

Abstract
The goal of this study was to determine the effects of PGZ and MK886 on the mRNA expression of PPARα and other associated genes in MDA-MB-231 cells, and the biological mechanisms induced by both drugs were also assessed. The levels of PPARα mRNA expression in PGZ-treated and MK886-treated MDA-MB-231 cells were determined using real-time PCR; the growth inhibitory effects of PGZ and MK886 were determined using the trypan blue exclusion assay; the induction of apoptosis by PGZ and MK886 was determined using DNA fragmentation assay and real-time PCR; and the invasion of PGZ-treated and MK886-treated MDA-MB-231 cells was determined using the wound healing and transwell migration assays. In addition, we correlated the expression of PPARα mRNA with other genes, including PPARγ, FGF4 and 5LOX, in drug-treated MDA-MB-231 cells. Our results demonstrated that the treatment of MDA-MB-231 cells with PGZ increased the expression of PPARα/γ mRNA and that this expression could be inhibited by treatment with MK886. Both drugs reduced the viability of MDA-MB-231 cells independently of PPARα/γ mRNA expression but did not induce apoptosis. The wound caused by invasion was not healed by PGZ-treated MDA-MB-231 cells, but it was healed by MK886-treated cancer cells, indicating that the reduction of invasion in PGZ-treated MDA-MB-231 cells was eliminated by treatment with MK886, and this finding was validated by the transwell migration assay. This phenomenon might also be associated with the expression of PPARα/γ, FGF4 and 5LOX mRNA in the treated cancer cells. This study provides useful information regarding the mRNA expression levels of PPARα and other related genes in MDA-MB-231 cells. These genes could be attractive targets for reducing the invasion of breast cancer.
Keywords: Pioglitazone, MK886, Cell invasion, PPARα, MDA-MB-231
Introduction
Peroxisome proliferator-activated receptor alpha (PPARα) plays pivotal roles in peroxisome proliferation, anti-inflammation, keratinocyte differentiation and proliferation, skin wound healing, lipid metabolism and homeostasis through the formation of a heterodimer with the retinoid X receptor (RXR) (Suchanek et al. 2002; Tachibana et al. 2008). This heterodimer binds specifically to peroxisome proliferator response elements (PPREs) in the promoter regions of peroxisome proliferator (PP)-regulated genes (Roberts et al. 1998). As a result, the activated genes promote an increase in the number and size of peroxisomes in addition to an increase in peroxisomal lipid metabolizing enzymes involved in the catalyzation of fatty acid oxidation (Peters et al. 1997). The important roles for PPARα in physiology and pathophysiology have remained elusive. PPARα could be significantly involved in specific stages of diseases, such as in breast cancer, which shows an abnormal expression of genes related to fatty acids or environmental xeno-oestrogens. Several studies have determined that essential fatty acids, including n-3 and n-6 fatty acids, play important roles in breast tumour growth (Bocca et al. 2008). The n-3 fatty acids protect human breast cancer cells against apoptosis (Wu et al. 2005), whereas n-6 fatty acids induce the growth and metastasis of human breast cancer cells (Lanson et al. 1990). In contrast, arachidonic acid (AA) induces apoptosis in human breast cancer cell lines at certain extents, e.g. AA induces apoptosis in MCF-7 and MDA-MD-231 cells via the inhibition of the cell proliferation signal transduction pathway and the activation of PPARs, particularly PPARα and PPARγ (Bocca et al. 2008). The involvement of PPARs during apoptosis was further demonstrated by the fact that the overexpression of a dominant negative PPARα abolished its apoptosis-promoting activity (Roberts et al. 1998).
PPARα is detectable at low levels in the mouse mammary gland during pregnancy and lactation, and at high levels in 2-week-old mice, suggesting the possible involvement of PPARα in hormonal regulation (Master et al. 2002). The relationship between PPARα and tumourigenic mammary glands was first determined by Roberts-Thomson and Snyderwine (2000), who demonstrated that the PPARα mRNA expression level was significantly increased in rat mammary gland carcinomas compared to normal rat mammary gland epithelial cells. A similar study showed that PPARα could be detected in MDA-MB-231 and MCF-7 human breast cancer epithelial cell lines (Suchanek et al. 2002). PPARα mRNA was detected at higher levels in MDA-MB-231 versus MCF-7 cells. Moreover, the dynamic regulation of PPARα was observed in the MDA-MB-231 cell line, but not in the MCF-7 cell line, indicating that MDA-MB-231 cells might undergo cell cycle-dependent and PPARα-dependent changes in gene expression. Although the level of PPAR expression was lower in MCF-7 cells, the activation of PPARα significantly induced the proliferation of both MDA-MB-231 and MCF-7 cells (Suchanek et al. 2002). However, the precise role of PPARα and its mechanism of action in breast cancer cells remain unclear.
Here, we studied the mechanisms involved when PPARα expression is induced or inhibited in a human breast cancer cell line. Our preliminary study found that PPARα was significantly up-regulated in pioglitazone (PGZ)-treated MDA-MB-231 cells, as assessed by real-time PCR. PGZ is a selective PPARγ ligand that has been demonstrated to reduce proliferation by inducing apoptotic cell death in a variety of cancer cells (Chaffer et al. 2006). In the present study, we aimed to determine the following: (1) the mRNA expression of PPARα in PGZ-treated and MK886-treated MDA-MB-231 cells using real-time PCR; (2) the growth inhibitory effects of PGZ and MK886 on MDA-MB-231 cells using the trypan blue exclusion assay; (3) the induction of apoptosis by PGZ and MK886 in MDA-MB-231 cells using the DNA fragmentation assay and real-time PCR; and (4) the invasion of PGZ-treated and MK886-treated MDA-MB-231 cells using the wound healing and transwell migration assays. We also correlated the expression of PPARα mRNA with the expression of other mRNAs, such as PPARγ, fibroblast growth factor 4 (FGF4) and 5-lipoxygenase (5LOX), in PGZ-treated and MK886-treated MDA-MB-231 using real-time PCR. MK886 was used in this study to inhibit the expression of PPARα mRNA, as the inhibition of PPAR activity by MK886 was quite pronounced for the α-subtype, while there was substantially less inhibition observed on the β and γ isoforms (Kehrer et al. 2001). This study provides useful information concerning the expression of PPARα mRNA in breast cancer cells, and PPARα could be an attractive target for gene-mediated therapy in human breast cancers and other malignancies.
Materials and methods
Culture of MDA-MB-231 cells
The MDA-MB-231 cell line used in this study was a kind gift from the Johns Hopkins Research Center (Baltimore, MD, USA). The cells were maintained in DMEM (Gibco BRL, Gaithersburg, MD, USA) supplemented with 10 % (v/v) FBS (Gibco BRL, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco BRL, USA). MycoKill (1 %) (PAA Laboratories GmbH, Chalfont St Giles, UK) was added to the growth medium occasionally to prevent mycoplasma contamination. The growth medium was changed every 2-3 days, and the cells were maintained at 37 °C in a humidified atmosphere of 5 % (v/v) CO2.
Drug preparation and cell treatment
Pioglitazone (PGZ, 97 % purity; Sigma-Aldrich, St. Louis, MO, USA) and MK886 (97 % purity; Sigma-Aldrich, USA) were dissolved in DMSO (Bio Basic Inc., Markham, ON, Canada) as a 10−2 M stock. The drug stocks were stored at −20 °C and further diluted to the working concentration with assay medium [DMEM supplemented with only 2 % (v/v) FBS and antibiotics]. Three groups of cell cultures (control DMSO-treated, PGZ-treated and MK886-treated) were prepared in triplicate for subsequent experiments. The treatments were performed by seeding the cells at a density of 1.0 × 103 cells/well in 24-well cell culture plates (TPP, Auckland, New Zealand) until the cells reached approximately 70 % confluence. The cells were then pre-incubated with assay medium for several hours before the application of the treatments. Subsequently, PGZ (30-50 μM) or MK886 (3–5 μM) were added to the cells, and the cells were incubated at 37 °C in 5 % (v/v) CO2 for 2, 4 and 6 days. Half of the drug-containing growth media for the cells was changed every 2–3 days. The trypan blue exclusion assay was performed to determine the viability of untreated and treated cells for each treatment time-point. DMSO (0.1 %) was used as the diluent control in this study. All of the control and experimental samples were cultured in triplicate. Following 2 days of treatment, the cells were harvested using a trypsin/EDTA solution (Gibco BRL, USA). The harvested cell suspension (0.9 mL) from each well was added to 0.1 mL of trypan blue solution (Gibco BRL, USA) and incubated at room temperature for 5 min. Live and dead cells were then counted using a haemocytometer, and the cell viability (%) was calculated using the following formula:
Live cells excluded the dye (unstained cells), while dead cells took up the blue dye. The same method was used to calculate the viability of MDA-MB-231 cells after 4 and 6 days of treatment. One-way ANOVA was used to compare the viability of MDA-MB-231 cells in different concentrations of drug to that of the DMSO control. The concentrations of PGZ and MK886 that promoted the highest and the lowest expression of PPARα mRNA, respectively, were selected for subsequent experiments. The effective concentration of PGZ (30 µM), MK886 (3 µM) or DMSO (0.1 %) was added to the cells as PGZ-treated, MK886-treated or control DMSO-treated cells, and the treated cells were incubated at 37 °C in 5 % (v/v) CO2 for 2, 4 or 6 days. Following the treatments, the samples were subjected to examination as described below.
RNA extraction and cDNA synthesis
The extraction of total RNA from PGZ-treated, MK886-treated and control DMSO-treated MDA-MB-231 cells was performed using the RNeasy® Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The integrity of the total RNA extracted was confirmed using agarose gel electrophoresis, while the purity and yield were measured using a spectrophotometer (Thermo Scientific, Vilnius, Lithuania). Subsequently, 1 μg of total RNA per sample was reverse transcribed to cDNA using the RevertAid™ First Strand cDNA Synthesis Kit (Thermo Scientific, Lithuania) according to the manufacturer’s instructions. The success of cDNA synthesis was validated by PCR amplification using the human β-actin housekeeping gene as an internal control.
Primer design and real-time PCR
Following cDNA synthesis, we used real-time PCR to assess the expression levels of genes that regulate invasive activity, including PPARα, PPARγ, cysteine–aspartic acid protease 9 (caspase-9), cysteine–aspartic acid protease 3 (caspase-3), FGF4, 5LOX, alkaline phosphatase (ALPI), interleukin 6 (IL6), hypoxia-inducible factor 1-alpha (HIF1α) and vascular endothelial growth factor (VEGF) in PGZ-treated, MK886-treated and control DMSO-treated MDA-MB-231 cells. Real-time PCR was performed using the 7500 Fast System (Applied Biosystems, Foster City, CA, USA). A total volume of 25 μl was set up in optical reaction tubes that included Applied Biosystems Power SYBR® Green Master Mix, 10 μM of each primer and template (cDNA or water for NTC). The reaction was initiated at 94 °C for polymerase activation, followed by 35 cycles of denaturation at 94 °C for 20 s, primer annealing at 55 °C for 20 s and extension at 72 °C for 30 s. A programme was added to generate the melt curve, and the dissociation curve programme was initiated at 95 °C for 15 s, followed by 60 °C for 1 min, 95 °C for 30 s and 60 °C for 15 s. The CT value of each unknown sample was normalised to β-actin, and the relative expression level and fold change in gene expression were calculated according to the manufacturer’s instructions. The experiments were repeated in triplicate for at least two independent experiments.
DNA fragmentation
The DNA from PGZ-treated, MK886-treated and control DMSO-treated MDA-MB-231 cells was extracted using the DNA Extraction Kit (Qiagen, USA). The extracted DNA and Lambda DNA/Hind III marker (Fermentas, Lithuania) were loaded into the respective wells on agarose gels. The gel was run at 70 V for 50 min, after which the gel was placed on a UV transilluminator (Syngene, Bengaluru, Karnataka, India) to visualise the fragmented DNA, and gel images were captured.
Wound healing migration assay
The wound healing migration assay was performed to determine the invasive activity of PGZ-treated, MK886-treated and control DMSO-treated MDA-MB-231 cells based on the repopulation of wounded cultures. MDA-MB-231 cells (1 × 105 cells/ml) were first seeded into cell culture dishes (Nunc, Germany), and the cancer cells were cultured in medium containing 10 % FBS for 24 h. The monolayer was then wounded using a blue pipette tip, and the cellular debris was removed by washing with phosphate-buffered saline (PBS). The wounded monolayers were incubated in assay media containing 30 µM PGZ, 3 µM MK886 or 0.1 % DMSO. Photographs of the exact initial wound areas were taken every 2 days.
Transwell migration assay
The same passage of MDA-MB-231 cells used for the wound healing migration assay was used for the transwell migration assay. Corning Transwell Inserts (Sigma-Aldrich, USA) with bottoms that were sealed with polycarbonate membranes with a 6.5-mm internal diameter and 8-μm pore size were used in this assay. The inserts were first coated with fibronectin (10 μg/ml, Invitrogen Corporation, United States) in PBS overnight at 4 °C and then air-dried. After coating, 100 cells/μl in growth medium containing 30 µM PGZ, 3 µM MK886 or 0.1 % DMSO was pipetted into the insert. The growth medium was supplemented with only 0.5 % FBS. The insert was transferred to a 24-wells culture plate as the upper compartment, and the bottom compartments of the plates were filled with 0.6 ml of growth medium supplemented with 10 % FBS as a chemo-attractant. The migration of cells from the upper compartment through the pores was allowed to proceed for 2, 4 and 6 days at 37 °C under serum-gradient conditions. The solution in the upper compartment was then removed, and the non-migratory cells on the top membrane surface were gently scraped with a cotton swab. The cells on the bottom surface, which had migrated through the pores, were fixed with freshly prepared 4 % paraformaldehyde in PBS for 10 min. The migrated cells were then stained with Diff-Quik Solutions (Merck, Germany) as recommended by the manufacturer. Five random fields per insert in triplicate were viewed under an inverted microscope at 40× magnification to obtain the average number of cells per field, and the number of cells per field was counted. The experiments were carried out in triplicate and repeated twice.
Statistical analysis
All graph preparations and statistical calculations were performed using GraphPad Prism 6.0 software (GraphPad Software Inc., La Jolla, CA, USA). The experiments were repeated several times to confirm the reproducibility of the results. All of the values were expressed as the mean ± standard deviation (SD) or standard error deviation (SEM). A p value of less than 0.05 was regarded as statistically significant.
Results
The mRNA expression levels of PPARα and PPARγ in PGZ-treated and MK866-treated MDA-MB-231 cells
Our results indicated that compared to control cells, PPARα mRNA expression was not significantly induced in MDA-MB-231 cells following treatment with 30 μM PGZ for 4 days (Fig. 1a), but was significantly induced following 6 days of 30 µM PGZ treatment (approximately 660-fold change, p < 0.001). Treatment with higher concentrations of PGZ (40 and 50 µM) also induced high levels of PPARα mRNA expression in MDA-MB-231 cells. However, a prolonged treatment period (more than 6 days) was required to induce the level of PPARα mRNA expression similar to that induced by 30 µM PGZ (data not shown). The treatment of MDA-MB-231 cells with 3 µM MK886 for 4 days also induced the expression of PPARα mRNA (approximately 330-fold change, p < 0.05) compared to the control cells (Fig. 1b). However, the mRNA expression decreased dranatically at day 6 of treatment, and there was a significant difference between the level of reduction in PPARα mRNA expression at day 6 compared to day 4 in MK886-treated MDA-MB-231 cells (p < 0.01). MK886 (at concentrations of 4 and 5 µM) also inhibited the expression of PPARα mRNA in MDA-MB-231 cells. However, a prolonged treatment period (more than 6 days) was required to inhibit the mRNA expression level of the target gene fully. As the higher concentrations of PGZ and MK886 did not effectively induce or inhibit PPARα mRNA expression, respectively, we selected 30 µM PGZ and 3 µM MK886 for use in subsequent experiments.
Fig. 1.

The expression of PPARs in MDA-MB-231 cells. The mRNA expression levels of PPARα in MDA-MB-231 cells treated with different concentrations of a PGZ and b MK886 for 2, 4 and 6 days. The mRNA expression levels of PPARγ in MDA-MB-231 cells treated with different concentrations of c PGZ and d MK886 for 2, 4 and 6 days. DMSO was used as a diluent control for the study. Data are shown as the mean ± SEM of triplicate cultures, *p < 0.05; **p < 0.01; ***p < 0.001
A similar mRNA expression profile for PPARγ was observed when MDA-MB-231 cells were treated with PGZ and MK886. The PGZ (30 µM) induced a high level of PPARγ mRNA expression (approximately 345-fold change, p < 0.01) compared to control cells in as early as 4 days following treatment (Fig. 1c). Although the level of mRNA expression was not as high as that of PPARα at the end of the experiment, the high expression level (approximately 400-fold change, p < 0.01) was sustained until day 6 of treatment. A high level of PPARγ mRNA expression was also observed in MDA-MB-231 cells treated with 3 µM MK886 for 4 days (approximately 290-fold change, p < 0.01) compared to the control cells. The expression of PPARγ mRNA was inhibited at day 6 following treatment with 3 µM MK886, indicating that the drug effectively inhibits both PPARα and PPARγ in MDA-MB-231 cells following 6 days of treatment (Fig. 1d). Interestingly, the expression of PPARγ mRNA in MDA-MB-231 cells at day 6 of MK886 treatment was significantly lower compared to day 4 of treatment (p < 0.01).
The viability of PGZ-treated and MK886-treated MDA-MB-231 cells
Figure 2a shows the viability of MDA-MB-231 cells treated with 30 µM PGZ for 2, 4 and 6 days, indicating a minimal inhibitory effect on the growth of the cells even after 6 days of treatment. In contrast, we observed a growth inhibitory effect in MK886-treated MDA-MB-231 cells in as early as 2 days, when 3 µM MK886 was observed to inhibit 13.7 % (p < 0.05) of the growth of MDA-MB-231 cells (Fig. 2b). The inhibitory effect of growth increased when the treatment was extended to 4 days (31.9 %, p < 0.01) and 6 days (32.6 %, p < 0.01). Our trypan blue exclusion test revealed that the viability of MDA-MB-231 cells following PGZ and MK886 treatments were likely independent of the mRNA expression of PPARα/γ. We selected the trypan blue exclusion assay to determine the viability of MDA-MB-231 cells following PGZ and MK886 treatments because a longer treatment duration (6 days) was required, and measurements of cell viability that access the enzymes in the conditioned medium using MTT or LDH were unsuitable for treatments that require an incubation time longer than 72 h.
Fig. 2.

The viability of MDA-MB-231 cells treated with a 30 µM PGZ and b 3 µM MK886 for 2, 4 and 6 days. Cells treated with DMSO were used as a control. Data are shown as the mean ± SEM of triplicate cultures, *p < 0.05; **p < 0.01
The DNA fragmentation in PGZ-treated and MK886-treated MDA-MB-231 cells
Our DNA fragmentation assay revealed that both 30 µM PGZ and 3 µM MK886 treatments of MDA-MB-231 cells did not yield the characteristic DNA laddering pattern in the cancer cells after 6 days (Fig. 3). This phenomenon was confirmed by the detection of the mRNA expression of caspase-9 (initiator caspase) and caspase-3 (executor caspase) using real-time PCR (Fig. 4). We could not detect these caspases in the PGZ-treated and MK886-treated MDA-MB-231 cells even after 6 days of treatment. It is possible that the cell death induced by PGZ and MK886 treatments was triggered by other cellular mechanisms.
Fig. 3.

The DNA fragmentation of MDA-MB-231 cells treated with various drugs for 2, 4 and 6 days. Lane M Lambda DNA/HindIII Marker; Lane 1 genomic DNA extracted from untreated cells; Lane 2 cells treated with 0.2 % DMSO; Lane 3 30 µM PGZ; Lane 4 3 µM MK886; Lane 5 30 µM PGZ + 3 µM MK886; Lane 6 0.25 µM camptothecin; Lane 7 5 µM tamoxifen
Fig. 4.

The expression of caspases in MDA-MB-231 cells. The mRNA expression levels of caspase-9 in MDA-MB-231 cells treated with different concentrations of a PGZ and b MK886 for 2, 4 and 6 days. The mRNA expression levels of caspase-3 in MDA-MB-231 cells treated with different concentrations of c PGZ and d MK886 for 2, 4 and 6 days. DMSO was used as a diluent control for the study. Data are shown as the mean ± SEM from triplicate cultures
The cell migration in PGZ-treated and MK886-treated MDA-MB-231 cells
We conducted the wound healing migration assay to study the in vitro cell migration or invasion rate of PGZ-treated and MK886-treated MDA-MB-231 cells. The assay revealed that the wound was not fully covered by PGZ-treated MDA-MB-231 cells via the healing process after 6 days of treatment (Fig. 5a). The wounds were only covered 20.5, 7.7 and 4.1 % at day 2, day 4 and day 6 of the healing process, respectively, by PGZ-treated MDA-MB-231 cells. However, the wound was observed to be covered at day 6 of the healing process by MK886-treated MDA-MB-231 cells, although the process did not occur as quickly as by control cells. The wounds were covered 28.6 % at day 2 and 31.4 % on days 4 and 6 of the healing process. The comparison of the uncovered and covered wounds in PGZ-treated and MK886-treated MDA-MB-231 cells showed a statistically significant difference (p < 0.05) (Fig. 5b). Our findings in the wound healing migration assay were validated by the transwell migration assay. This assay revealed that the number of PGZ-treated MDA-MB-231 cells that migrated through the pores of the insert was 103.2 ± 23.93, which was significantly different (p < 0.001) compared to the DMSO-treated MDA-MB-231 cells (169.8 ± 24.54) (Fig. 6b). We observed a significant increase in the number of migrating MK886-treated MDA-MB-231 cells (124.8 ± 5.19, p < 0.01) compared to the control, suggesting that the reduction in PPARα mRNA expression restored the invasion of the MDA-MB-231 cells. A smaller number of migratory PGZ-treated MDA-MB-231 cells compared to DMSO-treated and MK886-treated MDA-MB-231 cells was also observed under an inverted microscope (Fig. 6a). This finding implies a similar possibility as proposed by Liu et al. (2003) that PGZ possesses an anti-invasive activity in MDA-MB-231 cells, and our study revealed that use of the PPARα/γ inhibitor MK886 could counter the anti-invasive activity of PGZ. However, we hypothesized that the restoration of migratory activity was not due to only the inhibition of PPARα/γ mRNA expression in the cancer cells. Therefore, we determined the mRNA expression levels of other target genes, such as FGF4, 5LOX, ALPI, IL6, HIF1α and VEGF, in the PGZ-treated and MK886-treated MDA-MB-231 cells using real-time PCR.
Fig. 5.
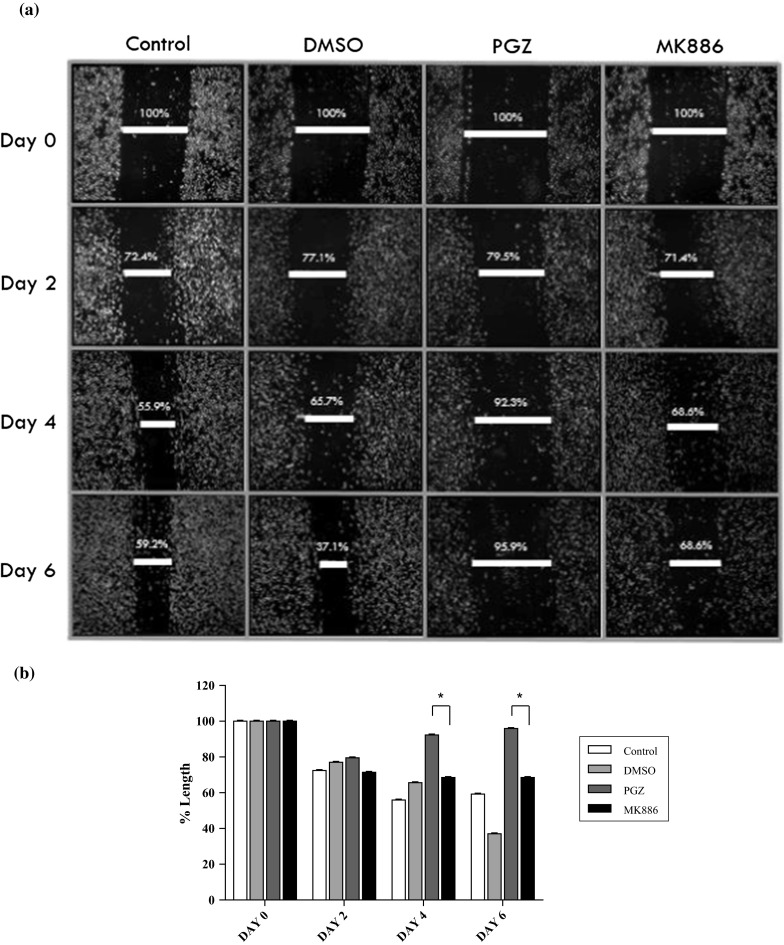
a The invasion of PGZ-treated and MK886-treated MDA-MB-231 cells as verified by the in vitro wound healing migration assay. Cell migration was estimated by measuring the gap within the wounded region. b Statistical analysis of the PGZ-treated and MK886-treated MDA-MB-231 cells for 2, 4 and 6 days. Data are shown as the mean ± SD of triplicate cultures, *p < 0.05
Fig. 6.

Validation of the invasion of PGZ-treated and MK886-treated MDA-MB-231 cells as determined by the wound healing migration assay by the transwell migration assay. a The cell migration was estimated by measuring the number of cells on the bottom surface that had migrated through the pores of the insert. b Statistical analysis of the number of cells that had migrated through the pores of the insert in MK886-treated MDA-MB-231 cells versus PGZ-treated MDA-MB-231 cells. Data are shown as the mean ± SD from triplicate cultures, **p < 0.01; ***p < 0.001
The mRNA expression of FGF4 and other genes in PGZ-treated and MK886-treated MDA-MB-231 cells
In addition to induce high level of PPARα/γ mRNA expression in MDA-MB-231 cells, treatment with 30 µM PGZ also induced the expression of other genes, including FGF4, 5LOX, ALPI, IL6, HIF1α and VEGF. The expression of FGF4 mRNA was not induced after 4 days of treatment with 30 µM PGZ. However, FGF4 mRNA expression was increased by approximately 260.0 % (p < 0.05) at day 6 post-treatment with 30 µM PGZ when compared to the control cells (Fig. 7a). A similar phenomenon was observed for the mRNA expression of 5LOX and ALPI, where their mRNA expressions were significantly induced by 471.5 % (p < 0.001) and 428.0 % (p < 0.001), respectively, in MDA-MB-231 cells treated with 30 µM PGZ for 6 days compared to the control cells (Fig. 7b, c). Moreover, high level of IL6 mRNA expression was also induced following 6 days of treatment with 30 µM PGZ, which led to a 484.5 % increase (p < 0.01) (Fig. 7d). High level of HIF1α mRNA expression was induced following treatment with 30 µM PGZ in as early as 4 days, resulting in a 340.5 % increase (p < 0.01) (Fig. 7e). The HIF1α mRNA expression was further induced by 367.5 % at day 6 of treatment (p < 0.01). In addition, the expression of VEGF mRNA was found to be increased by 340 % (p < 0.01) at day 6 post-treatment with 30 µM PGZ compared to cells treated with the same concentration of PGZ for 2 days (Fig. 7f). The comparison of VEGF mRNA expression levels in MDA-MB-231 cells treated with 30 µM PGZ for 6 days to control cells treated with DMSO for the same treatment duration did not reveal any significant changes.
Fig. 7.

The mRNA expression of a FGF4 b 5LOX c ALPI d IL6 e HIF1α and f VEGF in PGZ-treated MDA-MB-231 cells. DMSO was used as a diluent control for the study. Data are shown as the mean ± SEM of triplicate cultures, *p < 0.05; **p < 0.01; ***p < 0.001
The treatment of MDA-MB-231 cells with 3 µM MK886 resulted in similar gene expression profiles for PPARα/γ, FGF4 and 5LOX, indicating that the induction or inhibition of PPARα/γ, FGF4 and 5LOX may regulate the same mechanism. We observed a 508.5 % increase in the expression of FGF4 mRNA (p < 0.01) in MK886-treated cells at day 4 post-treatment with 3 µM MK886 compared to the control cells (Fig. 8a). The increase in FGF4 mRNA expression was reduced to 307.0 % (p < 0.05) at day 6 post-treatment with 3 µM MK886, although this level remained higher than control cells. There was a significant difference in the level of FGF4 mRNA expression at day 6 of MK886 treatment compared to day 4 of treatment (p < 0.05). A similar mRNA expression profile (induced at a higher level at day 4 compared to day 6 of treatment) was also observed for 5LOX, where its mRNA expression showed a 433.0 % increase (p < 0.001) and 226.0 % increase in cells treated with 3 µM MK886 for 4 days and 6 days, respectively, compared to the control cells (Fig. 8b). The mRNA expression levels of ALPI showed a 391.0 % increase (p < 0.001) at 4 days and a 345.5 % increase (p < 0.01) at 6 days following MK886 treatment compared to the control cells (Fig. 8c). However, the only statistically significant difference was observed between the expression of 5LOX mRNA at day 6 of MK886 treatment compared to cells treated with MK886 for 4 days (p < 0.01). The mRNA expression of IL6 was increased in MK886-treated MDA-MB-231 cells in as early as 2 days after treatment, resulting in a 373 % increase (p < 0.001) (Fig. 8d). The increase in mRNA expression levels were approximately 391.0 % (p < 0.001) at day 4 and 345.5 % (p < 0.01) at day 6 after treatment compared to the control cells. We did not observe similar expression profiles for HIF1α (Fig. 8e) or VEGF (Fig. 8f) in the MK886-treated MDA-MB-231 cells, indicating that the inhibition of PPARα/γ by MK886 was likely correlated with FGF4 and 5LOX but not with the expression of IL6, ALPI, HIF1α and VEGF mRNA.
Fig. 8.

The mRNA expression of a FGF4 b 5LOX c ALPI d IL6 e HIF1α and f VEGF in MK886-treated MDA-MB-231 cells. DMSO was used as a diluent control for the study. Data are shown as the mean + SEM of triplicate cultures, *p < 0.05; **p < 0.01; ***p < 0.001
Discussion
Our results showed that the treatment of MDA-MB-231 cells with PGZ increased the expression of PPARα/γ mRNA, while PPARα/γ mRNA expression was inhibited by the treatment with MK886 for 6 days. Both PGZ and MK886 reduced the viability of MDA-MB-231 cells through a mechanism that was independent of PPARα/γ mRNA expression. Moreover, we did not observe any induction of apoptosis in the cancer cells following treatment with either PGZ or MK886. MK886-treated MDA-MB-231 cells, but not PGZ-treated MDA-MB-231 cells, were able to heal a cellular culture wound via cell migration, indicating that the reduction in cell invasion resulting from PGZ treatment could be eliminated by MK886 treatment. We further validated this finding using a transwell migration assay. This phenomenon was also associated with the expression of PPARα/γ mRNA in the treated cancer cells. FGF4 and 5LOX showed similar mRNA expression profiles to PPARα/γ and was involved in the regulation of wound healing in the PGZ-treated and MK886-treated MDA-MB-231 cells. Therefore, PPARα/γ, FGF4 and 5LOX might play important roles in the mechanism underlying the invasion of MDA-MB-231 cells. Our study provides useful information about the mRNA expression profiles of PPARα and other related genes in MDA-MB-231 cells, and these genes could be attractive targets for the treatment of human breast cancer.
As previously mentioned, PGZ is a selective PPARγ ligand, and the extent of its effects is more pronounced for PPARγ compared to PPARα (Gillies and Dunn 2000; Smith 2001). However, our present study showed that the level of PPARα mRNA expression was higher than that of PPARγ in MDA-MB-231 cells. The differences in our study compared to others (Suchanek et al. 2002) could be due to an increased incubation time and the dynamic regulation of PPARα in MDA-MB-231 cells adopted in our study. The correlation of PGZ with the expressions of PPARα and PPARγ have been widely demonstrated in MDA-MB-231 cells and have been linked to many important processes of cancer cells. Our results showed that the treatment of MDA-MB-231 cells with PGZ increased the expression of PPARα/γ mRNA and that this expression could be inhibited by treating the cancer cells with MK886 for 6 days whereby we observed a greater effect of treatments on PPARα expression versus PPARγ expression. These findings are supported by those of Kehrer et al. (2001). To further investigate the aforementioned mechanism, we studied the expressions of FGF4, 5LOX, IL6, ALPI, HIF1α and VEGF mRNA, which were demonstrated to have similar profiles to PPARα/γ.
Our study demonstrated that both PGZ and MK886 reduced the viability of MDA-MB-231 cells independent of the expression of PPARα/γ mRNA. Moreover, apoptosis was not induced by either PGZ or MK886 treatment. Our results are consistent with a study by Zhou et al. (2009), which demonstrated that MDA-MB-231 cells did not undergo apoptosis when treated with sub-saturation doses of the synthetic PPARγ ligands troglitazone (TGZ) and rosiglitazone (RGZ). Consistent with this, our study demonstrated that treatment of MDA-MB-231 cells with PGZ, which is an analogue of glitazones, did not induce apoptosis in the cancer cells. Our results revealed that treatment of MDA-MB-231 cells with both PGZ and MK886 for 6 days did not yield the characteristic DNA laddering pattern in the cancer cells as evaluated by the DNA fragmentation assay. This phenomenon was confirmed by the detection of caspase-9 (initiator caspase) and caspase-3 (executor caspase) mRNA expression in the treated cells using real-time PCR whereby we were unable to significantly detect the pool of caspases in the PGZ-treated and MK886-treated MDA-MB-231 cells. Although it has been demonstrated that PPARγ ligands inhibit cellular proliferation and induce cell death in various cancer cell types, including breast cancer (Elstner et al. 1998; Clay et al. 1999; Nwankwo and Robbins 2001; Martelli et al. 2002; Shimada et al. 2002; Toyoda et al. 2002; Wang et al. 2002; Yoshizawa et al. 2002; Zhou et al. 2009), our study and others have found that not all PPARγ ligands induce apoptosis in MDA-MB-231 cells. It is now possible to conclude that treatment with PGZ and other glitazones does not induce apoptosis in MDA-MB-231 cells as found in ours and in Yamamoto et al. (2001) studies, and that the DNA fragmentation induced by apoptosis might be independent of the expression of PPARα/γ. Additional studies should investigate the induction of apoptosis in MDA-MB-231 cells by other non-glitazone selective and synthetic PPARγ ligands. MK886 has been reported to induce apoptosis, and its ability to induce apoptosis at higher concentrations (10–20 µM) is often linked with the expression of 5LOX (Lim et al. 2010). However, in our study, treatment with MK886 and the induction of 5LOX expression did not induce apoptosis in MDA-MB-231 cells. The expression of 5LOX might not correlate with the apoptosis mechanism induced by MK886, as the dose required for apoptosis is approximately 100-fold higher than that needed to inhibit the expression of 5LOX (Datta et al. 1999). Indeed, both MK886 and 5LOX could correlate with the inhibition of cancer cell invasion.
Cellular proliferation, invasion, metastasis, apoptosis, autophagy and angiogenesis are the primary biological pathways involved in cancer progression. Targeting these pathways is the goal for developing new diagnostic and therapeutic agents for cancers (Zhou et al. 2009). The wound healing assay, which we used to study in vitro cell migration as a measurement of invasion rate, revealed that the wound in PGZ-treated MDA-MB-231 cells was never covered by the process of healing after 6 days. This finding supports a hypothesis proposed by Liu et al. (2003) that states the PGZ possesses an anti-invasive activity in MDA-MB-231 cells. However, the anti-invasive activity could be blocked by inhibiting PPARα/γ mRNA expression in the cancer cells. This is supported by the finding that the wound in MK886-treated MDA-MB-231 cells was observed to be covered during the day 6 of healing process, although the process did not occur as quickly as in the control cells. We further validated this finding using the transwell migration assay. Notably, other genes, such as FGF4 and 5LOX, might also be involved in this mechanism, as similar mRNA expression profiles were observed in the PGZ-treated and MK886-treated MDA-MB-231 cells.
Currently, there are few studies regarding the correlation between FGF4 and PGZ that help to elucidate the pattern of FGF4 expression induced by PGZ treatment. A study on FGF expression following ciglitazone (CGZ) and PGZ treatments revealed that an enhancement in FGF expression compared to that of VEGF (Yasudaa et al. 2005). The same study suggested that GW9662, an inhibitor of PPARγ, reduced the effects of glitazones. In our study, the expression of FGF4 expression was significantly reduced by MK886. In addition to its inhibition by a PPARγ/α inhibitor, the inhibition of FGF also occurs in a dose-dependent manner (Zhao et al. 2011). Thus, a higher dose or a longer incubation time might be required for an effective inhibitory effect on FGF4. The expression of 5LOX, which is activated by five lipoxygenase activating protein (FLAP), was greatly increased following treatment with PGZ, and its expression was reduced following MK886 treatment. It has been reported that MK886 inhibits FLAP and lipoxygenase activities in addition to PPARα (Kehrer et al. 2001). This indicates that the lipoxygenase activities are closely related to the expression of PPARα, and when the expression of PPARα is elevated, 5LOX expression is also increased. A study by Avis et al. (2001) showed that the disruption of 5LOX induced growth arrest and apoptosis in breast cancer cells. Although our study did not find evidence of apoptosis in the drug-treated cells when 5LOX was inhibited, we found that the reduced expression of 5LOX eliminated the anti-invasive activity of the drug-treated cancer cells. Indeed, the growth inhibitory effect on MDA-MB-231 cells was more potent when the level of 5LOX expression was decreased.
ALPI has been closely linked to various malignancies and reported to be widely circulating in breast cancer patients (Singh et al. 2013). ALPI activity has been reported to increase following the administration of a PPARα agonist (Stunes et al. 2011). However, high levels of ALPI expression were observed in both PGZ-treated and MK886-treated MDA-MB-231 cells throughout the experiments. It is possible that the expression of ALPI is independent of PPARα expression. Another target gene that showed a high level of expression in cells treated with both drugs was IL6. Previous studies have reported that the activation of PPARα negatively regulated IL6 expression (Srinivasan et al. 2004), but other studies have shown that other factors could increase the expression of IL6. For instance, reduced TNFα expression has been shown to contribute to the activation of PPARα and to increase serum IL6 levels (Alwayn et al. 2006). In another study by Patzer et al. (2008), it was reported that PPARγ activation only managed to suppress the expression level of IL6 at 48 h prior to treatment. This was supported by our current study, which showed decreased IL6 expression at day 2 that gradually increased at day 6. This effect was reversible upon the administration of MK886 to the cells, and an additional study has also shown that MK886 serves as an anti-inflammatory agent and can decrease serum IL6 levels (Al-Amran et al. 2011).
Meanwhile, HIF1α and VEGF were significantly expressed in MDA-MB-231 cells following treatment with PGZ, while treatment of the same cancer cells with MK886 did not induce the expression of both genes throughout the experiment. Based on the findings of our study, the activation of PPARs, which increases the production of VEGF (Biscetti et al. 2008), could not be fully linked to the expression of PPARα/γ. However, our results indicate that this gene mRNA expression could be reversed by MK886 treatment. The ability of MK886 to decrease the expression of VEGF has been previously reported in a study on human pancreatic cancer cells (Zhou et al. 2011), whereas a recent study conducted by Papi et al. (2013) showed that PPARα and HIF1α have functional interplays in which their expressions are enhanced by each other, resulting in pro-inflammatory responses. This would support the current finding that HIF1α positively correlates with the expression of PPARα. However, this phenomenon was demonstrated to be reversed by MK886. It is possible that PGZ treatment induces autophagy rather than apoptosis in MDA-MB-231 cells. The reduction in cell viability could also be related to the increased expression of HIF1α, which triggers the autophagy process (Zhu et al. 2013).
Conclusions
Treatment of MDA-MB-231 cells with PGZ and MK886 reduced the viability of the cancer cells independent of PPARα/γ mRNA expression. The reduced invasion activity of PGZ-treated cells could be due to changes in the mRNA expression levels of PPARα/γ, FGF4 and 5LOX in cancer cells. Other possible mechanisms, such as autophagy, should also be considered in future studies.
Acknowledgments
The PPARα study was supported by a Fundamental Research Grant Scheme (FRGS) Fasa 2/2010 (Grant No. 203/CIPPM/6711162). The study using MDA-MB-231 cells was funded by the Universiti Sains Malaysia for Research University (Grant No. 1001/CIPPM/811200). The first author would like to thank the Graduate Assistant scheme (GA) from USM, MoHe for MyMasters scheme under MyBrain15 and Dr. Chew Ai Lan, who proofread the manuscript prior to its submission for publication.
Abbreviations
- PGZ
Pioglitazone
- mRNA
Messenger RNA
- PPARα
Peroxisome proliferator activated receptor alpha
- PPARγ
Peroxisome proliferator activated receptor gamma
- FGF4
Fibroblast growth factor 4
- 5LOX
5-Lipoxygenase
- Caspase-9
Cysteine–aspartic acid protease 9
- Caspase-3
Cysteine–aspartic acid protease 3
- ALPI
Alkaline phosphatase
- IL6
Interleukin 6
- HIF1α
Hypoxia-inducible factor 1-alpha
- VEGF
Vascular endothelial growth factor
References
- Al-Amran FG, Hadi NR, Hashim AM. Leukotriene biosynthesis inhibition ameliorates acute lung injury following hemorrhagic shock in rats. J Cardiothorac Surg. 2011;6:81. doi: 10.1186/1749-8090-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwayn IP, Andersson C, Lee S, Arsenault DA, Bistrian BR, Gura KM, Nose V, Zauscher B, Moses M, Pude M. Inhibition of matrix metalloproteinases increase PPAR-alpha and IL-6 and prevents dietary-induced hepatic steatosis and injury in a murine model. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1011–G1019. doi: 10.1152/ajpgi.00047.2006. [DOI] [PubMed] [Google Scholar]
- Avis I, Hong SH, Martinez A, Moody T, Choi YH, Trepel J, Das R, Jett M, Mulshine JL. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 2001;15:2007–2009. doi: 10.1096/fj.00-0866fje. [DOI] [PubMed] [Google Scholar]
- Biscetti F, Gaetani E, Flex A, Aprahamian T, Hopkins T, Straface G, Pecorini G, Stigliano E, Angelini F, Castellot JJ, Jr, Pola R. Selective activation of peroxisome proliferator-activated receptor (PPAR)alpha and PPARgamma induces neoangiogenesis through a vascular endothelial growth factor-dependent mechanism. Diabetes. 2008;57:1394–1404. doi: 10.2337/db07-0765. [DOI] [PubMed] [Google Scholar]
- Bocca C, Bozzo F, Martinasso G, Canuto RA, Miglietta A. Involvement of PPARα in the growth inhibitory effect of arachidonic acid on breast cancer cells. Br J Nutr. 2008;100:739–750. doi: 10.1017/S0007114508942161. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Thomas DM, Thompson EW, Williams ED. PPARγ-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer. 2006;6:53. doi: 10.1186/1471-2407-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay CE, Namen AM, Atsumi G, Willingham MC, High KP, Kute TE, Trimboli AJ, Fonteh AN, Dawson PA, Chilton FH. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis. 1999;20:1905–1911. doi: 10.1093/carcin/20.10.1905. [DOI] [PubMed] [Google Scholar]
- Datta K, Biswal SS, Kehrer JP. The 5-lipoxygenase-activating protein (FLAP) inhibitor, MK886, induces apoptosis independently of FLAP. Biochem J. 1999;340:371–375. doi: 10.1042/bj3400371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, Shintaku P, Said JW, Heber D, Koeffler HP. Ligands for peroxisome proliferator-activated receptor gamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA. 1998;95:8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies PS, Dunn CJ. Pioglitazone. Drugs. 2000;60:333–345. doi: 10.2165/00003495-200060020-00009. [DOI] [PubMed] [Google Scholar]
- Kehrer JP, Biswal SS, La E, Thuillier P, Datta K, Fischer SM, Heuvel JPV. Inhibition of peroxisome-proliferator-activated receptor (PPAR)alpha by MK886. Biochem J. 2001;356:899–906. doi: 10.1042/bj3560899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanson M, Bougnoux P, Besson P, Lansac J, Hubert B, Couet C, Le Floch O. n-6 Polyunsaturated fatty acids in human breast carcinoma phosphatidylethanolamine and early relapse. Br J Cancer. 1990;61:776–778. doi: 10.1038/bjc.1990.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JY, Oh JH, Jung JR, Kim SM, Ryu CH, Kim HT, Jeun SS. MK886-induced apoptosis depends on the 5-LO expression level in human malignant glioma cells. J Neurooncol. 2010;97:339–346. doi: 10.1007/s11060-009-0036-9. [DOI] [PubMed] [Google Scholar]
- Liu H, Zang C, Fenner MH, Possinger K, Elstner E. PPARgamma ligands and ATRA inhibit the invasion of human breast cancer cells in vitro. Breast Cancer Res Treat. 2003;79:63–74. doi: 10.1023/A:1023366117157. [DOI] [PubMed] [Google Scholar]
- Martelli ML, Iuliano R, Le Pera I, Sama’ I, Monaco C, Cammarota S, Kroll T, Chiariotti L, Santoro M, Fusco A. Inhibitory effects of peroxisome poliferator-activated receptor gamma on thyroid carci- noma cell growth. J Clin Endocrinol Metab. 2002;87:4728–4735. doi: 10.1210/jc.2001-012054. [DOI] [PubMed] [Google Scholar]
- Master SR, Hartman JL, D’Cruz CM, Moody SE, Keiper EA, Ha SI, Cox JD, Belka GK, Chodosh LA. Functional microarray analysis of mammary organogenesis reveals a developmental role in adaptive thermogenesis. Mol Endocrinol. 2002;16:1185–1203. doi: 10.1210/mend.16.6.0865. [DOI] [PubMed] [Google Scholar]
- Nwankwo JO, Robbins ME. Peroxisome proliferator-activated receptor-gamma expression in human malignant and normal brain, breast and prostate-derived cells. Prostaglandins Leukot Essent Fatty Acids. 2001;64:241–245. doi: 10.1054/plef.2001.0266. [DOI] [PubMed] [Google Scholar]
- Papi A, Storci G, Guarnieri T, Carolis DS, Bertoni S, Avenia N, Sanguinetti A, Sidoni A, Santini D, Ceccarelli C, Tafurelli M, Orlandi M, Bonafe M. Peroxisome proliferator activated receptor-α/Hypoxia inducible factor-1α interplay sustains carbonic anhydrase IX and apoliprotein E expression in breast cancer stem cells. PLoS One. 2013;8:e54968. doi: 10.1371/journal.pone.0054968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzer A, Zhao Y, Stock I, Gohike P, Herdegen T, Culman J. Peroxisome proliferator-activated receptors gamma (PPARgamma) differently modulate the interleukin-6 expression in the peri-intact cortical tissue in the acute and delayed phases of cerebral ischaemia. Eur J Neurosci. 2008;28:1786–1794. doi: 10.1111/j.1460-9568.2008.06478.x. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPARα in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- Roberts RA, James NH, Woodyatt NJ, Macdonald N, Tugwood JD. Evidence for the suppression of apoptosis by the peroxisome proliferator activated receptor alpha (PPARα) Carcinogenesis. 1998;19:43–48. doi: 10.1093/carcin/19.1.43. [DOI] [PubMed] [Google Scholar]
- Roberts-Thomson SJ, Snyderwine EG. Characterization of peroxisome proliferator-actiaved receptor alpha in normal rat mammary gland and 2-amino-1-methyl-6-phenylimidazol[4,5-b]pyridine-induced mammary gland tumors from rats fed high and low fat diets. Toxicol Lett. 2000;118:79–86. doi: 10.1016/S0378-4274(00)00265-4. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut. 2002;50:658–664. doi: 10.1136/gut.50.5.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Pandey A, Tewari M, Kumar R, Sharma A, Singh KA, Pandey HP, Shukla HS. Advanced stage of breast cancer hoist alkaline phosphatase activity: risk factor for females in India. 3 Biotech. 2013;3:517–520. doi: 10.1007/s13205-012-0113-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith U. Pioglitazone: mechanism of action. Int J Clin Pract Suppl. 2001;121:13–18. [PubMed] [Google Scholar]
- Srinivasan S, Hatley ME, Reilly KB, Danziger EC, Hedrick CC. Modulation of PPARalpha expression and inflammatory interleukin-6 production by chronic glucose increases monocyte/endhothelial adhesion. Arterioscler Thromb Vasc Biol. 2004;24:851–857. doi: 10.1161/01.ATV.zhq0504.2260. [DOI] [PubMed] [Google Scholar]
- Stunes AK, Westbroek I, Gustafsson BI, Fossmark R, Waarsing JH, Eriksen EF, Petzold C, Reseland JE, Syversen U. The peroxisome proliferator-activated receptor (PPAR)alpha agonist fenofibrate maintains bone mass, while the PPARgamma agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr Disord. 2011;11:11. doi: 10.1186/1472-6823-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchanek KM, May FJ, Robinson JA, Lee WJ, Holman NA, Monteith GR, Roberts-Thomson SJ. Peroxisome proliferator-actiavted receptor α; in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog. 2002;34:165–171. doi: 10.1002/mc.10061. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Yamasaki D, Ishimoto K, Doi T (2008) The role of PPARs in cancer. PPAR Res 2008:102737. doi:10.1155/2008/102737 [DOI] [PMC free article] [PubMed]
- Toyoda M, Takagi H, Horiguchi N, Kakizaki S, Sato K, Takayama H, Mori M. A ligand for peroxisome proliferator activated receptor gamma inhibits cell growth and induces apoptosis in human liver cancer cells. Gut. 2002;50:563–567. doi: 10.1136/gut.50.4.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidine-dione activation of peroxisome proliferator-activated receptor gamma can enhance mitochondrial potential and promote cell survival. J Biol Chem. 2002;277:31781–31788. doi: 10.1074/jbc.M204279200. [DOI] [PubMed] [Google Scholar]
- Wu M, Harvey KA, Ruzmetov N, Welch ZR, Sech L, Jackson K, Stillwell W, Zaloga GP, Siddiqui RA. Omega-3 polyunsaturated fatty acids attenuate breast cancer growth through activation of a neutral sphingomyelinase-mediated pathway. Int J Cancer. 2005;117:340–348. doi: 10.1002/ijc.21238. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Nakajima M, Yamazaki H, Yokoi T. Cytotoxicity and apoptosis produced by troglitazone in human hepatoma cells. Life Sci. 2001;70:471–482. doi: 10.1016/S0024-3205(01)01432-1. [DOI] [PubMed] [Google Scholar]
- Yasudaa E, Tokuda H, Ishisaki A, Hirade K, Kanno Y, Hanai Y, Nakamura N, Noda T, Katagiri Y, Kozawa O. PPAR-gamma ligands up-regulate basic fibroblast growth factor-induced VEGF release through amplifying SAPK/JNK activation in osteoblasts. Biochem Biophys Res Commun. 2005;328:137–143. doi: 10.1016/j.bbrc.2004.12.163. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K, Cioca DP, Kawa S, Tanaka E, Kiyosawa K. Peroxisome proliferator-activated receptor gamma ligand troglitazone induces cell cycle arrest and apoptosis of hepatocellular carcinoma cell lines. Cancer. 2002;95:2243–2251. doi: 10.1002/cncr.10906. [DOI] [PubMed] [Google Scholar]
- Zhao G, Li WY, Chen D, Henry JR, Li HY, Chen Z, Zia-Ebrahimi M, Bloem L, Zhai Y, Huss K, Peng SB, McCann DJ. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol Cancer Ther. 2011;10:2200–2210. doi: 10.1158/1535-7163.MCT-11-0306. [DOI] [PubMed] [Google Scholar]
- Zhou J, Zhang W, Liang B, Casimiro MC, Whitaker-Menezes D, Wang M, Lisanti MP, Lanza-Jacoby S, Pestell RG, Wang C. PPARgamma activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol. 2009;41:2334–2342. doi: 10.1016/j.biocel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Zhu C, Ding X, Zhang H, Zhang H, Cao W, Iang QH, Xu Z. Inhibitory effect of MK886 and celecoxib on the growth of pancreatic cancer cell line SW1990 and angiogenesis. Chin J Pancreatol. 2011;11:407–409. [Google Scholar]
- Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F, Zhou Y, Wu Y, Yu M, Zhang Z, Shao G. Role of the hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013;13:119. doi: 10.1186/1475-2867-13-119. [DOI] [PMC free article] [PubMed] [Google Scholar]