

Abstract
Key points
Physiologically relevant rodent models of non‐alcoholic steatohepatitis (NASH) that resemble the human condition are limited.
Exercise training and energy restriction are first‐line recommendations for the treatment of NASH.
Hyperphagic Otsuka Long–Evans Tokushima fatty rats fed a western diet high in fat, sucrose and cholesterol for 24 weeks developed a severe NASH with fibrosis phenotype.
Moderate intensity exercise training and modest energy restriction provided some improvement in the histological features of NASH that coincided with alterations in markers of hepatic stellate cell activation and extracellular matrix remodelling.
The present study highlights the importance of lifestyle modification, including exercise training and energy restriction, in the regulation of advanced liver disease.
Abstract
The incidence of non‐alcoholic steatohepatitis (NASH) is rising but the efficacy of lifestyle modifications to improve NASH‐related outcomes remain unclear. We hypothesized that a western diet (WD) would induce NASH in the Otsuka Long–Evans Tokushima Fatty (OLETF) rat and that lifestyle modification would improve this condition. Eight‐week‐old Long–Evans Tokushima Otsuka (L) and OLETF (O) rats consumed a control diet (10% kcal fat, 3.5% sucrose) or a WD (45% kcal fat, 17% sucrose, 1% cholesterol) for 24 weeks. At 20 weeks of age, additional WD‐fed OLETFs were randomized to sedentary (O‐SED), food restriction (O‐FR; ∼25% kcal reduction vs. O‐SED) or exercise training (O‐EX; treadmill running 20 m min–1 with a 15% incline, 60 min day–1, 5 days week–1) conditions for 12 weeks. WD induced a NASH phenotype in OLETFs characterized by hepatic fibrosis (collagen 1α1 mRNA and hydroxyproline content), as well as elevated inflammation and non‐alcoholic fatty liver disease activity scores, and hepatic stellate cell activation (α‐smooth muscle actin) compared to Long–Evans Tokushima Otsuka rats. FR and EX modestly improved NASH‐related fibrosis markers (FR: hydroxyproline content, P < 0.01; EX: collagen 1α1 mRNA, P < 0.05; both: fibrosis score, P < 0.01) and inflammation (both: inflammation score; FR: interleukin‐1β and tumor necrosis factor α) vs. O‐SED. FR reduced hepatic stellate cell activation markers (transforming growth factor‐β protein and α‐smooth muscle actin mRNA), whereas EX increased the hepatic stellate cell senescence marker CCN1 (P < 0.01 vs. O‐SED). Additionally, both FR and EX normalized extracellular matrix remodelling markers to levels similar to L‐WD (P > 0.05). Although neither EX nor FR led to complete resolution of the WD‐induced NASH phenotype, both independently benefitted liver fibrosis via altered hepatic stellate cell activation and extracellular matrix remodelling.
Key points
Physiologically relevant rodent models of non‐alcoholic steatohepatitis (NASH) that resemble the human condition are limited.
Exercise training and energy restriction are first‐line recommendations for the treatment of NASH.
Hyperphagic Otsuka Long–Evans Tokushima fatty rats fed a western diet high in fat, sucrose and cholesterol for 24 weeks developed a severe NASH with fibrosis phenotype.
Moderate intensity exercise training and modest energy restriction provided some improvement in the histological features of NASH that coincided with alterations in markers of hepatic stellate cell activation and extracellular matrix remodelling.
The present study highlights the importance of lifestyle modification, including exercise training and energy restriction, in the regulation of advanced liver disease.
Abbreviations
- CON
control diet
- EX
exercise training
- FR
food restriction
- HSC
hepatic stellate cell
- IL
interleukin
- L
Long–Evans Tokushima Otsuka
- LETO
Long–Evans Tokushima Otsuka
- MMP
matrix metalloproteinase
- NAFLD
non‐alcoholic fatty liver disease
- NAS
NAFLD activity scores
- NASH
non‐alcoholic steatohepatitis
- O
Otsuka Long–Evans Tokushima Fatty
- OLETF
Otsuka Long–Evans Tokushima Fatty
- PDGF
platelet‐derived growth factor
- PDGFr
platelet‐derived growth factor receptor
- SED
sedentary
- αSMA
α‐smooth muscle actin
- TG
triglyceride
- TGF
transforming growth factor
- TIMP
tissue inhibitor of metalloproteinase
- TUNEL
terminal deoxynucleotide transferase‐mediated dUTP nick end labelling
- WD
western diet
Introduction
Over 60% of the adult population in the USA is considered overweight or obese (Flegal et al. 2012) and this is accompanied by the growing incidence of non‐alcoholic fatty liver disease (NAFLD). NAFLD is a progressive liver disease ranging from simple steatosis (triglyceride storage ≥5% by weight), non‐alcoholic steatohepatitis (NASH), fibrosis and cirrhosis in the absence of excess alcohol consumption (< 20 g day–1) (Rector et al. 2008 b). NAFLD affects ∼30% of adults in the USA, with ∼20% of obese individuals developing hepatic inflammation, fibrosis and/or cirrhosis (Younossi et al. 2002). Habitual physical inactivity is also associated with NAFLD (Perseghin et al. 2007) and currently >90% of US adults do not meet physical activity recommendations (Troiano et al. 2008). Therefore, it is important to determine whether lifestyle modifications such as exercise training can effectively treat NASH.
Currently, appropriate animal models to assess mechanisms by which lifestyle modifications may improve NASH are lacking. Methionine choline deficient diets (Ota et al. 2007; Vetelainen et al. 2007; Staels et al. 2013) or carbon tetrachloride (Baeck et al. 2012; Staels et al. 2013) induce fibrosis, although these models do not mimic the pathology observed in humans because they cause weight loss (Ota et al. 2007; Vetelainen et al. 2007), lack insulin resistance (Vetelainen et al. 2007) or promote fibrosis without hepatic steatosis (Baeck et al. 2012; Staels et al. 2013). Diet‐induced obesity models are more physiologically relevant to human NASH, although most rodents do not develop significant liver fibrosis on a high‐fat diet alone (Kohli et al. 2010; Ishimoto et al. 2013; Savard et al. 2013). Dietary modifications in which fructose, sucrose and/or cholesterol are added to high‐fat diets appear to induce NASH in rodents (Kohli et al. 2010; Ishimoto et al. 2013; Savard et al. 2013; Mells et al. 2015) but may be limited by the duration of feeding. In the present study, we utilized non‐hyperphagic Long–Evans Tokushima Otsuka (LETO) rats and hyperphagic Otsuka Long‐Evans Tokushima Fatty (OLETF) rats to determine whether they could serve as physiologically relevant rodent models of NASH when they were fed a western diet (WD) high in fat, sucrose and cholesterol.
Lifestyle modifications that induce weight loss (exercise training, reduced energy intake) are recommended for individuals with NAFLD (Caldwell & Lazo, 2009) and have been shown to improve simple steatosis (Larson‐Meyer et al. 2008; Johnson et al. 2009; Elias et al. 2010; Lee et al. 2012; Linden et al. 2014; Linden et al. 2015); however, the effect of exercise training on NASH is not well understood. Exercise training has been shown to attenuate cardiac fibrosis (Emter & Baines, 2010; Kwak et al. 2011) and alter matrix metalloproteinase (MMP; proteins associated with extracellular matrix remodelling) content/activity within skeletal muscle (Rullman et al. 2009; Scheede‐Bergdahl et al. 2014). To our knowledge, no studies have assessed the effects of exercise training on hepatic MMPs in a model of NASH.
The OLETF rat is a commonly studied model of type 2 diabetes, in which animals are bred for null expression of the cholecystokinin‐1 receptor (Moran & Bi, 2006). Hyperphagic OLETF rats develop NAFLD on a standard chow diet and, by 32–40 weeks of age, develop hepatocellular inflammation and mild hepatic perivenular fibrosis that is not observed in non‐hyperphagic LETO control rats (Rector et al. 2011; Linden et al. 2015). These conditions can be prevented when OLETF rats are allowed to voluntary exercise on running wheels (Rector et al. 2008 a; Rector et al. 2011; Linden et al. 2015). Yet, it remains unclear whether treatment with treadmill exercise training following the consumption of a diet high in fat, sucrose and cholesterol can attenuate NASH, including fibrosis. The present study aimed to: (i) determine whether feeding LETO and OLETF rats a WD high in fat, sucrose and cholesterol can serve as a relevant model to human NASH and (ii) to test the hypothesis that aerobic exercise training can attenuate NASH, including fibrosis, in OLETF rats.
Methods
Animal protocol
The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Missouri and complied with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. The investigators understand the ethical principles under which the Journal of Physiology operates and the work presented complies with the animal ethics checklist.
Male LETO and OLETF rats (Tokushima Research Institute, Otsuka Pharmaceutical, Tokushima, Japan) were used to assess WD‐induced advancement of liver disease (i.e. hepatic fibrosis and inflammation). Animals (8 weeks old) were randomized to the groups (n = 10 per group): LETO fed a control diet (L‐CON), LETO fed WD (L‐WD), OLETF‐CON (O‐CON) or O‐WD for 24 weeks (up to 32 weeks of age). At 20 weeks of age, additional O‐WD animals were assigned to sedentary (O‐SED, n = 12), food restriction (O‐FR, n = 9) or exercise training (O‐EX, n = 10) conditions for 12 weeks to determine the therapeutic effects of lifestyle modification on NASH. Animals were individually housed in standard conditions (0600/1800 h light/dark cycle at 21°C). Body weight was measured weekly and type 2 diabetes associated‐weight loss, which typically begins at ∼32 weeks of age when provided standard chow, was assessed (Song et al. 2013). At 32 weeks of age, animals were anaesthetized by i.p. injection of sodium pentobarbital (100 mg kg–1) and killed by exsanguination following a 12 h fast.
Diet
The WD (D09071604; Research Diets Inc., New Brunswick, NJ, USA) contained 44.9% kcal fat, 35.1% kcal carbohydrate and 20% kcal protein, with 1% weight/weight from cholesterol and 17% kcal sucrose. CON contained 10% kcal fat, 70% kcal carbohydrate and 20% kcal protein, with 3.5% kcal sucrose (D12110704; Research Diets Inc.). Diet and water were provided ad libitum (except O‐FR) and caloric intake was determined.
Moderate intensity exercise training
At 20 weeks of age, O‐EX began treadmill running 5 days week–1 as described previously (Linden et al. 2014). The speed and duration of the treadmill exercise were gradually increased over the first 4 weeks of training until the animals could maintain a running speed of 20 m min–1 for 60 min day–1. By the fifth week of training, animals ran at 20 m min–1, 60 min day–1, on a 15% incline and maintained this until 32 weeks of age. Animals in the O‐SED and O‐FR groups were placed on the non‐moving treadmill weekly.
Food restriction
O‐FR underwent ∼25% reduction in kcal day–1 (vs. O‐SED) to match the body weights of O‐EX. Food was weighed and animals fed between 15.00 h and 18.00 h daily.
Dual‐energy X‐ray absorptiometry
Body composition was assessed before death as described previously (Linden et al. 2014; Linden et al. 2015).
Serum and whole blood measures
Fasting serum glucose (Thermo Scientific, Waltham, MA, USA) and insulin (Alpco, Salem, NH, USA) were assessed using commercially available assays. Haemoglobin A1c (HbA1c) was determined using a a DCA Vantage analyser (Siemens AG, Munich, Germany).
Tissue collection and preparation procedure
Livers were flash frozen in liquid nitrogen or placed in 10% formalin. The retroperitoneal, epididymal and omental fat pads were excised and weighed.
Intrahepatic lipid content, liver morphology and apoptosis
Formalin‐fixed, paraffin‐embedded livers were stained with haematoxylin and eosin or trichrome stain (IDEXX RADIL, Columbia, MO, USA). Biochemical intrahepatic triglyceride (TG) content was determined as described previously (Rector et al. 2008 a). NAFLD activity scores (NAS) were assessed as defined by Kleiner et al. (2005). Terminal deoxynucleotide transferase‐mediated dUTP nick end labelling (TUNEL; Roche Applied Science, Indianapolis, IN, USA) was used to determine apoptotic cell death.
Hepatic hydroxyproline content and gelatin zymography
Hepatic hydroxyproline content was assessed using a commercially available assay (Sigma, St Louis, MO, USA) and gelatinase activity was determined by gelatin zymography using commercially available reagents (Life Technologies, Grand Island, NY, USA).
Quantitative RT‐ PCR
Gene expression was quantified using the ABI 7500 Fast Sequence Detection System and Software (Applied Biosystems, Carlsbad, CA, USA) as described previously (Linden et al. 2014). Ppib served as the housekeeping gene. L‐CON was the referent group when determining whether WD could induce NASH in the normophagic LETO or the hyperphagic OLETF. Because we hypothesized that 12 weeks of lifestyle modification would not completely resolve NAFLD in animals with advanced liver disease, LETOs fed the western diet (L‐WD) were used as the referent group when assessing the effects of FR or EX.
Western blot analysis
Western blot analyses were conducted as described previously (Rector et al. 2008 a). CD68 antibody was from Santa Cruz Biotechnology (Dallas, TX, USA). Interleukin (IL)‐1β, α smooth muscle actin (αSMA), MMP‐2, MMP‐12 and CCN1 antibodies were obtained from Abcam (Cambridge, MA, USA). Transforming growth factor (TGF)‐β and cleaved caspase‐3 antibodies were obtained from Cell Signaling (Beverly, MA, USA). MMP‐9 and tissue inhibitor of metalloproteinase (TIMP)‐1 antibodies were obtained from EMD Millipore (Billerica, MA, USA). Protein bands were quantified using a densitometer (Bio‐Rad, Hercules, CA, USA).
Statistical analysis
Two‐way ANOVA was used to determine significant main effects between rat strain (LETO vs. OLETF) and diet (CON vs. WD), as well as significant strain × diet interactions. Fisher's least significant difference post hoc comparisons were used to determine differences when significant main effects and interactions were observed. To determine differences in the treatment arm of the study, a one‐way ANOVA with Fisher's least significant difference post hoc comparisons was performed (SPSS, version 22.0; IBM Corp., Armonk, NY, USA). Values are reported as the mean ± SE. P < 0.05 was considered statistically significant.
Results
Animal characteristics following 24 weeks of western diet
OLETF rats weighed significantly more and had a greater energy intake and greater adiposity than LETO rats (P < 0.001) (Table 1). Ad libitum access to the WD led to increased body weight (P < 0.01), energy intake (P < 0.001) and total fat pad mass (P < 0.001) compared to the CON diet for LETOs and OLETFs. The hyperphagic OLETF rats were more susceptible to increases in total fat pad mass with both the CON diet (P < 0.05 L‐CON vs. O‐CON) and the WD (P < 0.001 L‐WD vs. O‐WD). Fat pad mass was further increased with the WD in the OLETF (∼65% increase vs. O‐CON; P < 0.001).
Table 1.
Western diet‐induced effects on animal characteristics and NAFLD activity and fibrosis scores
| L‐CON | L‐WD | O‐CON | O‐WD | ||
|---|---|---|---|---|---|
| Body weight (g) | 526.8 ± 11.9 | 567.7 ± 12.9† | 648.3 ± 25.3*, ¶ | 746.9 ± 30.8† | |
| Weight loss from peak body weight (%) | –0.5 ± 0.2 | –0.1 ± 0.1 | –5.5 ± 1.9* | –3.3 ± 1.3* | |
| Mean energy intake (kcal week–1) | 516.5 ± 9.9 | 557.1 ± 10.2† | 749.4 ± 18.7* | 831.7 ± 8.9* † | |
| Percentage body fat (%) | 19.7 ± 1.3 | 22.5 ± 1.2 | 33.1 ± 3.4* | 38.2 ± 3.0* | |
| Fat pad mass (g) | 24.0 ± 1.7 | 37.2 ± 3.4†, § | 68.5 ± 6.7*, ¶ | 111.9 ± 10.3*, †, ‡, § | |
| NAS | Steatosis | 0.3 ± 0.1 | 2.6 ± 0.2† | 2.2 ± 0.2*, ‡ | 3.0 ± 0*, † |
| Inflammation | 0.1 ± 0.1 | 0.9 ± 0.2†, § | 0.8 ± 0.1*, ‡ | 2.9 ± 0.9*, †, ‡, § | |
| Ballooning | 0 ± 0 | 1.5 ± 0.2†, § | 1.4 ± 0.2*, ‡ | 2.0 ± 0*, †, ‡, § | |
| Total | 0.4 ± 0.2 | 5.0 ± 0.5† | 4.4 ± 0.4* | 7.9 ± 0.1*, † | |
| Fibrosis | Fibrosis score | 0 ± 0 | 0.9 ± 0.1† | 1.0 ± 0.2* | 2.3 ± 0.2*, † |
All values are the mean ± SE; n = 10/group. * P < 0.05, significant main effect of strain. † P < 0.05, significant main effect of diet. ‡ P < 0.05 for interaction (OLETF vs. LETO within diet). § P < 0.05 for interaction (CON vs. WD within strain, P < 0.05). ¶ P < 0.05 for interaction (L‐WD vs. O‐CON).
Liver phenotype following consumption of the western diet
WD contributed to the progression of liver disease in both LETOs and OLETFs (Fig. 1 A, trichrome stained images). O‐WD developed hallmark characteristics of NASH, including hepatocyte ballooning, nuclear displacement and bridging fibrosis (Fig. 1 A, arrows), which contributed to clinically significant NAS (P < 0.001 vs. O‐CON) (Table 1). OLETF rats also had increased relative liver weight (P < 0.001) (Fig. 1 B) and hepatic TG content (P < 0.001) (Fig. 1 C) compared to the LETOs. WD increased these variables in OLETF and LETO rats (P < 0.001), with greater susceptibility to WD‐induced hepatomegaly in OLETF compared to LETO rats (P < 0.001 L‐WD vs. O‐WD). O‐CON animals had relative liver weights that were ∼20% greater liver than L‐WD (P < 0.01), although biochemical liver TG content and steatosis score did not differ between L‐WD and O‐CON (P < 0.05) (Table 1). Although the WD increased collagen 1α1 mRNA expression, hepatic hydroxyproline content and the fibrosis score (Table 1) in both LETO and OLETF rats, the hyperphagic OLETF rat had greater WD‐induced injury, as indicated by an ∼2.5 fold increase in collagen 1α1 expression (P < 0.001 vs. O‐CON) (Fig. 1 D) and ∼80% increase in hepatic hydroxyproline content (P < 0.001 vs. O‐CON) (Fig. 1 E). Hepatic stellate cell (HSC) activation is a key contributor to fibrogenesis, and OLETF rats had greater hepatic protein expression of TGF‐β (P < 0.001) (Fig. 1 F) and αSMA (P < 0.01) (Fig. 1 F) compared to LETO rats. The WD also induced increases in hepatic TGF‐β (P < 0.05), with similar responses in both the OLETF and LETO rat strains. The OLETF rats demonstrated a marked WD‐induced increase in hepatic αSMA protein content (P < 0.001) (Fig. 1 F), which was not observed in the LETO rats. Additionally, CCN1, a protein associated with HSC senescence, was markedly lower in OLETF compared to LETO rats (P < 0.001) (Fig. 1 G) regardless of diet.
Figure 1. Western diet‐induced NASH liver phenotype .
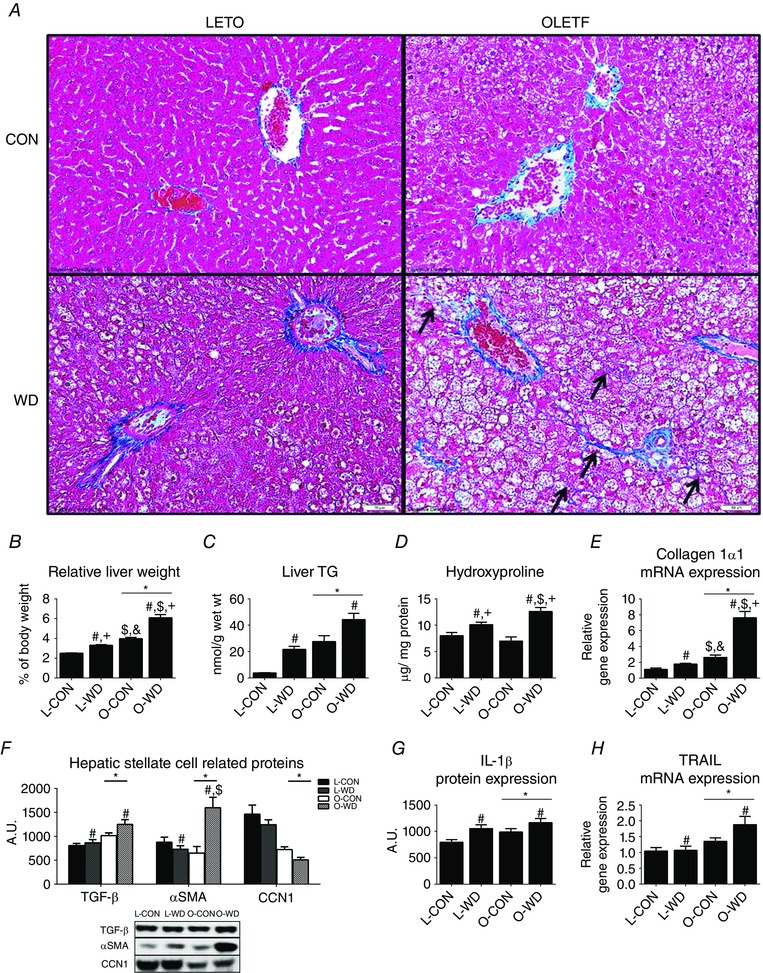
Representative trichrome stained images (A), relative liver weight (B), biochemical TG analysis (C), hepatic hydroxyproline content (D), collagen 1α1 mRNA expression (E), activated HSC related proteins (F), IL‐1β protein content (G) and TRAIL mRNA expression (H). All data are the mean ± SEM (n = 10 per group). * P < 0.05, significant main effect of strain. # P < 0.05, significant main effect of diet. $ P < 0.05 for interaction (OLETF vs. LETO within diet). + P < 0.05 for interaction (CON vs. WD within strain, P < 0.05). &P < 0.05 for interaction (L‐WD vs. O‐CON).
Inflammation and hepatocyte apoptosis are other hallmark characteristics of NASH. WD increased inflammation scores (P < 0.001) (Table 1) and hepatic IL‐1β protein expression (P < 0.01 vs. CON) (Fig. 1 G) in the LETO and the OLETF, with inflammation scores significantly elevated in O‐WD vs. L‐WD (P < 0.001) (Table 1). Cellular apoptosis was assessed by counting DAPI stained nuclei that were associated with TUNEL staining and no differences were observed among groups for the number of apoptotic cells per field of vision (data not shown) despite WD‐induced increases in hepatic TRAIL mRNA expression (P < 0.05) (Fig. 1 H).
Markers of extracellular matrix remodelling were assessed because of their importance in the regulation of fibrosis. WD feeding increased MMP‐2 gelatinase activity by ∼70% (P < 0.01) (Fig. 2 A) and MMP‐9 gelatinase activity by ∼140% (P < 0.01) (Fig. 2 A) despite a similar protein content for these MMPs among groups (Fig. 2 A). WD feeding also increased the hepatic protein expression of MMP‐12 (P < 0.01 vs. CON) (Fig. 2 B) and lowered TIMP‐1 protein content (P < 0.001 vs. CON) (Fig. 2 B), with O‐WD showing greater responses in both (P < 0.05 vs. L‐WD). Interestingly, there was a WD‐induced increase of ∼3.5 fold in MMP12: TIMP‐1 (P < 0.001), an indicator of extracellular matrix remodelling, with this being ∼40% lower in the OLETF vs. the LETO rats (P < 0.01) (Fig. 2 B).
Figure 2. Liver extracellular matrix remodelling markers .
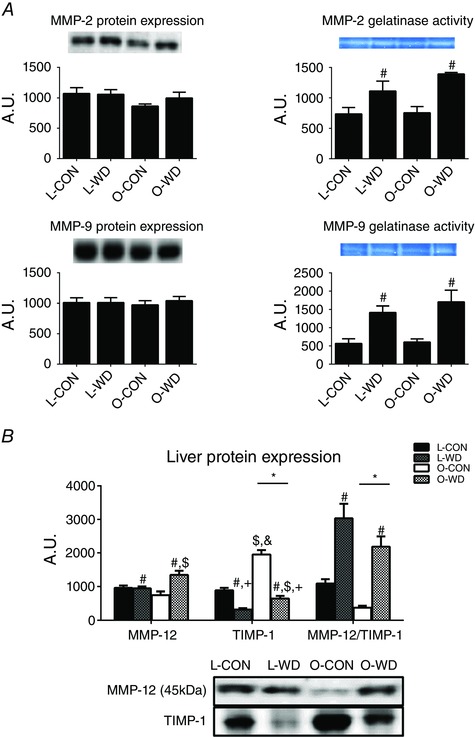
Hepatic MMP‐2 and MMP‐9 protein content and gelatinase activity (A) and MMP‐12 and TIMP‐1 protein expression (B). All values are the mean ± SEM (n = 10 per group). * P < 0.05, significant main effect of strain. # P < 0.05, significant main effect of diet. $ P < 0.05 for interaction (OLETF vs. LETO within diet). + P < 0.05 for interaction (CON vs. WD within strain, P < 0.05). &P < 0.05 for interaction (L‐WD vs. O‐CON). [Colour figure can be viewed at wileyonlinelibrary.com]
Alterations in body weight, adiposity and markers of type 2 diabetes with exercise training or food restriction
Having demonstrated that WD feeding induced a NASH phenotype in OLETF rats, we next tested the efficacy of FR or EX treatments on NASH‐related outcomes. Because it was considered improbable that either moderate intensity exercise training or modest food restriction would completely resolve NASH and restore the liver phenotype to that seen in L‐CON rat during a 12 week treatment period when maintained on a WD, L‐WD fed animals were used as control reference groups for these assessments. O‐SED animals weighed ∼25% more than L‐WD animals (P < 0.001) and this was associated with increased energy intake (P < 0.001), percentage body fat (P < 0.001) and fat pad mass (P < 0.001) (Table 2). No differences in body weight or fat pad mass were observed between O‐SED, O‐FR or O‐EX. O‐SED had lower percentage body fat than O‐FR (P < 0.05) despite a significantly greater weekly energy intake (P < 0.001) and this was probably a result of the ∼5% weight loss from peak body weight observed in O‐SED (P < 0.001) (Table 2). Consistent with our previous studies (Rector et al. 2010; Rector et al. 2011; Linden et al. 2014), O‐SED rats were hyperglycaemic and exercise training partially attenuated fasting blood glucose (P < 0.01 vs. O‐EX) (Table 2). Interestingly, fasting insulin concentrations were similar between O‐SED, O‐EX and O‐FR groups (Table 2), which was a result of the transition to frank type 2 diabetes and a loss of β‐cell function in the sedentary OLETF rats, whereas hyperinsulinaemia was prevented with EX and FR. This is supported by the significantly elevated HbA1c levels observed in O‐SED animals compared to EX and FR rats (P < 0.001 vs. O‐FR and O‐EX) (Table 2). The loss of body mass in the O‐SED rats with uncontrolled type 2 diabetes progression is consistent with the clinical literature, where type 2 diabetics lose muscle and fat mass as the condition worsens (Park et al. 2009).
Table 2.
Animal characteristics and NAFLD activity and fibrosis scores with FR or EX
| O‐SED | O‐FR | O‐EX | ||
|---|---|---|---|---|
| Body weight (g) | 706.5 ± 20.4 | 739.8 ± 8.3 | 720.4 ± 10.6 | |
| Weight loss from peak body weight (%) | −5.0 ± 1.0a | −0.2 ± 0.1b | −1.2 ± 0.8b | |
| Mean energy intake (kcal week–1) | 857.3 ± 40.4a | 687.6 ± 0.0b | 758.1 ± 9.7c | |
| Percent body fat (%) | 34.2 ± 2.4a | 39.6 ± 1.4b | 38.1 ± 1.3a , b | |
| Fat pad mass (g) | 97.1 ± 1.3 | 107.4 ± 4.4 | 102.1 ± 5.4 | |
| Glucose (mg dl–1) | 343.8 ± 21.5a | 296.6 ± 11.5a,b | 253.4 ± 13.8b | |
| Insulin (ng ml–1) | 2.8 ± 0.7 | 4.3 ± 0.3 | 2.7 ± 0.4 | |
| HbA1c (% glycosylated) | 9.7 ± 0.01a | 4.1 ± 0.01b | 4.3 ± 0.03b | |
| NAS | Steatosis | 3.0 ± 0 | 3.0 ± 0 | 3.0 ± 0 |
| Inflammation | 2.5 ± 0.2a | 1.5 ± 0.3b | 1.8 ± 0.3b | |
| Ballooning | 2.0 ± 0 | 2.0 ± 0 | 2.0 ± 0 | |
| Total | 7.5 ± 0.2a | 6.5 ± 0.3b | 6.8 ± 0.3b | |
| Fibrosis | Fibrosis score | 2.7 ± 0.2a | 1.7 ± 0.2b | 1.9 ± 0.3b |
All values are the mean ± SE; n = 9‐12/group except HbA1c (n = 3 per group). Values with different superscript letters are significantly different (P < 0.05).
Changes in liver phenotype with exercise training or food restriction
O‐SED developed progressive liver disease that was partially attenuated with FR or EX, as indicated by reduced fibrosis (less trichrome staining) (Fig. 3 A) and lower NAS (P < 0.05) (Table 2). Both FR and EX reduced relative liver weight by ∼25–50% (P < 0.001 vs. O‐SED) (Fig. 3 B), although no differences were observed among OLETF groups in hepatic TG content (Fig. 3 C) or steatosis score (Table 2). Hepatic hydroxyproline content was reduced with FR (P < 0.01 vs. O‐SED) (Fig. 3 D), normalizing it to levels similar to L‐WD, whereas EX lowered hepatic mRNA expression of collagen 1α1 (Col1α1; P < 0.05 vs. O‐SED) (Fig. 3 E). Both FR and EX similarly reduced fibrosis scores vs. O‐SED (P < 0.01) (Table 2), although neither completely reversed the condition.
Figure 3. NASH liver phenotype with EX or FR .
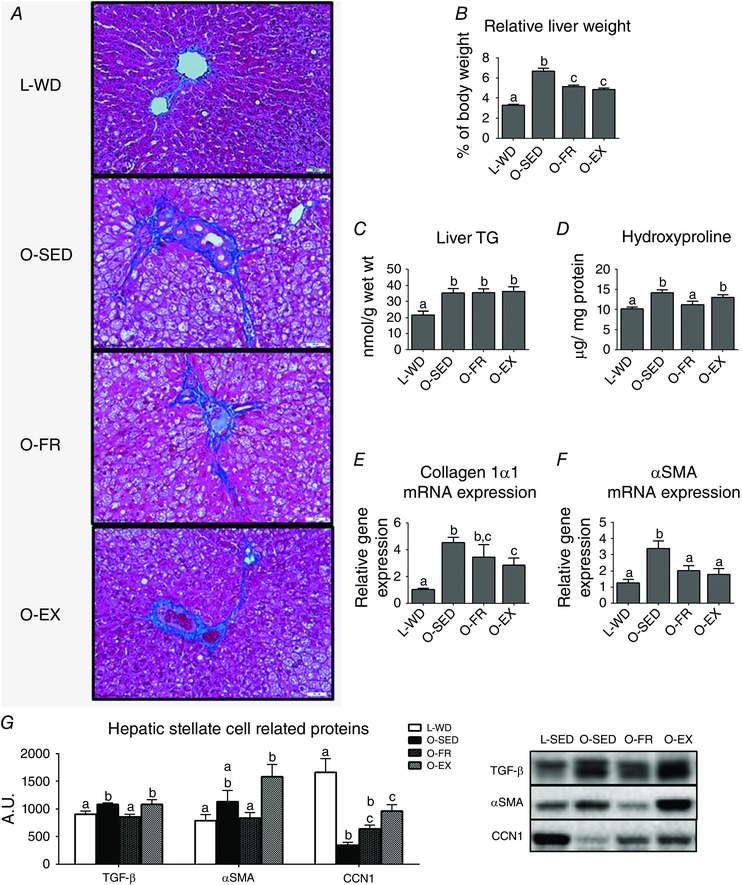
Representative trichrome stain images (A), relative liver weight (B), biochemical TG analysis (C), hepatic hydroxyproline content (D), collagen 1α1 mRNA expression (E), α smooth muscle actin mRNA expression (F) and protein content of HSC markers (G). All values are the mean ± SEM (n = 9–12 per group). Values with different superscripts are significantly different, P < 0.05.
When assessing measures related to HSC activation, both FR and EX lowered hepatic αSMA mRNA expression by ∼40–50% compared to O‐SED (P < 0.05) (Fig. 3 F), although these reductions in mRNA did not coincide with lower αSMA protein content (Fig. 3 G). FR also attenuated hepatic protein expression of TGF‐β (P < 0.05 vs. O‐SED) (Fig. 3 G). Interestingly, only EX increased CCN1 protein expression (P < 0.01 vs. O‐SED), partially restoring it to the level of L‐WD (P < 0.01) (Fig. 3 G).
Both FR and EX lowered hepatic inflammation scores compared to O‐SED (P < 0.01 vs. O‐SED) (Table 2). FR also lowered the hepatic mRNA expression of MCP‐1 (P < 0.001) (Fig. 4 A) and protein content of CD68 (P < 0.01) (Fig. 4 B) vs. O‐SED, restoring them to levels of L‐WD. Additionally, FR attenuated hepatic IL‐1β protein (P < 0.001) (Fig. 4 C) and tumor necrosis factor α mRNA (P < 0.01) (Fig. 4 D) compared to O‐SED.
Figure 4. Liver macrophage markers with FR or EX .
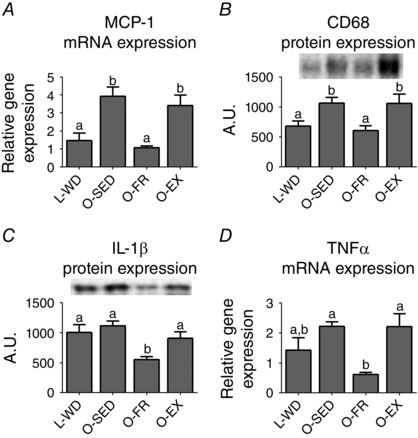
Liver monocyte chemoattractant protein‐1 mRNA expression (A), CD68 protein expression (B), IL‐1β protein expression (C) and tumor necrosis factor α mRNA expression (D). All values are the mean ± SEM (n = 9–12 per group). Values with different superscripts are significantly different, P < 0.05.
Finally, extracellular matrix remodelling‐related markers were assessed. MMP‐2 and MMP‐9 gelatinase activity were elevated in O‐SED rats (P < 0.01 vs. L‐WD for both) (Fig. 5 A) despite no differences in hepatic MMP‐2 protein expression and ∼10% lowering of MMP‐9 protein expression (P < 0.05 vs. L‐WD) (Fig. 5 A). EX and FR normalized MMP‐2 activity and FR normalized MMP‐9 activity to levels similar to L‐WD. Additionally, both EX and FR attenuated hepatic MMP‐12 protein expression by ∼15–20% (P < 0.05) and the MMP‐12: TIMP‐1 ratio by ∼25‐30% vs. O‐SED (P < 0.01) (Fig. 5 B).
Figure 5. Liver extracellular matrix remodelling markers with EX or FR .

Hepatic MMP‐2 and MMP‐9 protein content and gelatinase activity (A) and MMP‐12 and TIMP‐1 protein expression (B). All values are the mean ± SEM (n = 9–12 per group). Values with different superscripts are significantly different, P < 0.05. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Food restriction and exercise training are known to prevent and improve NAFLD related outcomes (Rector & Thyfault, 2011); however, the efficacy of these lifestyle modifications in the treatment of liver fibrosis remains unclear. Although chemical inducers and methionine‐choline deficient diet models are often used to quickly induce NASH in rodents, these models do not result in a pathology consistent with that observed in patients with NASH (Ota et al. 2007; Vetelainen et al. 2007; Baeck et al. 2012; Staels et al. 2013). In the present study, we aimed to utilize a more pathologically and physiologically relevant model of NASH and demonstrated that OLETF rats fed a WD containing high fat, high sucrose and high cholesterol for 24 weeks developed not only frank type 2 diabetes, but also hepatic inflammation, significant fibrosis and elevated NAS consistent with human NASH. Although FR provided better protection in some characteristics of NASH, both FR and EX beneficially affected markers of HSC activation and extracellular matrix remodelling , as well as reduced fibrosis.
The prevalence of NASH is growing at alarming rates, with ∼20% of obese individuals developing liver disease that includes inflammation, fibrosis and/or cirrhosis (Younossi et al. 2002), although the assessment of therapies for NASH in humans is difficult because it requires liver biopsies. Physiologically relevant animal models are limited but, recently, the addition of cholesterol to rodent diets was shown to increase the number of inflammatory cell foci by ∼1.5 fold (Liang et al. 2014; Mells et al. 2015) and also induce mild to severe hepatic fibrosis (Ichimura et al. 2015). OLETF rats fed rodent chow develop mild perivenular fibrosis (Linden et al. 2015), which was shown to be enhanced with the WD in the present study, in which O‐WD developed bridging fibrosis, severe biliary fibrosis, elevated hepatic hydroxyproline content and hepatocellular inflammation. Because the observed liver phenotype more closely resembles human NASH, this model was used to better understand the therapeutic effects of lifestyle interventions on liver fibrosis.
One of the first recommendations for those diagnosed with NAFLD/NASH is to undergo a weight management programme (energy restriction, exercise training) and recent evidence suggests that ∼10% reductions in body weight are needed for histological improvements in NAFLD patients (Thoma et al. 2012). More severe weight loss induced from gastric bypass surgery has been shown to provide some degree of NASH resolution, with improvements in hepatic inflammation, fibrosis and NAS score (Liu et al. 2007), whereas more modest reductions in body weight (–5.8%) from lifestyle modifications improved fibrosis in ∼50% of the participants (Hickman et al. 2004). Exercise training may also contribute to improved liver histology in NASH, with human cross‐sectional data suggesting that vigorous exercise has greater benefit (Kistler et al. 2011). We demonstrate that moderate intensity EX and modest FR reduced histological fibrosis without weight reduction or lower liver TG content in the OLETF rat, highlighting the importance of lifestyle modification in NASH. Reductions in fibrosis were accompanied by ∼25% reduction in col1α1 mRNA expression with EX and ∼10% reduction in hepatic hydroxyproline content with FR. Although fibrosis was attenuated with these treatments, significant hepatic fibrosis was still present. Future studies are needed to determine whether a vigorous or high‐intensity exercise training regimen may be more efficacious for the treatment of NASH‐related fibrosis.
Another hallmark characteristic of NASH is inflammation. Importantly, hepatic pro‐inflammatory immune responses promote activation and the survival of HSCs (Pradere et al. 2013) and TGF‐β related fibrogenesis (Hellerbrand et al. 1999). Hyperphagia in combination with WD increased inflammatory cell infiltration in the present study, whereas the WD had a more modest effect on hepatic inflammation in the non‐hyperphagic LETO. Both FR and EX improved hepatic inflammation scores, whereas FR lowered the protein content of CD68 and IL‐1β and tumor necrosis factor α mRNA expression. These data highlight the importance of limiting poor dietary choices because diet‐induced injury may contribute to HSC activation and fibrogenesis.
Once activated, HSCs promote fibrogensis by up‐regulating TGF‐β, col1α1 and PDGF‐β (Czochra et al. 2006; Giraudi et al. 2015). In the present study, ad libitum access to WD promoted HSC activation in both the LETO and the OLETF, although hyperphagic animals were more susceptible to increases in TGF‐β and αSMA. Lifestyle modification effectively lowered markers of HSC activation, with FR lowering hepatic TGF‐β protein content, and both FR and EX lowering hepatic αSMA mRNA expression by ∼40–50%, probably managing the initial step in fibrogenesis and limiting fibrosis. This potential regulation of initiation of fibrogenesis with lifestyle modification is further supported by alterations in CCN1. CCN1 expression is increased within hepatocytes and HSCs during early liver injury and cirrhosis (Bian et al. 2013; Kim et al. 2013; Borkham‐Kamphorst et al. 2014) and may have protective effects. Gene deletion of CCN1 is associated with a reduction in senescent HSCs and induces bridging fibrosis (Kim et al. 2013). Conversely, overexpression of CCN1 decreases αSMA and col1α1 mRNA (Borkham‐Kamphorst et al. 2014), supporting its beneficial effects on NASH‐related fibrosis. CCN1 can bind to TGF‐β and lower SMAD signalling to prevent collagen deposition (Borkham‐Kamphorst et al. 2014) and has been associated with endoplasmic reticulum stress‐induced apoptosis of HSCs (Borkham‐Kamphorst et al. 2016). In the present study, CCN1 expression was lower in the OLETF compared to LETO rats and exercising training increased it by ∼2.8 fold compared to O‐SED. These effects on CCN1 were not observed with modest FR. These data indicate that exercise training may have therapeutic effects, in part by increasing CCN1, perhaps reducing the potential for HSC activation and limiting fibrogenesis. This concept warrants further, more mechanistic investigations.
Hepatocyte apoptosis is often elevated in NASH patients and these increases can contribute to HSC activation and liver fibrosis (Canbay et al. 2002; Ribeiro et al. 2004). The induction of apoptosis can occur from an up‐regulation of the apoptotic factor TRAIL. In the present study, hepatic TRAIL mRNA was increased with WD in OLETF rats; however, TUNEL staining was not affected despite the NASH phenotype. Although some apoptosis is probably present, this does not account for the dramatically different liver phenotypes observed between O‐WD and L‐WD or the partial resolution of NASH with FR or EX.
Another means by which NASH related fibrosis may be regulated is via alterations in extracellular matrix remodelling proteins because of their ability to regulate collagen turnover. Circulating MMPs are increased in NASH (D'Amico et al. 2010), whereas models of liver fibrosis have increased the hepatic expression of MMPs (Wanninger et al. 2011; Nunes de Carvalho et al. 2013). MMPs do not respond uniformly during pathology and some MMPs promote fibrogenesis, whereas others contribute to fibrolysis. MMP‐12 deficiency is associated with a lowered liver hydroxyproline content and increased gelatinase (MMP‐2 and MMP‐9) activity during acute injury (Madala et al. 2010), supporting its contribution to fibrogenesis. Overexpression of hepatic PDGF‐β increases MMP‐2 and MMP‐9 activity (Czochra et al. 2006), perhaps serving as a protective mechanism during HSC activation to limit fibrogenesis. In the present study, increased hepatic fibrosis in O‐WD corresponded to elevations in MMP‐12 protein expression, consistent with MMP‐12 playing an important role in fibrogenesis. The WD triggered active remodelling in both L‐WD and O‐WD rats, with an increased ratio of MMP‐12:TIMP‐1 and MMP‐2 collagenase activity, although it was the hyperphagic condition in the OLETF that resulted in greater hepatic collagen deposition.
Additionally, we determined the efficacy of EX or FR on these markers of extracellular matrix remodelling aiming to determine whether these therapies could contribute to fibrosis resolution. Exercise training has been shown to alter the content and activity of MMPs in skeletal muscle (Rullman et al. 2009; Scheede‐Bergdahl et al. 2014). Previous work has also shown that apoptosis of HSCs (Oakley et al. 2005) or the injection of CCN1 (Kim et al. 2013) was associated with increased gelatinase expression/activity and decreased TIMP‐1 expression. Taken together, this indicates that an overall change in HSC activation may alter gelatinase activity and limit liver fibrosis. In the present study, FR and EX training lowered markers of HSC activation and EX increased CCN1 but alterations in HSC activation were not associated with MMP‐2 activity because MMP‐2 activity was suppressed in O‐FR and O‐EX vs. O‐SED. Interestingly, both MMP‐9 activity and TIMP‐1 were suppressed with FR; similar findings were seen in hepatic macrophage markers, suggesting that macrophages may influence MMP‐9 activity in the OLETF rat, although further studies are needed to better elucidate these mechanisms. It is important to note that both FR and EX suppressed active MMP‐12 protein expression compared to O‐SED because MMP‐12 can have inhibitory effects on the anti‐fibrotic MMP‐13 (Madala et al. 2010). Overall, these findings suggest that FR and EX training have modest and independent effects on ECM remodelling proteins and that the differential responses in the ECM remodelling proteins may result from the decelerated progression of liver disease with these therapies.
In summary, both LETO and OLETF rats had maladaptive health outcomes when provided ad libitum access to a WD high in sucrose, fat and cholesterol for 24 weeks but the hyperphagic OLETF rat developed a more severe NASH phenotype with inflammation and bridging fibrosis. Moreover, although neither exercise training, nor food restriction therapy resulted in complete resolution of the WD‐induced NASH phenotype, both EX and FR had independent beneficial effects on reducing liver fibrosis that occurred without significant reductions in body weight or improvements in hepatic steatosis. These improvements probably resulted from lowering HSC activation and altering extracellular matrix remodelling proteins. These findings highlight the beneficial effects of lifestyle modification on liver fibrosis and should be considered when designing therapeutic regimens for NASH patients.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
MAL, RDS, FWB, JAK, VJV, JRS, JAI, JPT, MHL and RSR were involved in the conception or design of the study. MAL, RDS, GMM, LCO, EMM, JPT and RSR were involved in the acquisition, analysis or interpretation of data. MAL, RDS, GMM, LCO, EMM, FWB, JAK, VJV, JRS, JAI, JPT, MHL and RSR were involved in drafting the work or revising it critically for important intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was partially supported by NIH DK088940 (JPT), RO1HL036088 (MHL), HL73101‐07 (JRS), HL107910‐03 (JRS), VA‐Merit System 0018 (JRS), 1I01BX002567‐01 (JPT) and VHA‐CDA2 IK2BX001299 (RSR), and University of Missouri Molecular Life Sciences Fellowship (RDS) and University of Missouri Research Council Grant (RSR).
Acknowledgements
The authors gratefully acknowledge the excellent technical assistance of David Bayless, Dr. T. Dylan Olver and Pam Thorne. This work was supported with resources and the use of facilities at the Harry S Truman Memorial Veterans Hospital in Columbia, MO.
This is an Editor's Choice article from the 15 September 2016 issue.
References
- Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, Trautwein C & Tacke F (2012). Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61, 416–426. [DOI] [PubMed] [Google Scholar]
- Bian Z, Peng Y, You Z, Wang Q, Miao Q, Liu Y, Han X, Qiu D, Li Z & Ma X (2013). CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res 54, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkham‐Kamphorst E, Schaffrath C, Van de Leur E, Haas U, Tihaa L, Meurer SK, Nevzorova YA, Liedtke C & Weiskirchen R (2014). The anti‐fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF‐beta signalling. Biochim Biophys Acta 1843, 902–914. [DOI] [PubMed] [Google Scholar]
- Borkham‐Kamphorst E, Steffen BT, Van de Leur E, Haas U, Tihaa L, Friedman SL & Weiskirchen R (2016). CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress‐related apoptosis. Cell Signal 28, 34–42. [DOI] [PubMed] [Google Scholar]
- Caldwell S & Lazo M (2009). Is exercise an effective treatment for NASH? Knowns and unknowns. Ann Hepatol 8 (Suppl 1), S60–S66. [PubMed] [Google Scholar]
- Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ & Gores GJ (2002). Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology 123, 1323–1330. [DOI] [PubMed] [Google Scholar]
- Czochra P, Klopcic B, Meyer E, Herkel J, Garcia‐Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle PR, Lohse AW & Kanzler S (2006). Liver fibrosis induced by hepatic overexpression of PDGF‐B in transgenic mice. J Hepatol 45, 419–428. [DOI] [PubMed] [Google Scholar]
- D'Amico F, Consolo M, Amoroso A, Skarmoutsou E, Mauceri B, Stivala F, Malaponte G, Bertino G, Neri S & Mazzarino MC (2010). Liver immunolocalization and plasma levels of MMP‐9 in non‐alcoholic steatohepatitis (NASH) and hepatitis C infection. Acta Histochem 112, 474–481. [DOI] [PubMed] [Google Scholar]
- Elias MC, Parise ER, de Carvalho L, Szejnfeld D & Netto JP (2010). Effect of 6‐month nutritional intervention on non‐alcoholic fatty liver disease. Nutrition 26, 1094–1099. [DOI] [PubMed] [Google Scholar]
- Emter CA & Baines CP (2010). Low‐intensity aerobic interval training attenuates pathological left ventricular remodelling and mitochondrial dysfunction in aortic‐banded miniature swine. Am J Physiol Heart Circ Physiol 299, H1348–H1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK & Ogden CL (2012). Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 307, 491–497. [DOI] [PubMed] [Google Scholar]
- Giraudi PJ, Becerra VJ, Marin V, Chavez‐Tapia NC, Tiribelli C & Rosso N (2015). The importance of the interaction between hepatocyte and hepatic stellate cells in fibrogenesis induced by fatty accumulation. Exp Mol Pathol 98, 85–92. [DOI] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER & Brenner DA (1999). The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 30, 77–87. [DOI] [PubMed] [Google Scholar]
- Hickman IJ, Jonsson JR, Prins JB, Ash S, Purdie DM, Clouston AD & Powell EE (2004). Modest weight loss and physical activity in overweight patients with chronic liver disease results in sustained improvements in alanine aminotransferase, fasting insulin, and quality of life. Gut 53, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura M, Kawase M, Masuzumi M, Sakaki M, Nagata Y, Tanaka K, Suruga K, Tamaru S, Kato S, Tsuneyama K & Omagari K (2015). High‐fat and high‐cholesterol diet rapidly induces non‐alcoholic steatohepatitis with advanced fibrosis in Sprague–Dawley rats. Hepatol Res 45, 458–469. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Rivard CJ, Roncal‐Jimenez CA, Orlicky DJ, Cicerchi C, McMahan RH, Abdelmalek MF, Rosen HR, Jackman MR, MacLean PS, Diggle CP, Asipu A, Inaba S, Kosugi T, Sato W, Maruyama S, Sanchez‐Lozada LG, Sautin YY, Hill JO, Bonthron DT & Johnson RJ (2013). High‐fat and high‐sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 58, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NA, Sachinwalla T, Walton DW, Smith K, Armstrong A, Thompson MW & George J (2009). Aerobic exercise training reduces hepatic and visceral lipids in obese individuals without weight loss. Hepatology 50, 1105–1112. [DOI] [PubMed] [Google Scholar]
- Kim KH, Chen CC, Monzon RI & Lau LF (2013). Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 33, 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler KD, Brunt EM, Clark JM, Diehl AM, Sallis JF & Schwimmer JB (2011). Physical activity recommendations, exercise intensity, and histological severity of nonalcoholic fatty liver disease. Am J Gastroenterol 106, 460–468; quiz 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp‐Arida A, Yeh M, McCullough AJ, Sanyal AJ & the Nonalcoholic Steatohepatitis Clinical Research Network (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, Woods SC & Seeley RJ (2010). High‐fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52, 934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak HB, Kim JH, Joshi K, Yeh A, Martinez DA & Lawler JM (2011). Exercise training reduces fibrosis and matrix metalloproteinase dysregulation in the aging rat heart. FASEB J 25, 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson‐Meyer DE, Newcomer BR, Heilbronn LK, Volaufova J, Smith SR, Alfonso AJ, Lefevre M, Rood JC, Williamson DA & Ravussin E (2008). Effect of 6‐month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity (Silver Spring) 16, 1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Bacha F, Hannon T, Kuk JL, Boesch C & Arslanian S (2012). Effects of aerobic versus resistance exercise without caloric restriction on abdominal fat, intrahepatic lipid, and insulin sensitivity in obese adolescent boys: a randomized, controlled trial. Diabetes 61, 2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Lindeman JH, Menke AL, Koonen DP, Morrison M, Havekes LM, van den Hoek AM & Kleemann R (2014). Metabolically induced liver inflammation leads to NASH and differs from LPS‐ or IL‐1beta‐induced chronic inflammation. Lab Invest 94, 491–502. [DOI] [PubMed] [Google Scholar]
- Linden MA, Fletcher JA, Morris EM, Meers GM, Kearney ML, Crissey JM, Laughlin MH, Booth FW, Sowers JR, Ibdah JA, Thyfault JP & Rector RS (2014). Combining metformin and aerobic exercise training in the treatment of type 2 diabetes and NAFLD in OLETF rats. Am J Physiol Endocrinol Metab 306, E300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden MA, Fletcher JA, Morris EM, Meers GM, Laughlin MH, Booth FW, Sowers JR, Ibdah JA, Thyfault JP & Rector RS (2015). Treating NAFLD in OLETF rats with vigorous‐intensity interval exercise training. Med Sci Sports Exerc 47, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lazenby AJ, Clements RH, Jhala N & Abrams GA (2007). Resolution of nonalcoholic steatohepatits after gastric bypass surgery. Obes Surg 17, 486–492. [DOI] [PubMed] [Google Scholar]
- Madala SK, Pesce JT, Ramalingam TR, Wilson MS, Minnicozzi S, Cheever AW, Thompson RW, Mentink‐Kane MM & Wynn TA (2010). Matrix metalloproteinase 12‐deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL‐13‐dependent fibrosis. J Immunol 184, 3955–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mells JE, Fu PP, Kumar P, Smith T, Karpen SJ & Anania FA (2015). Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J Nutr Biochem 26, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran TH & Bi S (2006). Hyperphagia and obesity in OLETF rats lacking CCK‐1 receptors. Philos Trans R Soc Lond B Biol Sci 361, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes de Carvalho S, Helal‐Neto E, de Andrade DC, Costa Cortez EA, Thole AA, Barja‐Fidalgo C & de Carvalho L (2013). Bone marrow mononuclear cell transplantation increases metalloproteinase‐9 and 13 and decreases tissue inhibitors of metalloproteinase‐1 and 2 expression in the liver of cholestatic rats. Cells Tissues Organs 198, 139–148. [DOI] [PubMed] [Google Scholar]
- Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, May MJ, Millward‐Sadler H, Wright MC & Mann DA (2005). Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology 128, 108–120. [DOI] [PubMed] [Google Scholar]
- Ota T, Takamura T, Kurita S, Matsuzawa N, Kita Y, Uno M, Akahori H, Misu H, Sakurai M, Zen Y, Nakanuma Y & Kaneko S (2007). Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 132, 282–293. [DOI] [PubMed] [Google Scholar]
- Park SW, Goodpaster BH, Lee JS, Kuller LH, Boudreau R, de Rekeneire N, Harris TB, Kritchevsky S, Tylavsky FA, Nevitt M, Cho YW, Newman AB, Health A & Body Composition S (2009). Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32, 1993–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perseghin G, Lattuada G, De Cobelli F, Ragogna F, Ntali G, Esposito A, Belloni E, Canu T, Terruzzi I, Scifo P, Del Maschio A & Luzi L (2007). Habitual physical activity is associated with intrahepatic fat content in humans. Diabetes Care 30, 683–688. [DOI] [PubMed] [Google Scholar]
- Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, Dragomir AC, Aloman C & Schwabe RF (2013). Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector RS & Thyfault JP (2011). Does physical inactivity cause nonalcoholic fatty liver disease? J Appl Physiol 111, 1828–1835. [DOI] [PubMed] [Google Scholar]
- Rector RS, Thyfault JP, Morris RT, Laye MJ, Borengasser SJ, Booth FW & Ibdah JA (2008. a). Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long‐Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol 294, G619–G626. [DOI] [PubMed] [Google Scholar]
- Rector RS, Thyfault JP, Wei Y & Ibdah JA (2008. b). Non‐alcoholic fatty liver disease and the metabolic syndrome: an update. World J Gastroenterol 14, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector RS, Uptergrove GM, Borengasser SJ, Mikus CR, Morris EM, Naples SP, Laye MJ, Laughlin MH, Booth FW, Ibdah JA & Thyfault JP (2010). Changes in skeletal muscle mitochondria in response to the development of type 2 diabetes or prevention by daily wheel running in hyperphagic OLETF rats. Am J Physiol Endocrinol Metab 298, E1179–E1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector RS, Uptergrove GM, Morris EM, Borengasser SJ, Laughlin MH, Booth FW, Thyfault JP & Ibdah JA (2011). Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am J Physiol Gastrointest Liver Physiol 300, G874–G883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro PS, Cortez‐Pinto H, Sola S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME & Rodrigues CM (2004). Hepatocyte apoptosis, expression of death receptors, and activation of NF‐kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol 99, 1708–1717. [DOI] [PubMed] [Google Scholar]
- Rullman E, Norrbom J, Stromberg A, Wagsater D, Rundqvist H, Haas T & Gustafsson T (2009). Endurance exercise activates matrix metalloproteinases in human skeletal muscle. J Appl Physiol (1985) 106, 804–812. [DOI] [PubMed] [Google Scholar]
- Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, Chait A, Yeh MM, Quinn LS & Ioannou GN (2013). Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 57, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheede‐Bergdahl C, Bergdahl A, Schjerling P, Qvortrup K, Koskinen SO & Dela F (2014). Exercise‐induced regulation of matrix metalloproteinases in the skeletal muscle of subjects with type 2 diabetes. Diab Vasc Dis Res 11, 324–334. [DOI] [PubMed] [Google Scholar]
- Song YS, Fang CH, So BI, Park JY, Lee Y, Shin JH, Jun DW, Kim H & Kim KS (2013). Time course of the development of nonalcoholic fatty liver disease in the Otsuka Long–Evans Tokushima Fatty rat. Gastroenterol Res Pract 2013, 342648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, Baron M, Lucas A, Tailleux A, Hum DW, Ratziu V, Cariou B & Hanf R (2013). Hepatoprotective effects of the dual peroxisome proliferator‐activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 58, 1941–1952. [DOI] [PubMed] [Google Scholar]
- Thoma C, Day CP & Trenell MI (2012). Lifestyle interventions for the treatment of non‐alcoholic fatty liver disease in adults: a systematic review. J Hepatol 56, 255–266. [DOI] [PubMed] [Google Scholar]
- Troiano RP, Berrigan D, Dodd KW, Masse LC, Tilert T & McDowell M (2008). Physical activity in the United States measured by accelerometer. Med Sci Sports Exerc 40, 181–188. [DOI] [PubMed] [Google Scholar]
- Vetelainen R, van Vliet A & van Gulik TM (2007). Essential pathogenic and metabolic differences in steatosis induced by choline or methione‐choline deficient diets in a rat model. J Gastroenterol Hepatol 22, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Wanninger J, Walter R, Bauer S, Eisinger K, Schaffler A, Dorn C, Weiss TS, Hellerbrand C & Buechler C (2011). MMP‐9 activity is increased by adiponectin in primary human hepatocytes but even negatively correlates with serum adiponectin in a rodent model of non‐alcoholic steatohepatitis. Exp Mol Pathol 91, 603–607. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Diehl AM & Ong JP (2002). Nonalcoholic fatty liver disease: an agenda for clinical research. Hepatology 35, 746–752. [DOI] [PubMed] [Google Scholar]