate glass (1.65/1.1, OD/ID; World Precision Instruments) using a Sutter Instruments P-97 puller and fire polished. Pipette solution contained (in mM): 140 KCl, 0.5 EGTA, 5 HEPES, 3 Mg-ATP, 10 dextrose; pH 7.30 with KOH (adjusted to 310 mOsm with sucrose). TTX was not used in the bath for current-clamp recordings. Pipette potential was zeroed before seal formation, and the voltages were not corrected for liquid junction potential.
Whole-cell configuration was obtained in voltage-clamp mode before proceeding to current-clamp mode. Recordings were performed from small neurons (20–30 μm) with ovoid cell bodies and neurite extensions that exhibited GFP fluorescence. All recordings were performed 40–50 h post-transfection at RT (20° ± 1°C) and within an hour after coverslips were transferred to the recording chamber. TRG neurons with stable resting membrane potential ( < 10% variation) more negative than –40 mV and overshooting action potentials ( > 85 mV peak to peak) were used for data analysis. Input resistance was determined by the slope of a linear fit to hyperpolarizing responses to current steps from –5 pA to –40 pA in 5 pA increments. Current threshold was determined from the first action potential elicited by a series of depolarizing current injections (200 ms) that increased in 5 pA increments. Action potential frequency was determined by depolarizing current injections (500 ms). Voltage threshold was determined by assessing the first derivative of the membrane potential (dV/dt) and when the slope reached 20 mV/ms.
Data Analysis
Data were analyzed offline with FitMaster (HEKA Electronik), Excel (Microsoft) and Origin 8.5 (Microcal Software) software and presented as mean ± standard error (SE). Statistical significance was determined by unpaired Student t test for voltage-clamp and current-clamp. Mann-Whitney test was used for comparison of firing frequencies between WT and Met136Val in response to stimuli ranging from 25 to 500 pA. Two-proportion z-test was used for comparison of spontaneous activity. Fisher’s exact test was used for comparing high/low firing groups of cells expressing Met136Val and WT channels.
RESULTS
Clinical Phenotype and Identification of Met136Val Mutation
The subject was a 64-year-old Caucasian female of European heritage. She presented with classical TN in the second branch of the nerve for more than one year, with both spontaneous and evoked pain paroxysms, and no family history of similar symptoms. A diagnosis monitoring committee consisting of three experts on TN (27) confirmed the presence of NVC on the affected side by MRI and the diagnosis of TN. Whole-exome sequencing with good coverage (40X) of a set of genes encoding voltage-gated sodium channels (NaV1.1, 1.2, 1.3, 1.6 and 1.7) and calcium channels (CaV2.1 and 2.2) identified a single nucleotide substitution c.A406G (reference sequence NM_0141913) in the second coding exon of SCN8A, the gene that encodes NaV1.6. This mutation has not been previously reported and is absent from the 1000 Genome database. This mutation substitutes methionine 136 by valine (Met136Val) in transmembrane segment 1 of domain 1 (DI/S1). This residue is highly conserved in all human NaVs except for NaV1.8, where it is leucine (Figure 1). No other mutations in neuronal sodium channels NaV1.1-1.3 and NaV1.7 or CaV2.1 and 2.2 were found in this individual.
Figure 1.

Sequence alignment of domain I transmembrane segment 1. Alignment of the sequence of DI/S1 from human NaV channels shows that Met136 is highly conserved in all human channels except NaV1.8, where it is leucine.
Voltage-Clamp Characterization of the Met136Val Channel
The location of the mutation and its conservation among sodium channels suggested that it can have an effect at the channel level. Sodium currents were recorded in the voltage-clamp mode from ND7/23 cells transiently transfected with either hNaV1.6R WT or Met136Val mutant channels. Figures 2A and B show representative traces of sodium current from WT and Met136Val mutant channels, respectively. The peak current density at –5 mV (Figure 2C) was significantly increased for Met136Val (294 ± 26.5 pA/pF, n = 14) by 1.5-fold compared with WT channels (189.4 ± 11.3 pA/pF, n = 15, p = 0.03). Met136Val did not alter the voltage-dependence of activation (V1/2: WT, –19.1 ± 0.2 mV, k = 6.5 ± 0.2, n = 9; Met136Val, –16.8 ± 0.1 mV, k = 6.9 ± 0.1, n = 10), fast inactivation (V1/2: WT, –62.5 ± 0.5 mV, k = 5.3 ± 0.3, n = 12; Met136Val, –64.2 ± 0.8 mV, k = 5.3 ± 0.4, n = 12) (Figure 2D), or slow inactivation (V1/2: WT, –47.3 ± 1.0, k = 10.1 ± 0.7, n = 10; Met136Val, –48.0 ± 1.5, k = 8.8 ± 1.1, n = 9) (Figure 2E).
Figure 2.

Met136Val mutation potentiates current density in ND7/23 cells. Representative current traces of sodium currents recorded from ND7/23 cells expressing either WT (A) or Met136Val (B) mutant channels. Cells were held at –120 mV and inward sodium currents were elicited by step depolarizations from –80 mV to +20 mV in 5 mV increments every 5 s. (C) Peak current-voltage relationship for WT (n = 15) and Met136Val (n = 14) channels showing significant increase in peak current density of M136V mutant channels. (D) Comparison of voltage-dependent activation and steady-state fast inactivation of WT and Met136Val mutant channels. (E) Comparison of steady-state slow inactivation of WT and Met136Val mutant channels. Cells were held at –120 mV and a series of depolarizing pulses (30 s) from –120 mV to +20 mV in 10 mV increments was applied, followed by 20 ms return to –120 mV and then a 20 ms pulse to –10 mV to elicit a test response. Data represent mean ± SE.
We examined the response of WT and Met136Val channels to slow depolarizations using a ramp stimulus from a holding potential of –120 mV to +20 mV over 600 ms for a ramp rate of 0.2 mV/ms. The ramp current was normalized to the maximal peak inward current recorded during the activation protocol for each cell. Peak ramp current of Met136Val measured 1.6 ± 1.3% (n = 6) of the peak current, and was not significantly different from WT (3.0 ± 2.2%, n = 6). Persistent current, measured 70 ms after the onset of a step depolarization to –10 mV and normalized to the peak inward current, also showed no significant difference (WT: 1.6 ± 0.2%, n = 15; Met136Val: 1.5 ± 0.3%, n = 14).
Resurgent Current of Met136Val Channel in TRG Neurons
NaV1.6 channels produce resurgent currents that facilitate repetitive firing of neurons, including DRG neurons (25, 28–30). To examine whether enhanced peak current density of NaV1.6 channels is paralleled by increased resurgent current in the native neuronal background, we expressed WT and Met136Val channels in NaV1.8-null TRG neurons. Representative traces of resurgent sodium currents are shown from WT (Figure 3A) and Met136Val mutant channels (Figure 3B). Resurgent currents were elicited by briefly depolarizing to +30 mV for 20 ms from a holding potential of –100 mV, followed by a range of repolarization pulses from –10 to –80 mV. Cells that showed slow rise and slow decay, typical of resurgent current and distinct from tail currents (25,28), were used for analysis.
Figure 3.
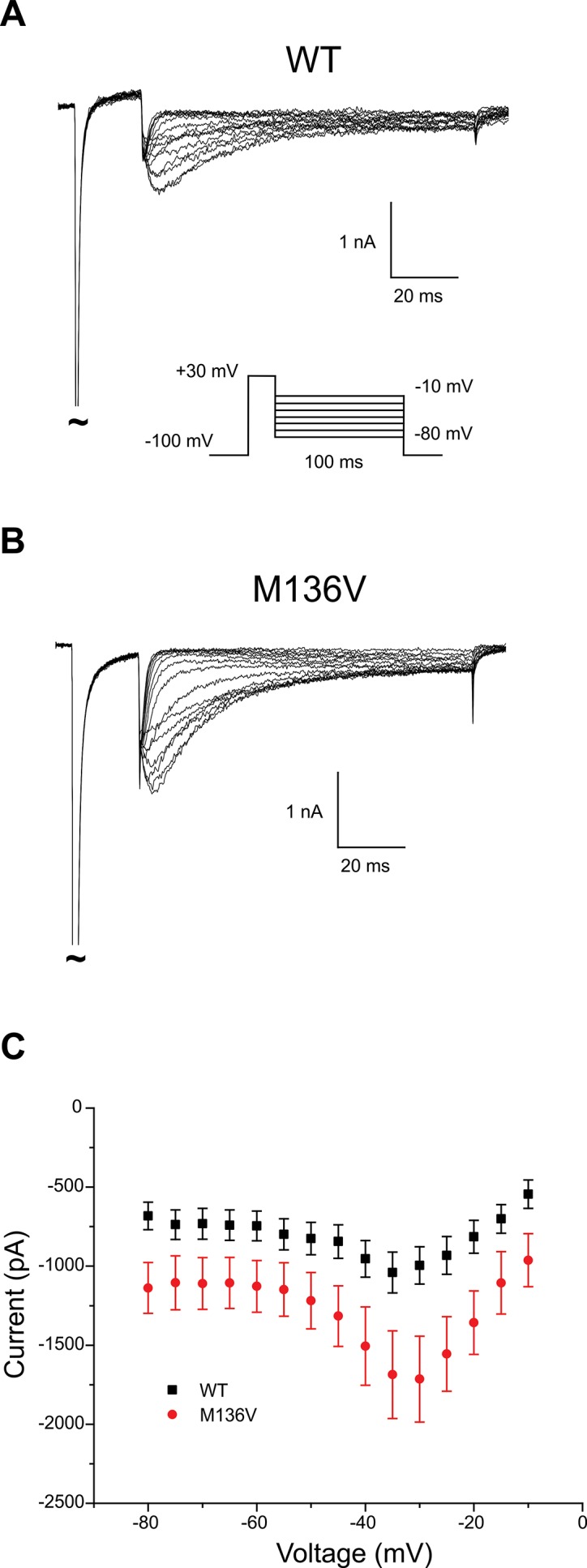
Resurgent sodium current increased in NaV1.8-null TRG neurons expressing Met136Val channels. Representative traces of resurgent currents recorded from NaV1.8-null TRG neurons expressing either WT (A) or Met136Val (B) mutant channels. TRG neurons were held at –100 mV and depolarized to +30 mV for 20 ms, followed by repolarization to a range of pulses (–10 mV to –80 mV) to elicit resurgent current. (C) Average peak amplitudes of resurgent current recorded from WT (n = 12) and Met136Val (n = 15) show an average 60% increase in NaV1.8-null TRG neurons expressing Met136Val mutant channels. Data represent mean ± SE.
Resurgent current was observed in 71% of NaV1.8-null TRG neurons transfected with Met136Val (15 of 21 cells), whereas 50% of neurons transfected with the WT channels (12 of 24 cells) showed resurgent currents. The range in peak amplitude of the resurgent current was larger in neurons transfected with Met136Val (0.7 to 4.8 nA, n = 15) compared with WT channels (0.3 to 2.0 nA, n = 12). The average peak amplitude of the resurgent current was significantly larger (1.6X) in neurons transfected with Met136Val compared with WT channels (Met136Val: 1.8 ± 0.3 nA, n = 15; WT: 1.1 ± 0.1 nA, n = 12; p < 0.05) (Figure 3C). Adjusting for cell capacitance, the peak resurgent current density was significantly increased in Met136Val mutant channels (89.8 ± 11.5 pA/pF, n = 15; p < 0.05) compared with NaV1.8-null TRG neurons transfected with WT channels (59.5 ± 6.1 pA/pF, n = 12).
Current-Clamp Analysis of TRG Neurons
To assess the effects of Met136Val on the excitability of adult TRG neurons, which express a full complement of sodium channels, current-clamp recordings were performed on TRG neurons transfected with Met136Val or WT channels. Input resistance was not significantly different among neurons expressing Met136Val (649 ± 75 MΩ, n = 29) and WT channels (601 ± 93 MΩ, n = 29). The resting potential of TRG neurons expressing Met136Val channels (–56.7 ± 1.2 mV, n = 29) was not significantly different from that of TRG neurons expressing WT channels (–57.1 ± 0.8 mV, n = 29).
To assess the effects of the variant channels on the current threshold of TRG neurons, we injected a series of depolarizing current pulses to evoke the first all-or-none action potential. TRG neurons that spontaneously fired were excluded from the analysis of evoked data. The current threshold of TRG neurons expressing Met136Val channels was significantly reduced compared with neurons expressing WT channels (Met136Val: 88.4 ± 10.2 pA, n = 29; WT: 128.3 ± 15.6 mV, n = 29; p < 0.05).
Voltage thresholds were determined by measuring the voltage at which the slope of the voltage trace (dV/dt) increases sharply at 20 mV/ms. Voltage threshold in TRG neurons expressing Met136Val channels (–20.3 ± 1.5 mV, n = 29) was similar to neurons expressing WT channels (–19.2 ± 1.5 mV, n = 29). Action potential amplitude was measured as the difference between the resting potential and the peak of the action potential. The action potential amplitude of TRG neurons expressing Met136Val channels (112.0 ± 2.5 mV, n = 29) was not significantly different from that in neurons expressing WT channels (112.4 ± 2.2 mV, n = 29). The half-width of action potentials evoked in TRG neurons expressing Met136Val channels (2.8 ± 0.2 ms, n = 29) was not significantly different from neurons expressing WT channels (WT: 3.0 ± 0.3 ms, n = 29).
The effect of the mutation on the excitability of TRG neurons was also assessed by analyzing the number of action potentials evoked by a series of depolarizing current injections (25–500 pA) for 500 ms. TRG neurons expressing Met136Val channels fired a greater number of action potentials compared with WT channels in response to similar stimuli. Figure 4 shows representative responses of TRG neurons expressing WT or Met136Val channels evoked at one, two and three times the current threshold. Repetitive spike firing significantly increased in neurons expressing Met136Val between 100 pA and 475 pA current injections compared with WT (Figure 4B, p < 0.05). Interestingly, a scatter plot of the number of maximal action potentials fired during the series of depolarizing current injections shows low- and high-firing groups for TRG neurons that express WT or Met136Val channels. Low-firing neurons (8 cells in each group) are comparable between the two groups, firing 1–6 APs. The majority of the high-firing neurons (10 cells) expressing WT channels fired 8–21 APs and only 2 neurons fired > 15 APs. In contrast, all high-firing neurons that expressed the Met136Val (14 cells) fired ≥ 15 APs. Using a threshold of 15 APs, there were significantly more high-firing cells in TRG neurons expressing Met136Val channels (14/22) compared with WT channels (2/18, p = 0.001; Table 1).
Figure 4.

Met136Val channels increase excitability in TRG neurons. (A) Representative responses of TRG neurons expressing WT or Met136Val channels to 500 ms sustained depolarizations in current steps one, two and three times (top, middle, bottom traces, respectively) the current threshold for action potential generation. (B) Comparison of repetitive action potential firing between TRG neurons expressing WT (black; n = 18) and Met136Val (red; n = 22) across a series of 500 ms current injections from 25–500 pA; p < 0.05.
Table 1.
Proportion of low- and high-firing TRG neurons expressing Met136Val or WT channels.
| hNaV1.6R | hNaV1.6R-M136V | |
|---|---|---|
| Low-firing | 16 | 8 |
| High-firing | 2 | 14* |
| P value 0.001 | ||
Neurons that are transfected with NaV1.6 channels demonstrate a propensity for spontaneous firing. Spontaneous action potentials recorded for 30 s in a representative TRG neuron expressing Met136Val channels is shown in Figure 5A, as well as an expansion of the time scale of the same trace illustrating a single action potential (Figure 5B). Spontaneous firing was observed in 41% and 48% of TRG neurons transfected with Met136Val and WT channels, respectively (Met136Val, 20/49 cells; WT, 27/56 cells, p = 0.40; Figure 5C). TRG neurons expressing Met136Val or WT channels fired spontaneously at comparable frequencies of 4.2 ± 0.9 Hz and 4.2 ± 0.8 Hz, respectively. A single action potential from each spontaneously firing cell was used to determine the voltage threshold. Although the voltage threshold of spontaneously firing cells was determined to be similar between TRG neurons expressing Met136Val or WT channels (Met136Val: –28.3 ± 1.2 mV, n = 20; WT: –28.7 ± 0.9 mV, n = 27), this was much lower than the voltage threshold measured for evoked action potentials by approximately –10 mV.
Figure 5.
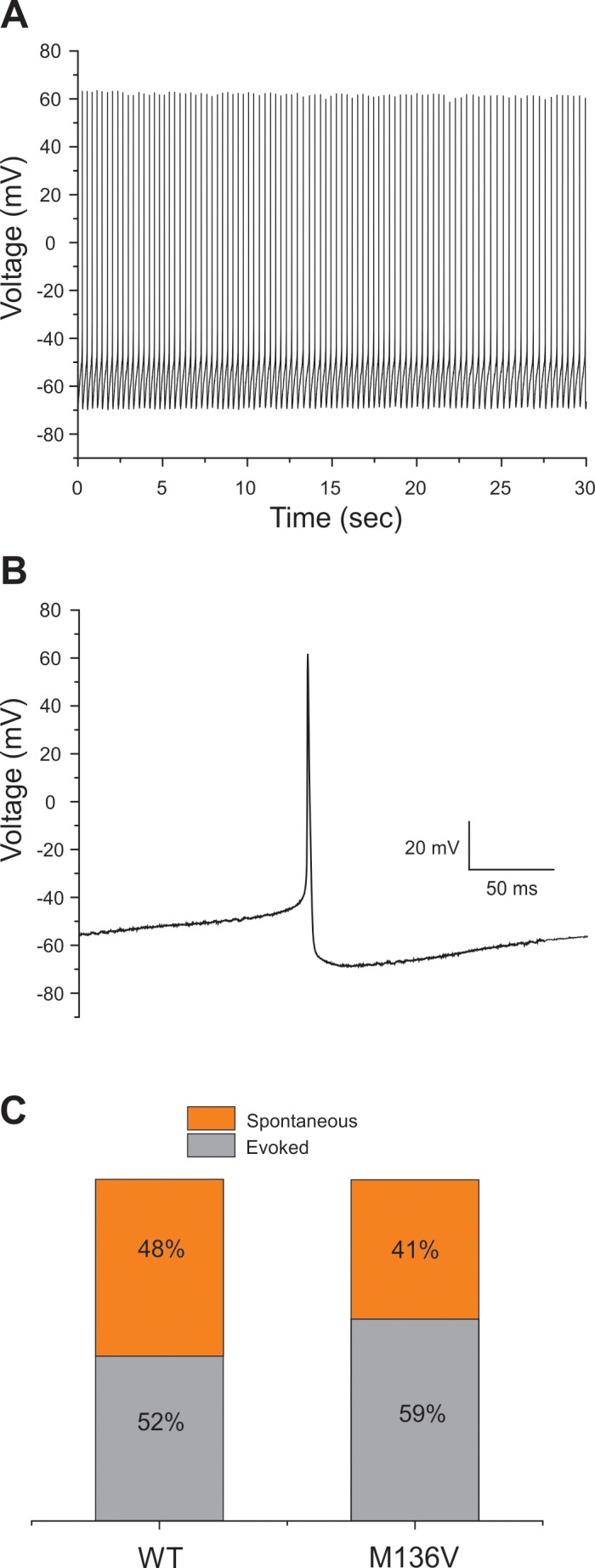
Spontaneous firing of TRG neurons transfected with Met136Val mutant channels. (A) Representative current-clamp traces of spontaneous firing in TRG neurons expressing Met136Val mutant channels. The traces were recorded for 30 s without current injection. (B) Expansion of a single action potential from a spontaneously firing cell expressing M136V channels shown in (A). (C) Bar graphs showing the proportion of spontaneous firing cells (orange) compared with evoked cells (gray) for TRG neurons expressing WT and Met136Val mutant channels. Numbers in the bar show the percentage values for WT and Met136Val mutant channels.
DISCUSSION
Genetic and functional evidence support a role for the SCN8A c.406A > G (p.Met136Val) mutation in TN. The mutation substitutes a highly conserved residue in DI/S1 of the channel, and has not been reported in the 1000 Genome database. The Met136Val substitution produces a significant increase in peak transient and resurgent currents of NaV1.6, reduces the threshold for action potential in TRG neurons, and enhances the neuronal evoked response and the fraction of neurons that fire at a higher rate than those expressing WT channels. Compared to other DI-S1 sodium channel mutations that have been linked to familial febrile seizures (31), erythromelalgia (32) and myotonia (33), the Met136Val mutation does not alter gating properties, which points to isoform-specific effects of mutations in this transmembrane segment. The increased peak transient and resurgent currents that confer hyperexcitability at the neuronal level combined with the rare occurrence of this mutation increase our confidence in concluding that Met136Val is a clinically significant variant that contributes to the pathophysiology of TN.
The paroxysmal nature of TN, the occurrence of intense afterdischarges and favorable response to carbamazepine are consistent with a role for voltage-gated sodium channels. NaV1.6 channels are predominant at axon initial segments (AISs) and mature nodes of Ranvier in myelinated fibers of the central and peripheral nervous system (34–37). NaV1.6 is also expressed in small DRG neurons that produce unmyelinated C-fibers, and total loss of this channel in DRG neurons slows C-fiber conduction in the sciatic nerve (38), suggesting that it contributes to the transmission of noxious signals in these fibers. We have previously shown that NaV1.6 is widely expressed in TRG neurons, giving rise to myelinated and unmyelinated fibers, and is localized in the nerve endings of unmyelinated fibers in the cornea (38), which are thought to be almost entirely nociceptive (39). Animal studies support a role for NaV1.6 in nerve injury models (40), toxin-induced pain (41), chemotherapy-induced pain (42) and local DRG inflammatory pain (43,44). Thus, NaV1.6 is poised to play an important role in the pathophysiology of chronic pain, including in TN.
NaV1.6 channels manifest distinctive gating properties consistent with a critical role in regulating firing in the different neurons in which they are expressed. NaV1.6 produces a rapidly repriming fast-inactivating TTX-S current (45–47) and a persistent current (47,48), which allow this channel to contribute to the overshoot of action potential in sensory neurons (49). Voltage-clamp recordings show that the Met136Val mutation significantly increases the peak current density. The increase in current density may have contributed to the reduction in current threshold for firing action potentials (~ 30%).
NaV1.6 produces a resurgent current in both CNS neurons and DRG neurons (28,29) as a result of channel reopening upon repolarization of the membrane (25). The resurgent current shortens refractory periods, thus supporting repetitive and burst firing of neurons (50), and has been implicated in human pain conditions caused by mutations in NaV1.7 (30,51,52) and in an animal model of radicular pain induced by local DRG inflammation (53). Our data show that Met136Val channels produce, on average, a 1.6-fold increase in the resurgent current in TRG neurons; the range of resurgent current amplitude in cells expressing WT channels is 0.3–2.4 nA, whereas resurgent currents in neurons that express the Met136Val channels range from 0.7–4.8 nA. We previously showed that roughly half of the small DRG neurons that are transfected with WT NaV1.6 channels produce resurgent currents (29). This is consistent with data showing that production of resurgent currents is cell-type-dependent (50), suggesting a cell-type-specific expression of an open channel blocker necessary for generating the resurgent current. The low-firing group of TRG neurons detected by the repetitive firing protocol possibly do not express an endogenous open channel blocker and do not produce a resurgent current, but such a blocker is likely present in the high-firing group of TRG neurons that produce a resurgent current. The increase in resurgent current of Met136Val channels provides the physiological basis for the enhanced evoked firing of TRG neurons that express these channels.
One possible mechanism that could link the mutation described here to TN involves ephaptic transmission, or electrotonic cross-talk between nearby axons, which has been proposed (54,55) as a mechanism contributing to hemifacial spasm. There have been multiple demonstrations of ephaptic interactions that can lead to ectopic impulse activity, not only between closely apposed uninjured axons (56) but also between injured axons (57,58) and between neighboring dysmyelinated axons (59). It has been suggested that demyelination due to compression is present in some cases of trigeminal neuralgia (2,60,61). It is well established that myelinated axons can produce large external electrical fields in regions where myelin is focally absent (62). Moreover, it has been demonstrated that increases in sodium channel density increase the likelihood of action potential generation in demyelinated axons (63).
The significant hyperexcitability of TRG neurons in which the Met136Val mutant channels are expressed is in agreement with the predictions of the ignition theory of TN. According to ignition theory, TN is triggered by ectopic afterdischarges that are either spontaneous or low-threshold evoked phenomena leading to paroxysmal pain (11,12). An increase in peak transient and resurgent current could be linked to the evoked repetitive firing, which would be consistent with the evoked pain phenomenon in TN. The high prevalence of spontaneous firing of TRG neurons overexpressing WT channels may have masked the real effect of mutant channels in rendering TRG neurons spontaneously firing when both WT and mutant channels were present at physiological levels. Additionally, we cannot formally discount the possible role of inflammatory mediator sensitization of primary afferents in TN due to NVC, which could lead to spontaneous excitability of TRG neurons.
CONCLUSION
The genetic and functional data presented in this study support a link of a gain-of-function mutation in NaV1.6 to human pain disorders, expanding the clinical-genetic spectrum of excitability disorders caused by this channel. Early-onset seizures and intellectual disabilities associated with human NaV1.6 mutations in infantile epileptic encephalopathy (64) may mask any pain phenotype in these individuals. Whole-exome screening of a number of relevant ion channel genes and functional testing in relevant cell background increase confidence in the utility of this approach for discovering pathogenic mutations in pain disorders of unknown etiology.
ACKNOWLEDGMENTS
We thank the subject for participating in this study. We thank Lawrence Macala and Palak Shah for technical support. This work was supported by the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs (SGW and SDH), and by funding to Yale University from Convergence Pharmaceuticals. The Center for Neuroscience and Regeneration Research is a collaboration of the Paralyzed Veterans of America with Yale University.
Footnotes
Online address: http://www.molmed.org
Disclosure
VM and ST are employees of Convergence Pharmaceuticals, a Biogen company.
Cite this article as: Tanaka BS, et al. (2016) A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol. Med. 22:338–48.
REFERENCES
- Cruccu G, Bonamico LH, Zakrzewska JM. Cranial neuralgias. Handb Clin Neurol. 2010;97:663–78. doi: 10.1016/S0072-9752(10)97056-5. [DOI] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R, Rappaport ZH. Mechanism of trigeminal neuralgia: an ultrastructural analysis of trigeminal root specimens obtained during microvascular decompression surgery. J Neurosurg. 2002;96:532–43. doi: 10.3171/jns.2002.96.3.0532. [DOI] [PubMed] [Google Scholar]
- Maarbjerg S, Wolfram F, Gozalov A, Olesen J, Bendtsen L. Significance of neurovascular contact in classical trigeminal neuralgia. Brain. 2015;138:311–19. doi: 10.1093/brain/awu349. [DOI] [PubMed] [Google Scholar]
- Antonini G, et al. Magnetic resonance imaging contribution for diagnosing symptomatic neurovascular contact in classical trigeminal neuralgia: a blinded case-control study and meta-analysis. Pain. 2014;155:1464–71. doi: 10.1016/j.pain.2014.04.020. [DOI] [PubMed] [Google Scholar]
- Lee A, McCartney S, Burbidge C, Raslan AM, Burchiel KJ. Trigeminal neuralgia occurs and recurs in the absence of neurovascular compression. J Neurosurg. 2014;120:1048–54. doi: 10.3171/2014.1.JNS131410. [DOI] [PubMed] [Google Scholar]
- Ko AL, et al. Trigeminal neuralgia without neurovascular compression presents earlier than trigeminal neuralgia with neurovascular compression. J Neurosurg. 2015;123:1519–27. doi: 10.3171/2014.11.JNS141741. [DOI] [PubMed] [Google Scholar]
- Bozkurt M, et al. Management of bilateral trigeminal neuralgia with trigeminal radiofrequency rhizotomy: a treatment strategy for the life-long disease. Acta Neurochir (Wien) 2012;154:785–91. doi: 10.1007/s00701-012-1311-7. discussion 791–2. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yu CY, Huang L, Riederer F, Ettlin D. Familial neuralgia of occipital and intermedius nerves in a Chinese family. J Headache Pain. 2011;12:497–500. doi: 10.1007/s10194-011-0350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer F, Sandor PS, Linnebank M, Ettlin DA. Familial occipital and nervus intermedius neuralgia in a Swiss family. J Headache Pain. 2010;11:335–8. doi: 10.1007/s10194-010-0207-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Otmani H, Moutaouakil F, Fadel H, Slassi I. [Familial trigeminal neuralgia] Rev Neurol (Paris) 2008;164:384–7. doi: 10.1016/j.neurol.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self-sustaining discharge in the trigeminal ganglion. Pain. 1994;56:127–38. doi: 10.1016/0304-3959(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Devor M, Amir R, Rappaport ZH. Pathophysiology of trigeminal neuralgia: the ignition hypothesis. Clin J Pain. 2002;18:4–13. doi: 10.1097/00002508-200201000-00002. [DOI] [PubMed] [Google Scholar]
- Burchiel KJ, Baumann TK. Pathophysiology of trigeminal neuralgia: new evidence from a trigeminal ganglion intraoperative microneurographic recording. Case report. J Neurosurg. 2004;101:872–3. doi: 10.3171/jns.2004.101.5.0872. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The NaV1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Waxman SG. Translational pain research: Lessons from genetics and genomics. Sci Transl Med. 2014;6:249sr244. doi: 10.1126/scitranslmed.3007017. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Black JA, Waxman SG. NaV1.9: a sodium channel linked to human pain. Nat Rev Neurosci. 2015;16:511–19. doi: 10.1038/nrn3977. [DOI] [PubMed] [Google Scholar]
- Blanchard MG, et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet. 2015;52:330–7. doi: 10.1136/jmedgenet-2014-102813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis. 2014;69:117–23. doi: 10.1016/j.nbd.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, et al. De Novo Pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–10. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling LC, et al. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain. 2005;113:27–36. doi: 10.1016/j.pain.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Wittmack EK, et al. Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J Neurosci. 2004;24:6765–75. doi: 10.1523/JNEUROSCI.1628-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci. 2005;25:6621–30. doi: 10.1523/JNEUROSCI.0541-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser A, et al. Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J Biol Chem. 2010;285:26149–61. doi: 10.1074/jbc.M109.098681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, et al. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain. 2011;7:92. doi: 10.1186/1744-8069-7-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–26. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, et al. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009;4:1118–26. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- Zakrzewska JM, et al. Novel design for a phase IIa placebo-controlled, double-blind randomized withdrawal study to evaluate the safety and efficacy of CNV1014802 in patients with trigeminal neuralgia. Trials. 2013;14:402. doi: 10.1186/1745-6215-14-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–91. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Letters. 2005;579:2166–70. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest. 2010;120:369–78. doi: 10.1172/JCI40801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures; Proc Natl Acad Sci USA; 2005. pp. 18177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Dib-Hajj SD, Tyrrell L, Waxman SG. Mutation I136V alters electrophysiological properties of the NaV1.7 channel in a family with onset of erythromelalgia in the second decade. Mol Pain. 2008;4:1. doi: 10.1186/1744-8069-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez S, et al. A novel dominant mutation of the Nav1.4 alpha-subunit domain I leading to sodium channel myotonia. Neurology. 2008;71:1669–75. doi: 10.1212/01.wnl.0000335168.86248.55. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Nav1.6 is localized at nodes of ranvier, dendrites, and synapses; Proc Natl Acad Sci USA; 2000. pp. 5616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Custer AW, Shrager P, Rasband MN. Early events in node of Ranvier formation during myelination and remyelination in the PNS. Neuron Glia Biol. 2006;2:69–79. doi: 10.1017/S1740925X06000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, et al. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Mol Brain Res. 1996;43:117–31. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–47. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Black JA, Renganathan M, Waxman SG. Sodium channel Nav1.6 is expressed along nonmyelinated axons and it contributes to conduction. Mol Brain Res. 2002;105:19–28. doi: 10.1016/s0169-328x(02)00385-6. [DOI] [PubMed] [Google Scholar]
- Chen X, Gallar J, Pozo MA, Baeza M, Belmonte C. CO2 stimulation of the cornea: a comparison between human sensation and nerve activity in polymodal nociceptive afferents of the cat. Eur J Neurosci. 1995;7:1154–63. doi: 10.1111/j.1460-9568.1995.tb01105.x. [DOI] [PubMed] [Google Scholar]
- Henry MA, Freking AR, Johnson LR, Levinson SR. Sodium channel Nav1.6 accumulates at the site of infraorbital nerve injury. BMC Neurosci. 2007;8:56. doi: 10.1186/1471-2202-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis JR, et al. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Na1.6 in peripheral pain pathways. Pain. 2013;154:1749–57. doi: 10.1016/j.pain.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, et al. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype NaV1.6-resurgent and persistent current; Proc Natl Acad Sci USA; 2012. pp. 6704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel Na1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain. 2013;154:1170–80. doi: 10.1016/j.pain.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Zhang JM. Local knockdown of the Na1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience. 2015;291:317–30. doi: 10.1016/j.neuroscience.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol (Lond) 2003;551:741–50. doi: 10.1113/jphysiol.2003.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangameswaran L, et al. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem. 1997;272:14805–9. doi: 10.1074/jbc.272.23.14805. [DOI] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurons. J Physiol (Lond) 2005;564:803–15. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol (Lond) 2007;579(Pt 1):1–14. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Resurgent current of voltage-gated Na(+) channels. J Physiol. 2014;592:4825–38. doi: 10.1113/jphysiol.2014.277582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CG, et al. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012a;71:26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- Han C, et al. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain. 2012b;135:2613–28. doi: 10.1093/brain/aws187. [DOI] [PubMed] [Google Scholar]
- Xie W, et al. Upregulation of the sodium channel NaVbeta4 subunit and its contributions to mechanical hypersensitivity and neuronal hyperexcitability in a rat model of radicular pain induced by local DRG inflammation. Pain. 2016;157:879–91. doi: 10.1097/j.pain.0000000000000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen VK. Pathophysiology of hemifacial spasm: I. Ephaptic transmission and ectopic excitation. Neurology. 1984;34:418–26. doi: 10.1212/wnl.34.4.418. [DOI] [PubMed] [Google Scholar]
- Sanders DB. Ephaptic transmission in hemifacial spasm: a single-fiber EMG study. Muscle Nerve. 1989;12:690–4. doi: 10.1002/mus.880120810. [DOI] [PubMed] [Google Scholar]
- Ramon F, Moore JW. Ephaptic transmission in squid giant axons. Am J Physiol. 1978;234:C162–69. doi: 10.1152/ajpcell.1978.234.5.C162. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Devor M. Ephaptic transmission in chronically damaged peripheral nerves. Neurology. 1979;29:1061–4. doi: 10.1212/wnl.29.7.1061. [DOI] [PubMed] [Google Scholar]
- Lisney SJ, Pover CM. Coupling between fibres involved in sensory nerve neuromata in cats. J Neurol Sci. 1983;59:255–64. doi: 10.1016/0022-510x(83)90043-6. [DOI] [PubMed] [Google Scholar]
- Rasminsky M. Ephaptic transmission between single nerve fibres in the spinal nerve roots of dystrophic mice. J Physiol. 1980;305:151–69. doi: 10.1113/jphysiol.1980.sp013356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Hilton DA, Coakham HB. Central demyelination of the Vth nerve root in trigeminal neuralgia associated with vascular compression. Brain Pathol. 1998;8:1–11. doi: 10.1111/j.1750-3639.1998.tb00126.x. discussion 11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagares A, Rivas JJ, Jimenez L, Cicuendez M, Avendano C. Central demyelination in the pathogenesis of trigeminal neuralgia associated with cerebellopontine angle tumors: case report with ultrastructural trigeminal root analysis. Neurosurgery. 2010;66:E841–42. doi: 10.1227/01.NEU.0000367550.30165.A7. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Pappas GD, Bennett MV. Morphological correlates of functional differentiation of nodes of Ranvier along single fibers in the neurogenic electric organ of the knife fish Stern archus. J Cell Biol. 1972;53:210–24. doi: 10.1083/jcb.53.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Wood SL. Impulse conduction in inhomogeneous axons: effects of variation in voltage-sensitive ionic conductances on invasion of demyelinated axon segments and preterminal fibers. Brain Res. 1984;294:111–22. doi: 10.1016/0006-8993(84)91314-3. [DOI] [PubMed] [Google Scholar]
- Wagnon JL, Meisler MH. Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Front Neurol. 2015;6:104. doi: 10.3389/fneur.2015.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]