

Abstract
Arterioles in the peripheral microcirculation are exquisitely sensitive to changes in in their environment: increases in cause vasoconstriction while decreases in result in vasodilatation. However, the cell type that senses O2 (the O2 sensor) and the signalling pathway that couples changes in to changes in arteriolar tone (the mechanism of action) remain unclear. Many (but not all) ex vivo studies of isolated cannulated resistance arteries and large, first‐order arterioles support the hypothesis that these vessels are intrinsically sensitive to with the smooth muscle, endothelial cells, or red blood cells serving as the O2 sensor. However, in situ studies testing these hypotheses in downstream arterioles have failed to find evidence of intrinsic O2 sensitivity, and instead have supported the idea that extravascular cells sense O2. Similarly, ex vivo studies of isolated, cannulated resistance arteries and large first‐order arterioles support the hypotheses that O2‐dependent inhibition of production of vasodilator cyclooxygenase products or O2‐dependent destruction of nitric oxide mediates O2 reactivity of these upstream vessels. In contrast, most in vivo studies of downstream arterioles have disproved these hypotheses and instead have provided evidence supporting the idea that O2‐dependent production of vasoconstrictors mediates arteriolar O2 reactivity, with significant regional heterogeneity in the specific vasoconstrictor involved. Oxygen‐induced vasoconstriction may serve as a protective mechanism to reduce the oxidative burden to which a tissue is exposed, a process that is superimposed on top of the local mechanisms which regulate tissue blood flow to meet a tissue's metabolic demand.
Keywords: arterioles, microcirculation, oxygen, oxygen sensing, vasoconstriction, vasodilatation
Abbreviations
- BKCa channels, large‐conductance
Ca2+‐activated K+ channels
- CaL channels
L‐type Ca2+ channels
- ClCa channels
Ca2+‐activated Cl− channels
- CYP450
cytochrome P450
- CysLTs
cysteinyl leukotrienes
- CysLTRs
CysLT receptors
- DDMS
N‐methylsulfonyl‐12,12‐dibromododec‐11‐enamide
- DIDS
4,4′‐diisothiocyano‐2,2′‐stilbenedisulfonic acid
- ETYA
eicosatetraynoic acid
- FLAP
5‐lipoxygenase‐activating protein
- 20‐HETE
20‐hydroxyeicosatetraenoic acid
- 5‐LO
5‐lipoxygenase
- LTA4
leukotriene A4
- LTC4
leukotriene C4
- LTD4
leukotriene D4
- LTE4
leukotriene E4
- 17‐ODYA
17‐octadecynoic acid
partial pressure of oxygen
- PSS
physiological salt solution
Introduction
Oxygen has been implicated in the local control of blood flow for more than a century (Sparks, 1980; Renkin, 1984; Kontos & Wei, 1985; Golub & Pittman, 2013). In tissues such as skeletal muscle, heart, brain, kidneys and the gut, blood flow (O2 supply) is directly proportional to tissue oxygen consumption (O2 demand) (Sparks, 1980; Renkin, 1984; Kontos & Wei, 1985; Golub & Pittman, 2013). Oxygen as one link between O2 demand and O2 supply provides a compact control system that would help ensure that O2 supply meets the tissues’ demand for O2. For O2 to participate in the local regulation of blood flow, arterioles in the microcirculation must be able to sense changes in the of the surrounding tissue, and then respond appropriately to either constrict, if O2 supply exceeds O2 demand and tissue increases, or dilate, if O2 supply is less than O2 demand and tissue decreases.
Consistent with a possible role for O2 in the local regulation of blood flow, there is consensus that O2 is vasoactive. Arterioles in the peripheral microcirculation constrict when exposed to increases in produced either by increases in inspired (Hutchins et al. 1974; Zhu et al. 1998; Demchenko et al. 2000; Vucetic et al. 2004; Sakai et al. 2007; Kisilevsky et al. 2008; Justesen et al. 2010) or increases in the of solutions flowing over microvascular beds (Duling, 1972; Prewitt & Johnson, 1976; Lombard & Duling, 1977, 1981; Tuma et al. 1977; Kontos et al. 1978; Lindbom et al. 1980; Proctor et al. 1981; Sullivan & Johnson, 1981 a,b; Proctor & Duling, 1982 a,b; Jackson & Duling, 1983; Jackson, 1986, 1987, 1988, 1989, 1993; Lombard et al. 1986, 1999, 2004; Kaul et al. 1995; Welsh et al. 1998; Hungerford et al. 2000; Kunert et al. 2001 a,b, 2009; Frisbee & Lombard, 2002; Drenjancevic‐Peric et al. 2003, 2004; Wang et al. 2009; Ngo et al. 2010, 2013). Conversely, decreases in , produced by lowering inspired , cause vasodilatation (see Marshall, 2000 for references). What remains unclear are the cellular location of the process that detects changes in (the O2 sensor) and the mechanism that transduces the changes in to changes in arteriolar tone (the mechanism of O2 action). This review will focus on the site and mechanism of action of O2 on arterioles in peripheral tissues at rest. Changes in arteriolar tone induced by systemic hypoxia or hyperoxia produced by changes in inspired will not be considered because the mechanisms involved may be different. Systemic hypoxia and hyperoxia are complicated by changes in neural outflow to arterioles (Marshall, 1994; Guyenet, 2000), changes in circulating hormones and vasoactive substances (Mazzeo et al. 1991), and changes in circulating cytokines released from macrophages in the lungs (Shah et al. 2003). Oxygen reactivity of microvessels in active tissue (contracting striated muscle, the heart, etc.) will also not be considered as there are probably interactions among mechanisms that have not been fully explored, and to maintain the focus on arteriolar O2 reactivity per se. Evidence will be presented that: (1) O2 acts as a vasoconstrictor on arterioles in the peripheral microcirculation, (2) there is regional heterogeneity in the specific vasoconstrictor pathway that mediates arteriolar O2 reactivity, and (3) arterioles may be tuned to detect changes in tissue , whereas upstream feed arteries may be tuned to respond to changes in blood .
Setting the stage
Microvascular architecture and the study of arteriolar O2 reactivity
The microcirculation is the business end of the cardiovascular system. It is here that exchange of O2, CO2, substrates, hormones, waste products, etc. occurs. This exchange function depends on a continuous, regulated flow of blood through the microvasculature. The microcirculation originates when a feed artery (100–200 μm internal diameter) that is external to the tissue branches into a first‐order arteriole (50–100 μm). Feed arteries provide up to half of the precapillary vascular resistance to blood flow in tissues such as skeletal muscle and importantly contribute to the regulation of total blood flow to the tissue (Segal & Duling, 1986). Arterioles are embedded in the parenchyma that they perfuse and have 1–2 layers of smooth muscle in the media of their walls. Arterioles contribute the remaining 50% of the precapillary vascular resistance to blood flow and determine the distribution of blood flow within the tissue. However, the primary effectors of local blood flow control to and within a tissue are the arterioles. Upstream feed arteries are external to the tissue, and are recruited to the process of blood flow regulation by indirect mechanisms such as conducted vasomotion (Segal & Duling, 1986) or flow‐dependent responses (Davis et al. 2011) that are initiated by changes in tone of the downstream arterioles. First‐order arterioles usually have ∼two layers of smooth muscle, while smaller arterioles are invested with only a single layer of smooth muscle. In striated muscle, for example, second‐ to fifth‐order arterioles divergently branch from the first‐order arterioles, ending in terminal arterioles that supply 10–20 capillaries each. Post‐capillary venules collect blood flow from two or more capillaries. These then drain into higher order venules, which converge on veins draining a tissue or organ. While capillaries are a major site of exchange of materials and heat between tissues and blood because of their large surface area, O2 and CO2 also readily diffuse across the wall of arterioles and venules contributing to the exchange of these respiratory gases in the microcirculation (Renkin, 1984).
Methods and approaches used to study arteriolar O2 reactivity
Two general approaches have been used to explore microvascular O2 reactivity: (1) pressure myography (Duling et al. 1981; Schjorring et al. 2015) and other techniques for the ex vivo study of isolated vessels, and (2) intravital microscopy of arterioles, in situ, in exteriorized, autoperfused tissues (Baez, 1973; Duling, 1973). Pressure myography involves the surgical removal of a vessel from a tissue. The vessels are then cannulated onto micropipettes and pressurized to a physiological pressure. The vessels are superfused with a physiological salt solution (PSS; to allow treatment with drugs, altering the , and temperature control), and imaged with a microscope to measure internal diameter. Intravital microscopy usually involves the exteriorization of a tissue from an anaesthetized animal retaining the tissue's blood and nerve supply. The tissue is superfused with PSS (to control the and temperature), and the vessels imaged using a microscope. Both approaches allow the in the environment around vessels to be measured, controlled and changed while the diameters of the vessels are measured, the main requirements to study vascular O2 reactivity.
The study of isolated vessels, ex vivo, offers the advantage of precise control of experimental conditions: the composition of the bathing solutions, perfusion solutions, pressure, etc. can be precisely controlled. In addition, the cell type responsible for O2‐mediated responses can be identified (Busse et al. 1983; Fredricks et al. 1994 a). However, this approach is suitable mainly for the study of feed arteries and first‐order arterioles due to the technical difficulties associated with the dissection and cannulation of vessels smaller than 50 μm. A major assumption of the ex vivo study of feed arteries is that the reactivity of these larger upstream vessels accurately models the reactivity of the smaller downstream arterioles, an assumption that has not been adequately tested. In addition, loss of input from hormones, extravascular cells and other vessels in the microcirculation may alter the function of isolated vessels. Trauma due to dissection of the vessels can also be problematic.
The primary advantage of the intravital microscopy approach is the ability to study arterioles of any size in their native environment. However, most studies have focused on smaller arterioles (third‐ to fifth‐order arterioles < 40 μm). As will be pointed out repeatedly in sections below, this difference between ex vivo and in situ studies (the study of arteries vs. arterioles) may contribute to the lack of consensus on the site and mechanism of action of O2 in the microcirculation. The main disadvantage of the in situ approach is the lack of control of the environment. The arterioles are embedded in a connected microvascular network that allows conduction of signals along the vessel wall. They are perfused with systemic blood and surrounded by parenchymal cells, mast cells, nerves, etc. Therefore, the signals to which a given vessel is exposed are not always apparent. As will be outlined below, there are strategies to circumvent some of these issues, but these approaches are technically challenging. Because of the requirement to control tissue and to observe vessel diameter, intravital microscopy is limited to the study of thin tissues such as the hamster cheek pouch or hamster, mouse, or rat cremaster muscles, although surface vessels can be studied in thicker tissues. The control of in superfused exteriorized tissues can also be problematic. Care must be taken to limit the depth of the superfusion solution to 0.5 mm or less, and to use relatively high superfusate flow rates (5–10 ml min−1 for cheek pouches or cremaster muscles) to obviate diffusional boundary layers and lack of the ability to accurately control at the surface of the preparations. As with the ex vivo study of isolated vessels, surgical trauma and the resultant inflammation can also alter microvascular reactivity in intravital preparations.
What is the relevant range over which arterioles are O2 sensitive?
Intravital microscopy studies have defined the range over which arterioles display O2 reactivity as illustrated in Fig. 1 A. Arterioles respond to changes in over a range from ∼10 to >70 mmHg in the tissue (Klitzman et al. 1982) and ∼30 to 150 mmHg at the surface of arterioles (Gorczynski & Duling, 1978; Jackson & Duling, 1983; Jackson, 1987) (Fig. 1 A). In Fig. 1 A, several scales are provided, because the cell type in which changes in are sensed, that will be termed the sensor, has yet to be firmly established. Thus, it is not clear what (tissue or arteriolar ) is particularly relevant to the vasomotor effect of O2 on the arterioles. However, these values (tissue and arteriolar ) should provide the lower and upper bounds, respectively, for defining the O2 sensitivity of the system.
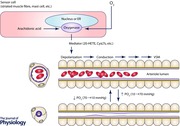
Figure 1. Arteriolar O2 reactivity in superfused microvascular preparations .
A, data from Klitzman et al. (1982). Data are mean ± SEM diameters of arterioles in hamster cremaster muscles when exposed to solutions equilibrated with gases containing different values and 5% CO2 (solution values, upper x‐axis labels). The values shown were measured with O2 microelectrodes in the free solution flowing over the preparations. The tissue values shown on the lower x‐axis were measured at the midpoint between the venous ends of two capillaries. The arteriolar values (top x‐axis labels) were estimated from haemoglobin oxygen saturation measurements in hamster cheek pouch (Jackson & Duling, 1983). B, typical O2‐induced vasoconstriction in a hamster cheek pouch preparation exposed to superfusion solutions with approximate values as indicated using methods as described (Jackson, 1987). C, same vessel as in B after exposure to ruthenium red (0.001%) to label mast cells (Shepherd & Duling, 1995). Scale bar in B, 25 μm.
Several points should be made regarding the relationship between O2 and arteriolar diameter (tone) in superfused microvascular preparations as depicted in Fig. 1. First, arterioles in intravital preparations display substantial myogenic tone (diameter at rest = 50–80% of the maximum diameter obtained with a vasodilator) with low in the superfusion solution (the solutions shown with = 12 mmHg were equilibrated with gases containing 0% O2 and 5% CO2). Arterioles in microvascular preparations such as hamster (Jackson, 1986), mouse (Hungerford et al. 2000; Figueroa et al. 2003) or rat cremaster muscle (Jackson, 1986), hamster cheek pouch (Jackson & Duling, 1983; Jackson, 1987), hamster retractor muscle (Lombard et al. 1999) rat spinotrapezius muscle (Marvar et al. 2007) or mouse gluteus maximus muscle (Sinkler & Segal, 2014) all retain substantial tone; they are not maximally dilated at the low values experienced by the preparations (note the right y‐axis scale in Fig. 1 A). Second, at rest, arterioles and the tissue surrounding them experience relatively ‘low’ values due to diffusional loss of O2 from precapillary vessels and consumption of O2 by the tissue as well as diffusion of O2 from the arterioles into the superfusion solution (for low equilibrated solutions) (Duling & Berne, 1970). Early studies using recessed‐tip O2 microelectrodes (Whalen et al. 1974) or multi‐point surface O2 electrodes (Lund et al. 1980 a,b) indicated that there was a broad range of values measured in the microcirculation of skeletal muscle, from < 1 to 100 mmHg. The distributions recorded by both methods were dominated by relatively low values with the means of the distributions being in the order of 17 mmHg in the anaesthetized cat (Whalen et al. 1974), 33 mmHg in the anaesthetized rat (Lund et al. 1980 b) and 15 mmHg in the conscious human (Lund et al. 1980 a). More recent measurements support the findings of these early studies (Smith et al. 2002, 2004; Johnson et al. 2005). The average minimum resting tissue (defined as the midway between the venous end of two capillaries away from arterioles or venules) in a relatively undisturbed striated muscle microvascular bed, in vivo, is ∼25 mmHg as measured with phosphorescence probes (Smith et al. 2002, 2004; Johnson et al. 2005). Thus, a superfused microvascular preparation allows the study of both the response to mild decreases in tissue (from 25 down to ∼10 mmHg, with the superfusate serving as a sink for O2) and increases in (>25 mmHg, with the superfusate serving as a source for O2). An additional weakness of the superfused intravital microvascular preparations is the lack of ability to study more severe hypoxia (below 10 mmHg). Tissue values below 10 mmHg can be attained during increases in metabolic activity (e.g. striated muscle contraction) (Lash & Bohlen, 1987; Boegehold & Bohlen, 1988; Smith et al. 2002), occlusion of blood flow (Lombard & Duling, 1977) or breathing gases with reduced O2 content (Shah et al. 2003). However, each of these approaches adds additional complexity beyond simple effects of O2 on the arterioles, and thus, will not be considered in this review.
Finally, it is important to recognize that elevated causes profound, maintained vasoconstriction of arterioles in well‐prepared microvascular beds (preparations with substantial resting arteriolar tone at physiological values as presented above and without leukocyte adherence to arteriolar endothelium or other signs of inflammation) (see Fig. 1 B in this review, Fig. 2 in Ngo et al. 2010 and Fig. 1 in Ngo et al. 2013, for examples). This point is particularly important when considering the data obtained from the ex vivo study of isolated arteries and arterioles.
Figure 2. Schematic diagram of O2 signalling in the microcirculation .
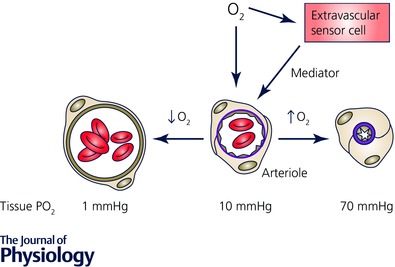
Oxygen in the environment of arterioles can act directly on the arteriolar wall or on cells in the lumen to produce a vasomotor effect (dilatation in the case of reduced , or constriction in the case of elevated ). Alternatively, changes in may be sensed by extravascular cells (parenchymal cells, mast cells, nerves, etc.), a mediator produced, which then acts on the vessel wall to produce the appropriate vasomotor effect. The values shown below the cross section of the arterioles refer to tissue in a superfused, intravital preparation measured at the midpoint between the venous ends of two capillaries as reference values, only.
Location of the sensor: what cell type senses changes in relevant to arteriolar O2 reactivity?
At least four cellular sites have been postulated to sense changes in and signal vascular smooth muscle cells to contract (elevated ) or relax (reduced ) in response to this change. These include the arteriolar wall (vascular smooth muscle or endothelial cells), red blood cells in the lumen of the vessels, and extravascular cells (parenchymal cells, nerves, mast cells, etc., see Fig. 2).
Arteriolar smooth muscle cells as an O2 sensor
Studies testing the hypothesis that arteriolar smooth muscle cells are intrinsically sensitive to changes in within the range of 10–150 mmHg have lead to equivocal results. Early organ bath studies of preparations from conduit arteries (as model systems), ex vivo, indicated that smooth muscle cells are intrinsically sensitive to changes in bath (Carrier et al. 1964; Detar & Bohr, 1968; Coburn et al. 1979; Chang & Detar, 1980). However, this intrinsic O2 sensitivity appears to result from the formation of an anoxic core in the tissue due to O2 consumption by the vascular smooth muscle cells and the multicellular thickness of these conduit artery preparations (Pittman & Duling, 1973). Thus, these studies do not appear to be relevant to arteriolar O2 reactivity except, perhaps, under conditions when the in the microcirculation falls to very low levels ( ≪ 10 mmHg).
Studies of the O2 reactivity of smooth muscle cells of resistance arteries and arterioles have lead to conflicting results. Endothelium‐denuded rat gracilis muscle feed arteries studied ex vivo by pressure myography display O2 reactivity in the appropriate range (10–150 mmHg), but the response is only 15–25% of that observed in vessels with intact endothelium (Frisbee et al. 2002). Low (∼12 mmHg) also inhibits vasoconstriction of first‐order rat cremaster arterioles constricted by activation of α2‐adrenoreceptors (Tateishi & Faber, 1995 a,b). In addition, noradrenaline‐induced contraction of isolated hamster cremaster arteriolar smooth muscle cells is inhibited by exposure to low (Jackson, 2000 b; Cohen & Jackson, 2003). In the latter studies, the low O2‐induced inhibition of noradrenaline‐induced contraction of hamster cremaster arteriolar smooth muscle cells was not due to the activation of K+ channels (Jackson, 2000 b), the inhibition of L‐type Ca2+ channels (Cohen & Jackson, 2003), nor diminution of noradrenaline‐induced Ca2+ transients (Cohen & Jackson, 2003). These data suggested that the low reduced the Ca2+ sensitivity of the contractile machinery, consistent with several studies of macrovascular smooth muscle cells (Gebremedhin et al. 1994; Aalkjaer & Lombard, 1995; Shimizu et al. 2000). On the other hand, patch clamp studies of isolated vascular smooth muscle cells from systemic arteries have demonstrated activation of ATP‐sensitive K+ (KATP) channels (Dart & Standen, 1995), activation of large‐conductance Ca2+‐activated K+ (BKCa) channels (Gebremedhin et al. 1994), or inhibition of L‐type voltage‐gated Ca2+ (CaL) channels (Franco‐Obregon et al. 1995; Franco‐Obregon & Lopez‐Barneo, 1996 a,b) when the cells are exposed to low (15–20 mmHg), any of which could contribute to O2 reactivity.
In contrast to the studies described above, removal of the endothelium from first‐order rat cremaster arterioles abolished O2 reactivity in ex vivo pressure‐myography studies (Messina et al. 1992, 1994). These data exclude smooth muscle as the O2 sensor in these isolated rat cremaster arterioles. Furthermore, several additional ex vivo studies of intact, endothelium‐replete rat first‐order cremaster arterioles have failed to demonstrate effects of changes in between 10 and 150 mmHg on myogenic tone (Tateishi & Faber, 1995 a,b; Kerkhof et al. 1999), again not supporting smooth muscle as the O2 sensor. In the studies by Tateishi and Faber (Tateishi & Faber, 1995 a) reduced O2 tensions ( < 6 mmHg) inhibited myogenic tone suggesting intrinsic O2 reactivity at much lower values.
Intravital microscopy studies performed in hamster cheek pouches have also failed to support smooth muscle, or other components of the vascular wall, as the O2 sensor (Duling, 1974; Jackson, 1987). Altering the of the wall of arterioles through the use of fluid‐filled micropipettes (Duling, 1974; Jackson, 1987) (Figs 3 and 4), or by in situ perfusion of segments of arterioles in the hamster cheek pouch (Jackson, 1987) (Fig. 5) failed to demonstrate arteriolar O2 reactivity intrinsic to the arteriolar wall. In contrast to these findings, elevation of the of the superfusion solution flowing over the entire cheek pouch produced typical O2‐induced vasoconstriction (Duling, 1974; Jackson, 1987) (Figs 3, 4, 5). The lack of effect of local changes in on arteriolar diameter (Figs 3, 4, 5) do not support the hypothesis that arteriolar smooth muscle cells are intrinsically sensitive to changes in between 10 and 150 mmHg.
Figure 3. Local increases in have no effect on arteriolar tone .
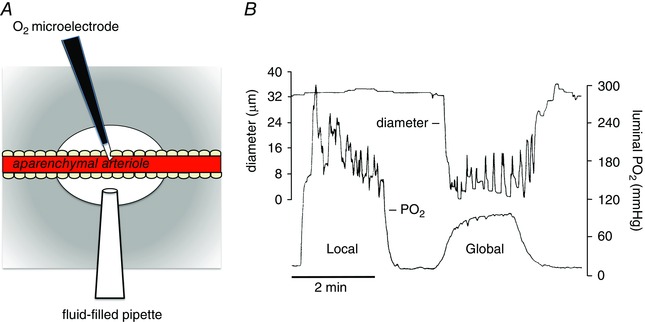
Data shown are modified from Jackson (1987). A, schematic diagram of a segment (1–8 mm) of an arteriole in a hamster cheek pouch from which the parenchyma has been surgically removed (aparenchymal arteriole) to obviate effects of local changes on parenchymal and other extravascular cells. A Whalen‐type recessed tip O2 microelectrode was inserted through the wall of the vessel into the lumen as shown to monitor luminal . A temperature‐controlled micropipette filled with PSS equilibrated with varied (fluid‐filled micropipette) was positioned opposite the O2 microelectrode. Pressurization of the fluid‐filled micropipette ejected the O2‐equilibrated solution onto the surface of the arteriole to produce a local change in . The entire cheek pouch preparation was superfused with PSS, the of which could be varied to produce global changes in that affected both the aparenchymal arteriole and all other vessels and cell types in the cheek pouch. The diameter of the arteriole was measured by intravital video microscopy. B, results from a typical experiment in which either local increases in were produced using the fluid‐filled pipette (Local) or global increases in were produced by changing the of the entire superfusate (Global) (replotted data are from Fig. 3 in Jackson, 1987). Local increases in that effectively changed the across the wall of the arteriole had no significant effect on arteriolar diameter, whereas a global increase in produced sustained vasoconstriction. These data suggest that components of the arteriolar wall (endothelial cells, smooth muscle cells or perivascular nerves) are not the sensor cells responsible for arteriolar O2 reactivity in the hamster cheek pouch. See Jackson (1987) for details.
Figure 4. Local increases in have no effect on arteriolar tone in occluded arterioles .
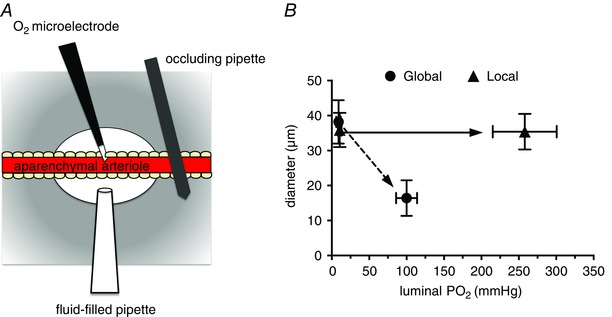
A, a preparation similar to that depicted in Fig. 3, but with the inclusion of an occluding pipette that was pressed down on the arteriole to eliminate blood flow through the aparenchymal segment. B, summary data from these experiments (data are means ± SEM, n = 5). Local changes in across the arteriolar wall produced by the fluid‐filled pipette (Local) were ineffective in producing arteriolar constriction. However, raising the of the superfusate over the entire preparation (Global) produced consistent, sustained arteriolar constriction. These data, along with those shown in Fig. 3, suggest that components of the arteriolar wall (endothelial cells, smooth muscle cells or perivascular nerves) are not the sensor cells responsible for arteriolar O2 reactivity in the hamster cheek pouch. Data are replotted from Fig. 4 B in Jackson (1987); see this reference for more details.
Figure 5. Perfusion of arterioles in situ with solutions equilibrated with high has no effect on arteriolar tone .
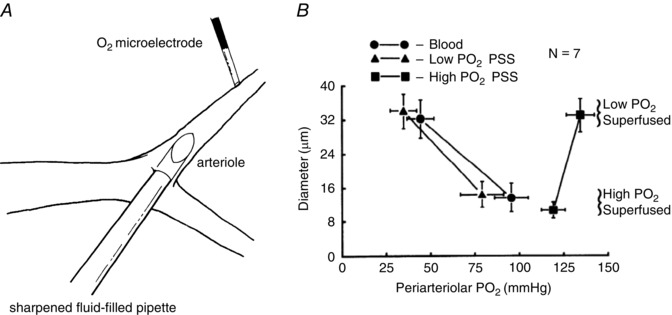
A and B are reproduced from Jackson (1987). A, schematic diagram of a hamster cheek pouch arteriole in which a sharpened fluid‐filled pipette has been inserted through the wall of an arteriole allowing perfusion of the arteriole with PSS equilibrated with varied . The entire cheek pouch preparation was superfused with PSS to allow global changes in . B, summary data (means ± SEM). Perfusion of the arterioles with solutions equilibrated with high or low had no significant effect on arteriolar diameter. Only when the global was elevated via the superfusate did the arterioles constrict (compare low superfusate points with high superfusate points). These data suggest that components of the arteriolar wall (endothelial cells, smooth muscle cells or perivascular nerves) are not the sensor cells responsible for arteriolar O2 reactivity in the hamster cheek pouch. See Jackson (1987) for details.
Thus, smooth muscle per se, in some vessels (resistance arteries and first‐order arterioles), under some conditions, may indeed sense changes in in the range of 10–150 mmHg. However, there does not appear to be significant support for smooth muscle cells as the O2 sensor, particularly based on in situ studies of arterioles in the intact microcirculation.
Arteriolar endothelial cells as an O2 sensor
There is considerable evidence from ex vivo, pressure‐myography studies of isolated vessels that endothelial cells may serve as an O2 sensor, at least in small arteries and large arterioles. Busse and coworkers first demonstrated endothelium‐dependent O2 reactivity in rat tail arteries, canine femoral artery branches and canine epicardial coronary arteries (Busse et al. 1983, 1984). These findings were subsequently confirmed in rabbit aortas and rabbit femoral arteries (Pohl & Busse, 1989), hamster carotid arteries (Jackson et al. 1987), first‐order rat cremaster arterioles (Messina et al. 1992, 1994), rat middle cerebral arteries (Fredricks et al. 1994 b), large arterioles from rat diaphragm (Ward, 1999), and rat gracilis muscle feed arteries (Fredricks et al. 1994 a; Frisbee et al. 2001 a,b,c, 2002).
In contrast, several pressure‐myography studies of endothelium‐intact first‐order rat cremaster arterioles have failed to demonstrate effects of changes on myogenic tone (Tateishi & Faber, 1995 a,b; Kerkhof et al. 1999), as noted above. The apparent difference between the studies showing endothelium‐dependent O2 reactivity (Busse et al. 1983, 1984; Messina et al. 1992, 1994; Fredricks et al. 1994 a,b; Ward, 1999; Frisbee et al. 2001 a,b,c, 2002) and those not displaying O2 reactivity (Tateishi & Faber, 1995 a,b; Kerkhof et al. 1999) is the presence of luminal flow in the former and not in the latter. The exception to this relationship is a pressure‐myography study by Dietrich et al. (2000) using endothelium‐intact rat cerebral arterioles. These investigators found that PSS or dextran–PSS‐perfused arterioles did not display O2 reactivity in the absence of luminal red blood cells.
Lack of evidence for endothelium‐dependent O2 reactivity also can be gleaned from the hamster cheek pouch studies already cited in the previous section (Duling, 1974; Jackson, 1987). In those studies, either blood (Duling, 1974; Jackson, 1987) or O2‐equilibrated PSS (Jackson, 1987) was flowing through the arterioles. In these studies, despite effectively changing the of endothelial cells, O2 reactivity was not observed when only the vessel wall was exposed to changes in . These data do not support an endothelial O2 sensor in arterioles in the hamster cheek pouch. They, along with the studies by Dietrich et al. (2000), suggest that shear stress is not the key to promoting endothelial cell O2 sensing.
Thus, there is evidence both for and against an endothelial cell O2 sensor. However, the extant data supporting this hypothesis stem from pressure‐myography studies of isolated arteries and large arterioles, ex vivo. No data from intravital preparations support endothelial cells as the sensors mediating arteriolar O2 reactivity.
As noted in the previous paragraph, all of the ex vivo studies of vascular O2 reactivity have been performed on arteries or large, first‐order arterioles (diameters > 50 μm). In contrast, most in situ intravital microscopy studies of arteriolar O2 reactivity have focused on small, third‐ to fifth‐order arterioles (diameters < 40 μm). Given the known differences in mechanisms regulating myogenic tone between small arteries and downstream arterioles (Westcott & Jackson, 2011; Westcott et al. 2012), it may be that there are different mechanisms (and hence, sites of action) coupling changes in with changes in vascular tone in different parts of the vascular tree. In addition, as noted above, in situ, elevated produces dramatic, long lasting constriction of arterioles (Fig. 1). This degree of O2 reactivity has not been described in the ex vivo study of isolated vessels, perhaps reflecting that mostly feed arteries and first‐order arterioles have been studied ex vivo. However, these data may also indicate that sites other than the arteriolar wall are more important for O2 reactivity of arterioles in the intact, living microcirculation.
Red blood cells as an O2 sensor
Red blood cells also have been proposed as the O2‐sensitive cell type responsible for arteriolar O2 reactivity. Three lines of evidence support this hypothesis. First, in vitro, red blood cells release ATP as is reduced from 85 mmHg down to 17 mmHg (Ellsworth et al. 1995). Second, at concentrations consistent with the amount of ATP released from red blood cells, intraluminal ATP produces conducted dilatation of arterioles in the hamster retractor muscle in situ, through a mechanism mediated by NO (McCullough et al. 1997). Intraluminal application of ATP into collecting venules also produces dilatation of upstream arterioles suggesting conduction of vasodilatation initiated by ATP from venules to arterioles through capillaries (Collins et al. 1998) (see below for more on conducted responses). Third, inclusion of red blood cells in the solution perfusing rat cerebral arterioles in pressure‐myography studies instills O2 reactivity to isolated arterioles that do not respond to changes in in their absence (Dietrich et al. 2000).
In contrast, the studies outlined in Figs 3, 4, 5 (Jackson, 1987) suggest that red blood cells may not be the O2 sensor that mediates arteriolar O2 reactivity in situ. In those studies, local changes in that effectively altered blood had no effect on arteriolar tone (Figs 3 and 4). Oxygen reactivity was not observed whether blood was flowing through the lumen of the arterioles (Fig. 3) or not (Fig. 4). In addition, if red blood cells were major O2 sensors coupling changes in with changes in vasomotor tone, then perfusion of arterioles in situ with PSS should eliminate arteriolar O2 reactivity. This was not the case in the hamster cheek pouch (Fig. 5) (Jackson, 1987) or in the mouse cremaster muscle (Ngo et al. 2010); in both tissues, arteriolar O2 reactivity was retained when the tissues were perfused with physiological salt solution and no red blood cells were present. Thus, in situ, red blood cells may not be a major site that senses changes in values greater than 10–15 mmHg.
Extravascular cells as an O2 sensor
To test the hypothesis that extravascular cells (parenchymal cells, etc.) are the location of the O2 sensor involved in arteriolar O2 reactivity in the hamster cheek pouch, Jackson and Duling surgically removed the parenchyma along long segments of blood‐perfused second‐order arterioles (Jackson & Duling, 1983) (Figs 3, 4 and 6). Surprisingly, this manoeuvre had no significant effect on O2 reactivity: the aparenchymal arteriolar segments continued to respond to changes in superfusate as the solution flowed over the entire preparation (Jackson & Duling, 1983). Furthermore, sealing the aparenchymal arteriolar segments under glass (to prevent superfusate access) and occluding the arterioles (to obviate convection of O2 in the blood to the segments) did not eliminate O2 reactivity (Fig. 6). To complicate matters, we found that some isolated, cannulated cheek pouch arterioles retained O2 reactivity. However, the responses of these isolated arterioles were ephemeral. Taken as a whole, the data from this study suggested that: (1) changes in are probably sensed at extravascular sites that are non‐uniformly distributed along the arterioles (to account for the lack of O2 reactivity in some isolated vessels), and (2) O2‐dependent vasoconstriction, once initiated, can be conducted along the arteriolar wall (to account for the maintained O2 reactivity of aparenchymal segments sealed under glass: Fig. 6) (see below for more on the topic of conducted O2 responses). Subsequent studies, outlined above (Fig. 4), in which segments of cheek pouch arterioles were perfused in situ with solutions with varied supported and extended these conclusions (Jackson, 1987).
Figure 6. Covering aparenchymal segments with glass and occluding them to eliminate blood flow does not eliminate arteriolar O2 reactivity .
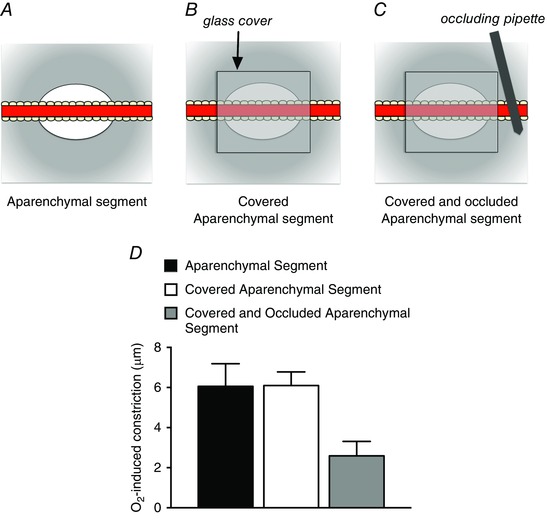
A, schematic diagram of an aparenchymal segment in which the parenchyma has been removed from a long segment of hamster cheek pouch arteriole as described by Jackson & Duling (1983). Elevation of the of the solution flowing over the preparation from 12 to 150 mmHg resulted in arteriolar constriction as depicted in D. As shown in B, subsequent covering of the aparenchymal segment with a piece of glass coverslip (sealed in place with silicone grease), to eliminate contact of the arteriole with the superfusate, had no effect on O2‐induced constriction as shown in D. To eliminate blood flow through the covered aparenchymal segments, an occluding pipette was used as shown in C. Despite the lack of access to the superfusate and flowing blood, these covered and occluded aparenchymal segments retained significant O2 reactivity as shown in D. These data suggest that the constriction induced by elevated can be conducted along the arteriolar wall. See Jackson & Duling (1983) for details.
Mast cells: a possible O2 sensor in the hamster cheek pouch
Arterioles in the hamster cheek are intimately associated with mast cells as shown in Fig. 1 C. These cells cluster, non‐uniformly, near the arterioles and they produce cysteinyl leukotrienes (CysLTs) (Storch et al. 2015), vasoconstrictors that mediate arteriolar O2 reactivity in the hamster cheek pouch (see below for more on this topic). Mast cells as the location of the O2 sensor may also explain why some isolated cannulated cheek pouch arterioles retained O2 reactivity in a previous study (Jackson & Duling, 1983). Dissection of these arterioles may remove or damage some/all of these perivascular cells accounting for the loss and the labile nature of O2 reactivity in the ex vivo study of cheek pouch arterioles.
Conduction of O2‐induced vasoconstriction: a complicating factor
As noted above, we (Jackson & Duling, 1983) found that even when arterioles in the hamster cheek pouch were sealed under glass (to prevent superfusion solution access) and occluded (to obviate convection of O2 into the segment by blood flow) the vessels retained O2 reactivity (Fig. 6). We hypothesized that O2‐induced vasoconstriction could be conducted along the arterioles over considerable distance (at least several millimetres). Mouse cremaster muscle arterioles also display conducted O2‐induced vasoconstriction (Ngo et al. 2010; Riemann et al. 2011), suggesting that this phenomenon is not restricted to the hamster cheek pouch.
Conduction of O2‐induced arteriolar responses implies that responses can be initiated away from the site of observation and, importantly, that responses initiated at several sites may summate, as has been observed for conducted vasomotion in response to other vasoactive substances (Segal et al. 1989). Based on conducted vasomotor responses induced by other vasoactive substances (Delashaw & Duling, 1991; Xia & Duling, 1995), the conduction of O2‐induced vasomotor responses implies that changes in vascular smooth muscle membrane potential are probably involved in the mechanism of action of O2 on arterioles. As will be discussed below, this prediction was verified in the hamster cheek pouch where O2‐induced vasoconstriction was preceded by depolarization of the smooth muscle cells (Welsh et al. 1998).
Conduction of O2‐induced changes in vascular tone along arterioles complicates identification of the location of the cell type that actually senses changes in because the sensor cells can be remote from the site of observation. Changes in , could, for example, be sensed in parenchyma around terminal arterioles or even capillaries or venules downstream from arterioles, a response initiated, and this response conducted upstream to produce the appropriate change in arteriolar tone as suggested by Jackson (1987) and later by Collins et al. (1998) and Ellsworth et al. (2016). Oxygen sensing in the vicinity of terminal arterioles, capillaries or post‐capillary venules might explain why, in situ, changes in that are restricted to segments of second‐ to third‐order arterioles, do not produce a change in arteriolar tone, whereas global changes in consistently produce an upstream arteriolar response (Duling, 1974; Jackson, 1987). However, in the hamster cheek pouch, conduction of vasoconstrictor‐induced depolarization occurs along the smooth muscle layer (Welsh & Segal, 1998). This constraint suggests that, in the cheek pouch, the O2‐sensing cells are probably adjacent to arteriolar smooth muscle cells and not in downstream capillaries or venules (see below for more on this topic). The pathway for conduction in skeletal muscle arterioles has not been as firmly established.
The hypothesis that changes in can be sensed in capillaries was tested in rat extensor digitorum longus muscles using a microfluidic device approach (Ghonaim et al. 2011). The of regions overlying capillary beds ranging from 100 μm circles (affecting 1 capillary and surrounding skeletal muscle fibres) to 200 μm × 1000 μm rectangles (affecting ∼3 capillaries and surrounding muscle fibres) were changed (Ghonaim, 2013), with capillary red blood cell haemoglobin O2 saturation (%Sat) and red blood cell flux measured. The authors found that while changes in applied through 100 or 200 μm circles (affecting only 1–2 capillaries) produced appropriate changes in red blood cell %Sat, they were without effect on red blood cells flux. In contrast, changes in applied to a 200 μm × 1000 μm area (affecting ∼3 capillaries and associated muscle fibres) produced changes in red blood cell flux through the affected capillaries. These results suggest that there may be summation of responses to altered that are required to initiate a conducted response sufficient to affect upstream arterioles that control red blood cell flux through the capillaries. Although the authors interpreted these findings as evidence for capillaries as the site where changes in are sensed (in the context of red blood cells as the O2 sensors), effects of changes in on striated muscle fibres, which are also putative O2 sensors (see below), were not excluded. Striated muscle fibres are much longer than capillaries, and they will contact arterioles in the network. Thus, it is also possible that the striated muscle fibres sensed changes in , a response initiated and transmitted to upstream arterioles to modulate arteriolar smooth muscle tone and hence capillary perfusion.
Striated muscle fibres as an O2 sensor
Frisbee and Lombard compared the O2 reactivity of first‐order arterioles in rat cremaster muscles, in situ, with similar arterioles studied by pressure myography, ex vivo (Frisbee & Lombard, 2002). They found that while these large arterioles retained O2 reactivity when studied ex vivo, the maximal O2‐induced diameter responses of the isolated vessels were only 50% of the responses observed in situ. These data support a role for striated muscle fibres as an O2 sensor in rat cremaster muscle. As will be outlined below, the location of cytochrome P4504A (CYP4504A) ω‐hydroxylase, a key enzyme involved in arteriolar O2 reactivity in striated muscle fibres (Kunert et al. 2001 a), also lends support for these cells as O2 sensors in striated muscle.
Summary of the location of the sensor
Ex vivo studies of isolated feed arteries and first‐order arterioles support the hypothesis that these vessels are intrinsically sensitive to changes in within the appropriate range (10–150 mmHg), with smooth muscle, endothelium, or red blood cells as the location of the sensor (see references above). In contrast, intravital microscopy studies (Duling, 1974; Jackson & Duling, 1983; Jackson, 1987), all in the hamster cheek pouch, and all directed at smaller, downstream arterioles, have failed to support the hypothesis that these smaller vessels are intrinsically sensitive to O2 in situ. Furthermore, local changes in that effectively changed the of the blood flowing through the arterioles in the cheek pouch were also without effect on arteriolar tone (Jackson, 1987). Microvascular beds perfused with PSS retain O2 reactivity (Jackson, 1987; Ngo et al. 2010). These data do not support the hypothesis that red blood cells are a major sensor mediating arteriolar O2 reactivity in the intact microcirculation of the hamster cheek pouch or mouse cremaster muscle. Instead, the data from intravital preparations suggest that changes in are sensed at some extravascular site. These conflicting conclusions may indicate that vascular O2 sensing varies along the vascular tree with upstream feed arteries possessing intrinsic mechanisms to respond to changes in blood , and downstream arterioles being tuned to detect local changes in tissue . However, tissue‐dependent heterogeneity also cannot be excluded as studies to evaluate the intrinsic O2 sensitivity of small arterioles (<30 μm), in situ, have not been evaluated in tissues other than the hamster cheek pouch. Conduction of O2‐induced changes in arteriolar tone also complicates identification of the microvascular O2 sensor that has not been adequately explored in preparations other than the hamster cheek pouch.
Mechanism of action
Several signalling pathways have been proposed to explain arteriolar O2 reactivity. These can be separated into two groups: (1) mechanisms in which low O2 results in increased production or decreased destruction of a vasodilator substance, and (2) mechanisms by which increased O2 results in production of a vasoconstrictor. As will be evident in the sections below, there appears to be significant regional heterogeneity that has complicated the study of the mechanisms responsible for arteriolar O2 reactivity. As discussed above, conduction of O2‐initiated responses also adds to the complexity of defining the mechanism of action of O2 on arterioles in the microcirculation.
Oxygen‐dependent inhibition of production of prostanoids
A number of ex vivo studies of isolated vessels have supported a role for prostaglandins in mediating dilatation of resistance arteries and large arterioles to a reduction in from 150 mmHg to 20–50 mmHg (Busse et al. 1983, 1984; Fredricks et al. 1994 a,b; Frisbee et al. 2001 b,c, 2002) or constriction of these vessels to increases in from 20 to 150–600 mmHg (Messina et al. 1994; Frisbee et al. 2001 a).
However, the relevance of these ex vivo studies to in situ arteriolar O2 reactivity remains unclear. Effective inhibition of cyclooxygenase has no effect on arteriolar O2 reactivity in intravital preparations of the hamster cheek pouch, hamster and rat cremaster muscles (Jackson, 1986), rat spinotrapezius muscle (Pries et al. 1995) and mouse cremaster muscle (Ngo et al. 2013). A weakness in these intravital studies is that only large changes in (from ∼12 to 150 mmHg) were examined. Thus, subtle effects of cyclooxygenase inhibition on the –response relation could have been missed. Nonetheless, the intravital studies do not support a sole, major role for prostaglandins in mediating arteriolar O2 reactivity in the intact microcirculation. However, the intravital studies do not exclude a role for prostaglandins in larger feed arteries outside of the tissue proper.
The mechanism by which elevated inhibits, and lowered enhances, vasodilator prostanoid formation also is not known. The enzymes catalysing oxygenation of arachidonic acid, the cyclooxygenases (COXs), are half‐maximally activated at a of about 3 mmHg (Lands et al. 1978; Mukherjee et al. 2007), with maximal activity at values greater than 18 mmHg (Lands et al. 1978; Mukherjee et al. 2007). Thus, formation of prostanoids should be independent over much of the range to which arterioles are O2 sensitive (Fig. 1). However, the COXs can undergo auto‐oxidation, which inhibits enzyme activity (Tsai & Kulmacz, 2010). Oxygen‐dependent oxidation and inhibition of COXs might explain O2‐dependent inhibition of production of vasodilator prostanoids, but this has not been established.
Oxygen‐dependent destruction of nitric oxide
Reducing the of solutions perfusing rabbit aortas and rabbit femoral arteries (Pohl & Busse, 1989) or hamster carotid arteries (Jackson et al. 1987), ex vivo, from 150 to 20–30 mmHg produces endothelium‐dependent vasodilatation mediated by NO. Nitric oxide has also been proposed to mediate arteriolar O2 reactivity in rat spinotrapezius muscle (Pries et al. 1995) and the O2 reactivity of rat intestinal arterioles (Nase et al. 2003) in situ. Ex vivo studies of rat gracilis muscle feed arteries also support a role for NO, but only at relatively high values (between 100 and 150 mmHg) (Frisbee et al. 2002). Pressure‐myography studies of rat first‐order cremaster muscle arterioles, ex vivo, found that NO mediated O2 reactivity, but only after application of the CYP4504A ω‐hydroxylase inhibitor, 17‐octadecynoic acid (17‐ODYA) (Kerkhof et al. 1999). In the absence of 17‐ODYA, these authors found that isolated rat cremaster arterioles did not respond to changes in suggesting that endogenous production of 20‐hydroxyeicosatetraenoic acid (20‐HETE) by CYP4504A ω‐hydroxylation of arachidonic acid inhibited O2‐dependent signalling by NO.
In contrast to the studies supporting a role for NO in arteriolar O2 reactivity, intravital studies in the hamster cheek pouch (Jackson, 1991) and in mouse cremaster muscle (Ngo et al. 2010, 2013; Riemann et al. 2011) found that effective inhibition of NO synthesis had no effect on arteriolar O2 reactivity. An earlier ex vivo study of rat gracilis feed arteries also did not support a role for NO in the reactivity of these vessels to changes in between 35 and 150 mmHg (Frisbee et al. 2001 a). These findings may indicate that there is heterogeneity in the mechanism of action of O2, which is dependent on the region, location in the vascular tree and possibly on the species studied. In addition, another study in the rat spinotrapezius muscle suggests a major role for CYP4504A ω‐hydroxylase and 20‐HETE in arteriolar O2 reactivity (see below) (Marvar et al. 2007). This observation is difficult to reconcile with the findings of Pries et al. cited above, who reported that inhibition of NO synthase abolished arteriolar O2 reactivity in the same tissue, from the same rat strain (Pries et al. 1995). It could be that multiple mechanisms are in play, as suggested by ex vivo studies of first‐order rat cremaster arterioles noted above (Kerkhof et al. 1999). Subtle methodological differences also cannot be excluded. Thus, the contribution of NO to arteriolar O2 reactivity remains unclear.
Oxygen‐dependent production of superoxide anion
If NO mediates arteriolar O2 reactivity, the relationship between O2 and NO production cannot be direct, because O2 is a substrate for NO synthase, with half‐maximal activation at about 2 mmHg for the isolated enzyme (Abu‐Soud et al. 2000), or as high as 38 mmHg in cell‐based assays (Whorton et al. 1997). Thus, NO production should increase or remain unchanged as O2 increases. This is opposite to what would be required for direct effects of O2 on NO production to mediate arteriolar O2 reactivity.
It has been suggested that O2‐dependent production of superoxide anion (O2 −•) and the subsequent destruction of NO may explain arteriolar O2 reactivity in situ (Golub & Pittman, 2013). However, previous direct tests of this hypothesis in striated muscle preparations using exogenous superoxide dismutase (SOD) both failed (Proctor & Duling, 1982 b; Pries et al. 1995). Golub & Pittman (2013) argue that the kinetics of the reaction between O2 −• and NO is so much faster than that between O2 −• and SOD, and that the tissue content of extracellular SOD is so high in striated muscle, the use of exogenous SOD does not adequately test for a role for O2 −•. In the rat brain, where there is lower expression of extracellular SOD, hyperbaric O2‐induced constriction of resistance arteries indeed does appear to be mediated by O2‐dependent production of O2 −• and destruction of NO, and can be inhibited by application of exogenous SOD (Demchenko et al. 2000). Similarly, superfusion with SOD and catalase or the SOD mimetic tempol abolishes O2‐induced constriction of rat sciatic epineural arterioles, supporting a role for O2 −• in these vessels (Sakai et al. 2007). Inhibitors of NADPH oxidase or xanthine oxidase also eliminated O2 reactivity, suggesting that these enzymes may be the O2 sensors in rat epineural arteriolar O2 reactivity (Sakai et al. 2007). However, unlike the studies in the brain (Demchenko et al. 2000), inhibition of NO synthase did not inhibit O2 reactivity of rat sciatic epineural arterioles suggesting that alterations in NO bioavailability do not explain O2 reactivity in this tissue. In addition, earlier studies demonstrated that O2 −• is a dilator in cat (Wei et al. 1996) and rabbit (Didion & Faraci, 2002) cerebral arterioles, and human coronary arterioles (Sato et al. 2003), supporting the idea that the O2 −•–NO destruction pathway is not a general mechanism accounting for arteriolar O2 reactivity. The lack of effect of effective inhibition of NO production on O2 reactivity in other preparations (Jackson, 1991; Ngo et al. 2010, 2013; Riemann et al. 2011) also argues that this is not a general mechanism explaining arteriolar O2 reactivity in all tissues and every species.
All of the mechanisms discussed above involve O2‐dependent modulation of a vasodilator: as increases, the dilator signal would decrease and as decreases, the dilator signal would increase. Recall that in the normal resting microcirculation of skeletal muscle, for example, the in the environment of arterioles is low. Thus, if arteriolar O2 reactivity involves modulation of a vasodilator, then when arterioles are isolated and cannulated for ex vivo study and exposed to ambient O2 conditions (21% O2), these arterioles should display substantially greater tone than they displayed in situ under low (∼10–15 mmHg) conditions. This simply has not been observed: the degree of myogenic tone developed by cannulated, pressurized arterioles ex vivo exposed to room air (21% O2, ∼150 mmHg) is similar to the tone observed when these same vessels are studied in situ by intravital microscopy under low conditions ( in the order of 10–15 mmHg) (for example, compare Jackson & Blair, 1998 with Burns et al. 2004). This observation alone suggests that arteriolar O2 reactivity in situ does not involve a vasodilator and also that it is unlikely that O2 is sensed directly by cells that comprise the wall of the arterioles.
Oxygen as a vasoconstrictor
In contrast to the mechanisms discussed thus far involving the O2‐dependent modulation of vasodilators, there is a substantial body of literature that O2‐dependent production of vasoconstrictors mediates arteriolar O2 reactivity in the peripheral microcirculation (see below for references). These vasoconstrictor mechanisms largely have been ignored when considering the role played by O2 in the local regulation of blood flow (Casey & Joyner, 2011; Marshall & Ray, 2012; Golub & Pittman, 2013; Reglin & Pries, 2014).
Oxygen‐dependent production of leukotrienes
The 5‐lipoxygenase and CysLTs appear to mediate arteriolar O2 reactivity in the epithelial portion of the hamster cheek pouch (Jackson, 1988, 1989, 1993) (Fig. 7). General lipoxygenase inhibitors (Jackson, 1988), selective 5‐lipoxygenase inhibitors (Jackson, 1989, 1993), a selective inhibitor of the 5‐lipoxygenase‐activating protein (FLAP) (Jackson, 1993), as well as antagonists of CysLT receptors (Jackson, 1989, 1993) all selectively and effectively inhibit O2‐induced arteriolar constriction in the cheek pouch. Importantly, these same inhibitors have no effect on the O2 reactivity of arterioles in hamster cremaster muscles, supporting their vascular bed selectivity (Jackson, 1993). These latter findings also strongly support the hypothesis that there are regional differences in the mechanisms coupling changes in with changes in arteriolar tone.
Figure 7. The site and mechanism of action of O2 in the hamster cheek pouch .
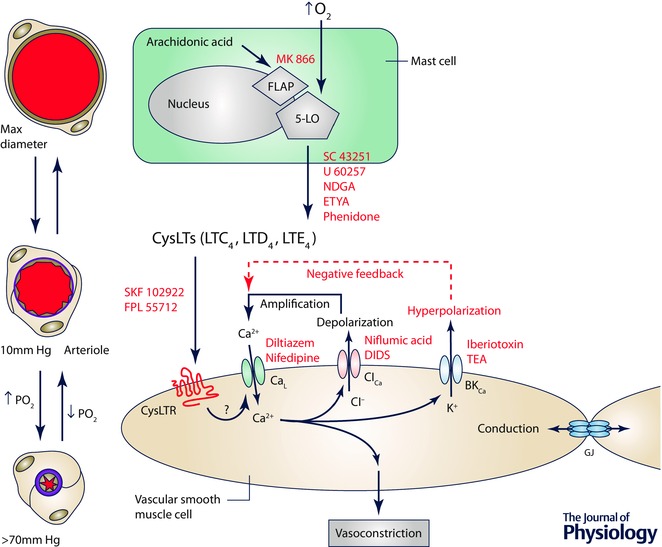
Schematic diagram depicting a mast cell (the proposed sensor site in the cheek pouch), a smooth muscle cell replete with receptors for cysteinyl leukotrienes (CysLTRs) and ion channels involved in arteriolar O2 reactivity in the cheek pouch. Elevated is sensed by the 5‐lipoxygenase (5‐LO) in the nuclear membrane of periarteriolar mast cells that decorate arterioles in this tissue (see Fig. 1). This results in conversion of arachidonic acid to cysteinyl leukotrienes (CysLTs) such as LTC4, LTD4 and LTE4 through a process that involves presentation of the arachidonic acid to the 5‐LO by the 5‐LO‐activating protein (FLAP). This process can be inhibited by drugs such as MK 866, that blocks interaction of FLAP with the 5‐LO, or SC 43251, U 60257, nordihydroguaiaretic acid (NDGA), eicosatetraynoic acid (ETYA) or phenidone, inhibitors of the 5‐LO. The CysLTs then bind to and activate CysLTRs on vascular smooth muscle cells to induce vasoconstriction. CysLTR antagonists such as SKF 102922 or FPL 55712 can inhibit this step in the process. Activation of CysLTRs results in activation of L‐type Ca2+ channels (CaL), Ca2+ influx, an increase in intracellular Ca2+ and vasoconstriction, which can be antagonized by CaL blockers such as diltiazem or nifedipine. The increase in Ca2+ activates Ca2+‐activated Cl− channels (ClCa). The resulting efflux of Cl− through these channels causes membrane depolarization, further activating CaL and amplifying the initial signal. Blockers of ClCa channels such as niflumic acid or DIDs can inhibit this step in the process. The increase in intracellular Ca2+ and the membrane depolarization due to activation of CaL and ClCa activates large conductance, Ca2+‐activated K+ channels (BKCa). The efflux of K+ through BKCa channels blunts the depolarizing effects of activation of CaL and ClCa providing a degree of negative feedback, and limiting membrane depolarization. This step in the process can be inhibited by iberiotoxin or tetraethylammonium ions (TEA). Oxygen‐induced smooth muscle depolarization can be conducted along the vessel wall by gap junctions (GJ) supporting the conduction of O2‐induced vasoconstriction that has been observed experimentally.
Leukotrienes are synthesized from arachidonic acid released from membrane phospholipid stores by a multistep process (Peters‐Golden, 1998; Storch et al. 2015). In activated cells, FLAP presents arachidonic acid to the 5‐lipoxygenase that catalyses the first step in this reaction sequence to form an unstable epoxide, leukotriene A4 (LTA4). The epoxide LTA4 is then conjugated with glutathione to form the CysLT leukotriene C4 (LTC4), which is converted to leukotriene D4 (LTD4) and leukotriene E4 (LTE4) (also CysLTs) by consecutive cleavage of peptides from the added glutathione. The K m for O2 of the 5‐lipoxygenase purified from porcine leukocytes is about 9 mmHg (Ibe & Raj, 1992). Given the nuclear membrane localization of the 5‐lipoxygenase (Peters‐Golden & Brock, 2001; Woods et al. 1995) and the steep intracellular gradients in O2 that arise from O2 consumption by mitochondria, oxidases and oxygenases within cells (Jones, 1981), leukotriene production by intact cells will be O2 dependent well within the physiological range (10–150 mmHg) required for a microvascular O2 sensor (see Fig. 1). Oxygen‐dependent synthesis of leukotrienes has been reported in several in vitro systems (Paterson, 1986; Ohwada et al. 1990; Ibe & Raj, 1992; Martin et al. 1992) providing support for the hypothesis that this pathway is involved in O2 sensing, at least in the hamster cheek pouch.
Oxygen‐dependent production of 20‐HETE
In contrast to the data from the epithelial portion of the cheek pouch, arteriolar O2 reactivity in striated muscle appears to be mediated by 20‐HETE produced by CYP4504A ω‐hydroxylase (Harder et al. 1996; Lombard et al. 1999; Hungerford et al. 2000; Kunert et al. 2001 a,b, 2009; Drenjancevic‐Peric et al. 2003, 2004; Marvar et al. 2007; Wang et al. 2009; Ngo et al. 2013) (Fig. 8). This pathway also has been implicated in O2‐induced cerebral vasoconstriction in fetal sheep (Ohata et al. 2010) and contributes to O2‐induced constriction of retinal arterioles (Zhu et al. 1998) produced by increases in inspired . Importantly, inhibitors of CYP4504A ω‐hydroxylase have no effect on the O2 reactivity of arterioles in the epithelial portion of the hamster cheek pouch (Lombard et al. 1999) indicating that the inhibition observed in striated muscle preparations is not simply some non‐specific effect. The lack of effect of CYP4504A ω‐hydroxylase inhibitors on O2 reactivity in the epithelial portion of the cheek pouch also further supports the idea that there are regional differences in the mechanism of action of O2. The K m for O2 for formation of 20‐HETE by CYP4504A ω‐hydroxylase by renal microvessels is in the order of 50 mmHg (Harder et al. 1996), well within the physiological range experienced by the microcirculation (see Fig. 1).
Figure 8. The site and mechanism of action of O2 in cremaster muscle .
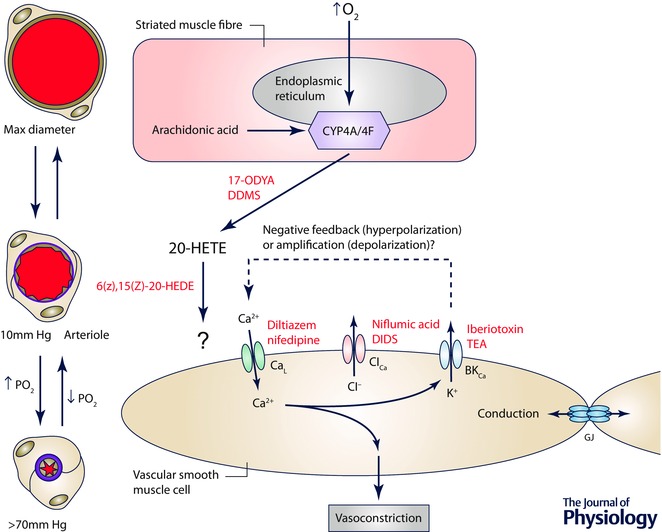
Schematic diagram of a striated muscle fibre (the proposed sensor cell in this tissue) and a smooth muscle cell replete with ion channels that may be involved in arteriolar O2 reactivity in this tissue. Elevated is sensed by cytochrome P4504A/4F ω‐hydroxylase (CYP4A/4F) located in the endoplasmic reticulum of striated muscle fibres, resulting in conversion of arachidonic acid into 20‐HETE, a process that can be inhibited by 17‐ODYA or DDMS. 20‐HETE then acts on smooth muscle cells to induce Ca2+ influx through L‐type Ca2+ channels (CaL), an increase in intracellular Ca2+ and vasoconstriction. As indicated by the ‘?’ next to the arrow connecting 20‐HETE and the smooth muscle cell, the precise receptor for 20‐HETE that is responsible for O2 reactivity is unclear because 20‐HETE has been proposed to close large conductance, Ca2+‐activated K+ channels (BKCa), which would lead to membrane depolarization activating CaL. In contrast, other studies suggest that BKCa serve a negative feedback role as they do in the cheek pouch. As in the cheek pouch it is proposed that O2‐induced depolarization of smooth muscle cells can be conducted along the vessel wall through gap junctions (GJ), consistent with the observed conducted vasoconstriction that has been observed experimentally. 6(Z),15(Z)‐20‐HEDE, 20‐hydroxy‐6Z,15Z‐eicosadienoic acid.
Studies in rat cremaster muscle have demonstrated expression of CYP4504A ω‐hydroxylase in both arteriolar smooth muscle cells and in the surrounding striated muscle (Kunert et al. 2001 a). This observation suggests that O2 may be sensed either by smooth muscle cells or by striated muscle fibres to mediate arteriolar O2 reactivity. In first‐order arterioles within the cremaster muscle, it appears that striated muscle cells serve as an important source of 20‐HETE that is responsible for the bulk of O2 reactivity in those large arterioles in situ (Frisbee & Lombard, 2002). Isolated first‐order rat cremaster arterioles studied ex vivo were demonstrated to display intrinsic O2 reactivity that is also mediated, in part, by O2‐dependent production of 20‐HETE by the smooth muscle (Frisbee & Lombard, 2002). However, this is not a universal finding as other investigators have failed to demonstrate intrinsic O2 reactivity in isolated, cannulated first‐order arterioles from the rat (Tateishi & Faber, 1995 a,b; Kerkhof et al. 1999), as noted above. This is in contrast to the large body of evidence from intravital preparations demonstrating 20‐HETE‐mediated O2 reactivity in small arterioles in a variety of striated muscle microvascular preparations (see references above).
Thus, it seems likely that striated muscle fibres and the CYP4504A ω‐hydroxylase contained within are the site where changes in are primarily sensed, with 20‐HETE serving as the mediator linking changes in with changes in arteriolar tone in the microcirculation of striated muscle. Additional experiments designed to critically test the hypothesis that vascular smooth muscle cell CYP4504A ω‐hydroxylase contributes to arteriolar O2 reactivity in situ are needed; perhaps conditional, cell‐specific knock‐out of this enzyme family in smooth muscle cells might help to resolve this issue. In addition, the location of CYP4504A ω‐hydroxylase in striated muscle fibres (and smooth muscle cells) favours O2 sensing in the muscle fibres rather than capillaries downstream from the arterioles as has been proposed (Ghonaim et al. 2011). Additional experiments will be required to positively identify where O2 is sensed in striated muscle to mediate arteriolar O2 reactivity.
While there is considerable evidence that CYP4504A ω‐hydroxylase serves as the main O2 sensor mediating arteriolar O2 reactivity in the microcirculation of striated muscle, this system is not immutable. For example, the CYP4504A ω‐hydroxylase antagonist N‐methylsulfonyl‐12,12‐dibromododec‐11‐enamide (DDMS) (Kroetz & Xu, 2005) reduces arteriolar O2 reactivity in cremaster muscles of Wistar–Kyoto (WKY) rats and spontaneously hypertensive rats (SHR) with established hypertension (Kunert et al. 2001 b). However, DDMS is without significant effect on O2 reactivity during the development of hypertension in the SHR (Kunert et al. 2001 b). Similarly, DDMS inhibited O2 reactivity in cremaster arterioles in sham‐operated controls and in rats with established Goldblatt hypertension, but was without effect during the early phase of hypertension development in this model (Kunert et al. 2009). Spinotrapezius arterioles of Sprague–Dawley rats fed a high salt diet lose CYP4504A ω‐hydroxylase‐mediated O2 reactivity, with no change in CYP4504A ω‐hydroxylase protein expression (Marvar et al. 2007). Conversely, in Dahl salt‐sensitive rats, high salt diet increases arteriolar O2 reactivity and the sensitivity to inhibition by DDMS that is associated with an increased expression of CYP4504A ω‐hydroxylase isoforms (Wang et al. 2009). These data indicate that the mechanism of action of O2 on arterioles can change and support the idea that there are multiple mechanisms coupling changes in with changes in arteriolar diameter. The fact that O2 reactivity remains but the mechanism of action of O2 can change suggests that this is an important process that is ‘protected’ by mechanism redundancy.
As demonstrated by Kerkhof et al. (1999), there may be substantial interactions among signalling pathways that complicate interpretation of data based on inhibition of single pathways. While such interactions have been explored in isolated vessels ex vivo (Kerkhof et al. 1999; Frisbee et al. 2001 a,b, 2002), there are no similar studies examining the interaction among all of the putative O2‐related signalling pathways in small arterioles in situ in the intact microcirculation.
Ionic basis of arteriolar O2 reactivity
Despite the differences in chemical mediators of O2 reactivity between the hamster cheek pouch and striated muscle preparations (CysLTs vs. 20‐HETE, respectively), a common downstream element in the signalling cascade appears to be activation of L‐type Ca2+ channels. Blockers of these channels eliminate O2 reactivity in both the cheek pouch and cremaster muscle models (Welsh et al. 1998; Jackson, 2012; Ngo et al. 2013) (Figs 7 and 8). However, the signalling pathways leading to activation of these Ca2+ channels may be different in the two tissues. In the hamster cheek pouch in situ, elevated causes arteriolar smooth muscle cell depolarization (Welsh et al. 1998) and a subsequent increase in intracellular Ca2+ (Brekke et al. 2006). Consistent with a role for CysLTs in arteriolar O2 reactivity, we have found that LTD4 depolarizes smooth muscle cells, constricts isolated, cannulated hamster cheek pouch arterioles and induces conducted vasoconstriction of arterioles in cheek pouches in situ (Jackson & Segal, 2005). Blockade of L‐type Ca2+ channels in situ obliterates both O2‐induced constriction and depolarization (Welsh et al. 1998). There are at least two, non‐mutually exclusive mechanisms that could be responsible for the O2‐dependent depolarization in the cheek pouch and that are consistent with the findings that L‐type Ca2+ channel blockers prevent both constriction and depolarization of the arteriolar muscle cells (Welsh et al. 1998). First, it is possible that the inward current of Ca2+ through L‐type Ca2+ channels causes the depolarization. However, this seems unlikely given the high resting K+ permeability of vascular muscle cells (Jackson et al. 1997, 1998, 2000 c), the density of voltage‐gated K+ (KV) and BKCa channels (which will activate as the membrane is depolarized) (Jackson et al. 1997, 1998, 2000 c), and the small currents through L‐type Ca2+ channels at physiological membrane potentials and Ca2+ gradients (Nelson et al. 1990; Gollasch et al. 1992). More likely, this depolarizing Ca2+ signal must be amplified. Calcium‐activated Cl− (ClCa) channels could provide a means of amplifying the Ca2+ signal and promoting depolarization. These channels are activated by elevation of intracellular Ca2+ and, given the electrochemical gradient for Cl− ions in most vascular muscle cells, result in Cl− efflux from the cells and hence depolarization (Large & Wang, 1996). Calcium‐activated Cl− channels mediate depolarization associated with vasoconstrictor‐induced contraction of vascular muscle in other systems (Large & Wang, 1996). Preliminary studies showed that O2‐induced constriction and membrane depolarization of smooth muscle cells in hamster cheek pouch arterioles could be reversed by the ClCa channel blockers niflumic acid and 4,4′‐diisothiocyano2,2′‐stilbenedisulfonic acid (DIDS) (Jackson, 2000 a), supporting this hypothesis. Additional research will be needed to fill in the gaps in this signalling pathway and to exclude alternative hypotheses.
The details of the signalling pathway by which elevated causes constriction of arterioles in striated muscle, between 20‐HETE and activation of L‐type Ca2+ channels, remain unclear (Fig. 8). It has been proposed that O2‐induced constriction of arterioles in rat spinotrapezius muscle involves 20‐HETE‐dependent closure of BKCa channels, membrane depolarization and subsequent opening of L‐type Ca2+ channels (Marvar et al. 2007). In preparations exposed to low , they found that the BKCa channel blocker, iberiotoxin, produced substantial vasoconstriction that was similar in magnitude to that produced by elevation of (Marvar et al. 2007). Iberiotoxin blunted both 20‐HETE‐ and O2‐induced vasoconstriction, while arteriolar constriction induced by noradrenaline remained intact. Studies of isolated rat gracilis arteries ex vivo also support this signalling pathway (Frisbee et al. 2001 a). In contrast, it was previously shown in hamster cremaster muscle that arteriolar BKCa channels are silent at rest under low conditions and that inhibition of these channels potentiated, rather than inhibited, O2‐induced vasoconstriction (Jackson, 1998) even though O2 reactivity in this preparation is also mediated by CYP4504A ω‐hydroxylase (Lombard et al. 1999). These data suggest that in the hamster microcirculation, BKCa channels participate in the negative feedback regulation of arteriolar tone and actually blunt O2‐induced vasoconstriction (Jackson & Blair, 1998). Blockade of BKCa channels also potentiates O2‐induced constriction of hamster cheek pouch arterioles (W. F. Jackson, personal observation) (Fig. 7). Thus, in hamster cremaster muscle and in the cheek pouch, BKCa channels appear to lie downstream from L‐type Ca2+ channels in the O2‐dependent signalling cascade, rather than upstream as was proposed by Marvar et al. (2007). In addition to inhibiting BKCa channels (Zou et al. 1996), 20‐HETE augments currents through L‐type Ca2+ channels (Gebremedhin et al. 1998), possibly through activation of protein kinase C (Lange et al. 1997) or tyrosine kinases (Sun et al. 1999). In porcine coronary resistance arteries, 20‐HETE‐induced vasoconstriction appears to involve activation of Rho kinase (Randriamboavonjy et al. 2003). Thus, alternative signalling pathways are possible that could reconcile these data. Additional research in which both membrane potential and diameter are measured before and after blockade of L‐type Ca2+ channels, BKCa channels and/or CYP4504A ω‐hydroxylase in striated muscle arterioles, in situ, will be required to resolve these apparent differences. It is also not known if 20‐HETE can induce conducted vasoconstriction, as does O2 in striated muscle arterioles. Nonetheless, as stated above, the observation that O2‐induced vasomotor responses can be conducted in mouse cremaster muscle (Ngo et al. 2010; Riemann et al. 2011) strongly suggests that changes in arteriolar smooth muscle membrane potential are part of the mechanism of action of O2 in this tissue and presumably other striated muscle beds.
Summary and conclusions
The location of the cell type responsible for arteriolar O2 reactivity, in situ, remains unclear, because there is regional heterogeneity in the site and mechanism of action of O2 in the peripheral microcirculation. As a target for future investigation, it is assumed that the models shown in Figs 7 and 8 apply to arteriolar O2 reactivity over the tissue range of 10–70 mmHg for tissues such as the hamster cheek pouch (Fig. 7), or striated muscle preparations such as the hamster, rat or mouse cremaster muscles (Fig. 8). Based largely (but not exclusively) on studies in the hamster cheek pouch, it is proposed that changes in are sensed, extravascularly, by O2‐dependent enzymes located in the membrane of the nucleus or endoplasmic reticulum in parenchymal or other cell types (e.g. mast cells in the case of the hamster cheek pouch). This results in the O2‐dependent production of a vasoconstrictor (CysLTs in hamster cheek pouch or 20‐HETE in striated muscle) that acts on arteriolar smooth muscle cells in the wall of arterioles. The vasoconstrictors produce membrane depolarization and activation of L‐type, voltage‐gated Ca2+ channels, leading to an increase in intracellular Ca2+ and local vasoconstriction at the site of action of the mediator. The membrane depolarization is conducted along the vessel via gap junctions to produce conducted vasoconstriction along the arteriole allowing for summation and integration of O2‐generated signals throughout the microvascular network. In the hamster cheek pouch, vasoconstrictors that depolarize smooth muscle cells produce conducted constriction transmitted along the smooth muscle cell layer (Welsh & Segal, 1998; Bartlett & Segal, 2000). These data suggest that, in this tissue, O2‐induced vasomotor effects are initiated near smooth muscle‐containing arterioles, rather than in downstream capillaries or venules, to provide an arteriolar smooth muscle pathway for conduction of the O2‐initiated response. A similar scenario is assumed in striated muscle with the understanding that the tremendous length of striated muscle fibres (relative to the length of arterioles, capillaries and venules) may also contribute to transmission of O2‐induced signals within a microvascular network. However, it is also recognized that additional research is required to exclude alternatives in the microcirculation of striated muscle. It should also be noted that detailed studies of arteriolar O2 reactivity have only been studied in rodents (hamsters, mice and rats). Whether similar mechanisms are functional in larger mammals (including humans) remains to be established.
While there is a plethora of ex vivo studies suggesting that smooth muscle cells, endothelial cells, red blood cells or some combination contribute to O2 reactivity, it is not clear that these ex vivo findings accurately represent the arteriolar O2 reactivity that is consistently observed, in situ. This may partly be due to the use of relatively large vessels (small arteries and larger arterioles) for the majority of ex vivo studies. Given the regional heterogeneity of mechanisms modulating myogenic tone between arteries and arterioles (Westcott & Jackson, 2011; Westcott et al. 2012), it seems likely that different mechanisms, and hence different O2 sensors and mechanisms, may exist for sensing and responding to changes in in different segments of the vascular tree. Resistance arteries upstream from the microcirculation may be tuned to respond primarily to changes in blood (systemic hypoxia/hyperoxia), whereas downstream arterioles may be more tuned to respond to changes in local tissue .
It is also possible that different mechanisms operate over different ranges, as suggested by the ex vivo studies of Frisbee et al. (Frisbee et al. 2002). Most studies have examined responses of vessels to relatively large changes in and have not carefully examined the concentration–response relationship between O2 and vessel tone. Nor have detailed concentration–response relationships been performed for antagonists and inhibitors to define their efficacy and potency on O2 reactivity. Thus, when a single concentration of an inhibitor is used and only partial inhibition is observed, is this because there are multiple mechanisms involved, or because of a lack of efficacy of the drug on its target? Interactions among signalling pathways (Kerkhof et al. 1999) also complicate the interpretation of inhibitor data. Signalling pathway interactions have not been adequately explored in the O2 reactivity of small arterioles in situ. Conduction of O2‐induced vasomotor responses also complicates the study of arteriolar O2 reactivity, and should be further examined.
It is clear that there are regional differences in the mechanism of action of O2 on arterioles in the peripheral microcirculation (e.g. hamster cheek pouch epithelium vs. hamster striated muscle). However, there is a remarkably consistent result: elevated in the environment of small arterioles in situ results in profound vasoconstriction. Oxygen‐induced arteriolar constriction may be a protective mechanism to defend against oxidative stress and excessive production of O2‐derived reactive oxygen species (ROS) as suggested by Golub and Pittman (Golub & Pittman, 2013). Oxygen‐induced arteriolar constriction may be part of a negative feedback mechanism to limit tissue exposure to O2 to help reduce the oxidative burden that is superimposed on the mechanisms that are involved in the regulation of blood flow to meet the metabolic demands of tissues. That different tissues have different mechanisms (e.g. cheek pouch vs. striated muscle) to accomplish the same end‐result (O2‐induced arteriolar constriction) suggests that this may be an important response to maintain homeostasis.
Additional information
Competing interests
None declared.
Funding
The author is supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH) under award numbers RO1 HL32469 and P01 HL070687. The content is solely the responsibility of the author and does not necessarily represent the official views of the NIH.
Biography
William F. Jackson is Professor of Pharmacology and Toxicology at Michigan State University. He has had a long‐term interest in identifying the site in tissues where changes in oxygen tension are sensed and how such changes alter the contractile function of arteriolar muscle cells. His research focuses on identifying the ion channels expressed by both arteriolar smooth muscle and endothelial cells, how these ion channels function in the regulation of myogenic tone and oxygen signalling in the microcirculation, and how ageing and disease states such as hypertension and obesity impact the expression and function of ion channels in the microcirculation.

References
- Aalkjaer C & Lombard JH (1995). Effect of hypoxia on force, intracellular pH and Ca2+ concentration in rat cerebral and mesenteric small arteries. J Physiol 482, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu‐Soud HM, Ichimori K, Presta A & Stuehr DJ (2000). Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric‐oxide synthase. J Biol Chem 275, 17349–17357. [DOI] [PubMed] [Google Scholar]
- Baez S (1973). An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res 5, 384–394. [DOI] [PubMed] [Google Scholar]
- Baron JF, Vicaut E, Hou X & Duvelleroy M (1990). Independent role of arterial O2 tension in local control of coronary blood flow. Am J Physiol 258, H1388–H1394. [DOI] [PubMed] [Google Scholar]
- Bartlett IS & Segal SS (2000). Resolution of smooth muscle and endothelial pathways for conduction along hamster cheek pouch arterioles. Am J Physiol Heart Circ Physiol 278, H604–H612. [DOI] [PubMed] [Google Scholar]
- Boegehold MA & Bohlen HG (1988). Arteriolar diameter and tissue oxygen tension during muscle contraction in hypertensive rats. Hypertension 12, 184–191. [DOI] [PubMed] [Google Scholar]
- Bongard O, Bounameaux H & Fagrell B (1992). Effects of oxygen inhalation on skin microcirculation in patients with peripheral arterial occlusive disease. Circulation 86, 878–886. [DOI] [PubMed] [Google Scholar]
- Bredle DL, Bradley WE, Chapler CK & Cain SM (1988). Muscle perfusion and oxygenation during local hyperoxia. J Appl Physiol (1985) 65, 2057–2062. [DOI] [PubMed] [Google Scholar]
- Brekke JF, Jackson WF & Segal SS (2006). Arteriolar smooth muscle Ca2+ dynamics during blood flow control in hamster cheek pouch. J Appl Physiol (1985) 101, 307–315. [DOI] [PubMed] [Google Scholar]
- Burns WR, Cohen KD & Jackson WF (2004). K+‐induced dilation of hamster cremasteric arterioles involves both the Na+/K+‐ATPase and inward‐rectifier K+ channels. Microcirculation 11, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Forstermann U, Matsuda H & Pohl U (1984). The role of prostaglandins in the endothelium‐mediated vasodilatory response to hypoxia. Pflugers Arch 401, 77–83. [DOI] [PubMed] [Google Scholar]
- Busse R, Pohl U, Kellner C & Klemm U (1983). Endothelial cells are involved in the vasodilatory response to hypoxia. Pflugers Arch 397, 78–80. [DOI] [PubMed] [Google Scholar]
- Carrier O Jr, Walker JR & Guyton AC (1964). Role of oxygen in autoregulation of blood flow in isolated vessels. Am J Physiol 206, 951–954. [DOI] [PubMed] [Google Scholar]
- Casey DP & Joyner MJ (2011). Local control of skeletal muscle blood flow during exercise: influence of available oxygen. J Appl Physiol (1985) 111, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AE & Detar R (1980). Oxygen and vascular smooth muscle contraction revisited. Am J Physiol 238, H716–H728. [DOI] [PubMed] [Google Scholar]
- Coburn RF, Grubb B & Aronson RD (1979). Effect of cyanide on oxygen tension‐dependent mechanical tension in rabbit aorta. Circ Res 44, 368–378. [DOI] [PubMed] [Google Scholar]
- Cohen KD & Jackson WF (2003). Hypoxia inhibits contraction but not calcium channel currents or changes in intracellular calcium in arteriolar muscle cells. Microcirculation 10, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DM, McCullough WT & Ellsworth ML (1998). Conducted vascular responses: communication across the capillary bed. Microvasc Res 56, 43–53. [DOI] [PubMed] [Google Scholar]
- Curtis TM & Scholfield CN (2000). Transient Ca2+‐activated Cl− currents with endothelin in isolated arteriolar smooth muscle cells of the choroid. Invest Ophthalmol Vis Sci 41, 2279–2285. [PubMed] [Google Scholar]
- Dart C & Standen NB (1995). Activation of ATP‐dependent K+ channels by hypoxia in smooth muscle cells isolated from the pig coronary artery. J Physiol 483, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Hill MA & Kuo L (2011). Local regulation of microvascular perfusion In Comprehensive Physiology, pp. 161–284. John Wiley & Sons, Inc. [Google Scholar]
- Delashaw JB & Duling BR (1991). Heterogeneity in conducted arteriolar vasomotor response is agonist dependent. Am J Physiol 260, H1276–H1282. [DOI] [PubMed] [Google Scholar]
- Demchenko IT, Boso AE, Bennett PB, Whorton AR & Piantadosi CA (2000). Hyperbaric oxygen reduces cerebral blood flow by inactivating nitric oxide. Nitric Oxide 4, 597–608. [DOI] [PubMed] [Google Scholar]
- Detar R & Bohr DF (1968). Oxygen and vascular smooth muscle contraction. Am J Physiol 214, 241–244. [DOI] [PubMed] [Google Scholar]
- Devillier P, Baccard N & Advenier C (1999). Leukotrienes, leukotriene receptor antagonists and leukotriene synthesis inhibitors in asthma: an update. Part I: synthesis, receptors and role of leukotrienes in asthma. Pharmacol Res 40, 3–13. [DOI] [PubMed] [Google Scholar]
- Didion SP & Faraci FM (2002). Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am J Physiol Heart Circ Physiol 282, H688–H695. [DOI] [PubMed] [Google Scholar]
- Dietrich HH, Ellsworth ML, Sprague RS & Dacey RG Jr (2000). Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol 278, H1294–H1298. [DOI] [PubMed] [Google Scholar]
- Drenjancevic‐Peric I, Frisbee JC & Lombard JH (2003). Skeletal muscle arteriolar reactivity in SS.BN13 consomic rats and Dahl salt‐sensitive rats. Hypertension 41, 1012–1015. [DOI] [PubMed] [Google Scholar]
- Drenjancevic‐Peric I, Greene AS, Kunert MP & Lombard JH (2004). Arteriolar responses to vasodilator stimuli and elevated P O2 in renin congenic and Dahl salt‐sensitive rats. Microcirculation 11, 669–677. [DOI] [PubMed] [Google Scholar]
- Duling BR (1972). Microvascular responses to alterations in oxygen tension. Circ Res 31, 481–489. [DOI] [PubMed] [Google Scholar]
- Duling BR (1973). The preparation and use of the hamster cheek pouch for studies of the microcirculation. Microvasc Res 5, 423–429. [DOI] [PubMed] [Google Scholar]
- Duling BR (1974). Oxygen sensitivity of vascular smooth muscle. II. In vivo studies. Am J Physiol 227, 42–49. [DOI] [PubMed] [Google Scholar]
- Duling BR & Berne RM (1970). Longitudinal gradients in periarteriolar oxygen tension. A possible mechanism for the participation of oxygen in local regulation of blood flow. Circ Res 27, 669–678. [DOI] [PubMed] [Google Scholar]
- Duling BR, Gore RW, Dacey RG Jr & Damon DN (1981). Methods for isolation, cannulation, and in vitro study of single microvessels. Am J Physiol 241, H108–H116. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG & Sprague RS (2016). Role of erythrocyte‐released ATP in the regulation of microvascular oxygen supply in skeletal muscle. Acta Physiol (Oxf) 216, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML, Forrester T, Ellis CG & Dietrich HH (1995). The erythrocyte as a regulator of vascular tone. Am J Physiol Heart Circ Physiol 269, H2155–H2161. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Paul DL, Simon AM, Goodenough DA, Day KH, Damon DN & Duling BR (2003). Central role of connexin40 in the propagation of electrically activated vasodilation in mouse cremasteric arterioles in vivo. Circ Res 92, 793–800. [DOI] [PubMed] [Google Scholar]
- Franco‐Obregon A & Lopez‐Barneo J (1996. a). Differential oxygen sensitivity of calcium channels in rabbit smooth muscle cells of conduit and resistance pulmonary arteries. J Physiol 491, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco‐Obregon A & Lopez‐Barneo J (1996. b). Low PO2 inhibits calcium channel activity in arterial smooth muscle cells. Am J Physiol 271, H2290–H2299. [DOI] [PubMed] [Google Scholar]
- Franco‐Obregon A, Urena J & Lopez‐Barneo J (1995). Oxygen‐sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proc Natl Acad Sci U S A 92, 4715–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredricks KT, Liu Y & Lombard JH (1994. a). Response of extraparenchymal resistance arteries of rat skeletal muscle to reduced PO2. Am J Physiol 267, H706–H715. [DOI] [PubMed] [Google Scholar]
- Fredricks KT, Liu Y, Rusch NJ & Lombard JH (1994. b). Role of endothelium and arterial K+ channels in mediating hypoxic dilation of middle cerebral arteries. Am J Physiol 267, H580–H586. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Krishna UM, Falck JR & Lombard JH (2001. a). Role of prostanoids and 20‐HETE in mediating oxygen‐induced constriction of skeletal muscle resistance arteries. Microvasc Res 62, 271–283. [DOI] [PubMed] [Google Scholar]
- Frisbee JC & Lombard JH (2002). Parenchymal tissue cytochrome P450 4A enzymes contribute to oxygen‐induced alterations in skeletal muscle arteriolar tone. Microvasc Res 63, 340–343. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Maier KG, Falck JR, Roman RJ & Lombard JH (2002). Integration of hypoxic dilation signaling pathways for skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol 283, R309–R319. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Roman RJ, Krishna UM, Falck JR & Lombard JH (2001. b). Relative contributions of cyclooxygenase‐ and cytochrome P450 omega‐hydroxylase‐dependent pathways to hypoxic dilation of skeletal muscle resistance arteries. J Vasc Res 38, 305–314. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Roman RJ, Murali Krishna U, Falck JR & Lombard JH (2001. c). Altered mechanisms underlying hypoxic dilation of skeletal muscle resistance arteries of hypertensive versus normotensive Dahl rats. Microcirculation 8, 115–127. [PubMed] [Google Scholar]
- Gebremedhin D, Bonnet P, Greene AS, England SK, Rusch NJ, Lombard JH & Harder DR (1994). Hypoxia increases the activity of Ca2+‐sensitive K+ channels in cat cerebral arterial muscle‐cell membranes. Pflugers Arch 428, 621–630. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER & Harder DR (1998). Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20‐HETE which enhances L‐type Ca2+ current. J Physiol 507, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonaim NW (2013). Investigating conducted microvascular response to localized oxygen delivery in vivo using a novel micro‐delivery approach. p. 214 PhD Thesis. University of Western Ontario, London, Ontario, Canada: Electronic thesis and dissertation repository. Paper 1643. http://ir.lib.uwo.ca/etd/1643. [Google Scholar]
- Ghonaim NW, Lau LW, Goldman D, Ellis CG & Yang J (2011). A micro‐delivery approach for studying microvascular responses to localized oxygen delivery. Microcirculation 18, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokina NI & Bevan JA (2000). Role of intracellular Ca2+ release in histamine‐induced depolarization in rabbit middle cerebral artery. Am J Physiol Heart Circ Physiol 278, H2105–H2114. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Hescheler J, Quayle JM, Patlak JB & Nelson MT (1992). Single calcium‐channel currents of arterial smooth‐muscle at physiological calcium concentrations. Am J Physiol 263, C948–C952. [DOI] [PubMed] [Google Scholar]
- Golub AS & Pittman RN (2013). Bang‐bang model for regulation of local blood flow. Microcirculation 20, 455–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczynski RJ & Duling BR (1978). Role of oxygen in arteriolar functional vasodilation in hamster striated muscle. Am J Physiol 235, H505–H515. [DOI] [PubMed] [Google Scholar]
- Guyenet PG (2000). Neural structures that mediate sympathoexcitation during hypoxia. Respir Physiol 121, 147–162. [DOI] [PubMed] [Google Scholar]
- Harder DR, Narayanan J, Birks EK, Liard JF, Imig JD, Lombard JH, Lange AR & Roman RJ (1996). Identification of a putative microvascular oxygen sensor. Circ Res 79, 54–61. [DOI] [PubMed] [Google Scholar]
- Hirakawa Y, Gericke M, Cohen RA & Bolotina VM (1999). Ca2+‐dependent Cl− channels in mouse and rabbit aortic smooth muscle cells: regulation by intracellular Ca2+ and NO. Am J Physiol 277, H1732–H1744. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Sessa WC & Segal SS (2000). Vasomotor control in arterioles of the mouse cremaster muscle. FASEB J 14, 197–207. [DOI] [PubMed] [Google Scholar]
- Hutchins PM, Bond RF & Green HD (1974). Participation of oxygen in the local control of skeletal muscle microvasculature. Circ Res 34, 85–93. [DOI] [PubMed] [Google Scholar]
- Ibe BO & Raj JU (1992). Metabolism of endogenous arachidonic acid by isolated lungs of neonatal and adult rabbits stimulated with calcium ionophore: effect of hypoxia. Exp Lung Res 18, 69–85. [DOI] [PubMed] [Google Scholar]
- Jackson WF (1986). Prostaglandins do not mediate arteriolar oxygen reactivity. Am J Physiol 250, H1102–H1108. [DOI] [PubMed] [Google Scholar]
- Jackson WF (1987). Arteriolar oxygen reactivity: where is the sensor? Am J Physiol 253, H1120–H1126. [DOI] [PubMed] [Google Scholar]
- Jackson WF (1988). Lipoxygenase inhibitors block O2 responses of hamster cheek pouch arterioles. Am J Physiol 255, H711–H716. [DOI] [PubMed] [Google Scholar]
- Jackson WF (1989). Arteriolar oxygen reactivity is inhibited by leukotriene antagonists. Am J Physiol 257, H1565–H1572. [DOI] [PubMed] [Google Scholar]
- Jackson WF (1991). Nitric oxide does not mediate arteriolar oxygen reactivity. Microcirc Endothelium Lymphatics 7, 199–215. [PubMed] [Google Scholar]
- Jackson WF (1993). Regional differences in mechanism of action of oxygen on hamster arterioles. Am J Physiol 265, H599–H603. [DOI] [PubMed] [Google Scholar]
- Jackson WF (1998). Potassium channels and regulation of the microcirculation. Microcirculation 5, 85–90. [PubMed] [Google Scholar]
- Jackson WF (2000. a). Cl− channel blockers dilate O2 constricted arterioles in the hamster cheek pouch. FASEB J 14, A31. [Google Scholar]
- Jackson WF (2000. b). Hypoxia does not activate ATP‐sensitive K+ channels in arteriolar muscle cells. Microcirculation 7, 137–145. [PMC free article] [PubMed] [Google Scholar]
- Jackson WF (2000. c). Ion channels and vascular tone. Hypertension 35, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WF (2012). Microcirculation In Muscle, ed. Olson JA. & Hill EN, chap. 89, pp. 1197–1206. Academic Press, Boston/Waltham. [Google Scholar]
- Jackson WF & Blair KL (1998). Characterization and function of Ca2+‐activated K+ channels in arteriolar muscle cells. Am J Physiol 274, H27–H34. [DOI] [PubMed] [Google Scholar]
- Jackson WF & Duling BR (1983). The oxygen sensitivity of hamster cheek pouch arterioles. In vitro and in situ studies. Circ Res 53, 515–525. [DOI] [PubMed] [Google Scholar]
- Jackson WF, Huebner JM & Rusch NJ (1997). Enzymatic isolation and characterization of single vascular smooth muscle cells from cremasteric arterioles. Microcirculation 4, 35–50. [DOI] [PubMed] [Google Scholar]
- Jackson WF, Pohl U & Busse R (1987). Endothelium‐dependent oxygen reactivity in hamster carotid arteries. Fed Proc 46, 1530. [Google Scholar]
- Jackson WF & Segal SS (2005). Cysteinyl leukotrienes depolarize arteriolar muscle cells and initiate conducted vasoconstriction. FASEB J 19, A722–A723. [Google Scholar]
- Johnson PC, Vandegriff K, Tsai AG & Intaglietta M (2005). Effect of acute hypoxia on microcirculatory and tissue oxygen levels in rat cremaster muscle. J Appl Physiol (1985) 98, 1177–1184. [DOI] [PubMed] [Google Scholar]
- Jones DP (1981). Hypoxia and drug metabolism. Biochem Pharmacol 30, 1019–1023. [DOI] [PubMed] [Google Scholar]
- Justesen BL, Mistry P, Chaturvedi N, Thom SA, Witt N, Kohler D, Hughes AD & Sjolie AK (2010). Retinal arterioles have impaired reactivity to hyperoxia in type 1 diabetes. Acta Ophthalmol 88, 453–457. [DOI] [PubMed] [Google Scholar]
- Kaul DK, Fabry ME, Costantini F, Rubin EM & Nagel RL (1995). In vivo demonstration of red cell‐endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J Clin Invest 96, 2845–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhof CJM, Bakker ENTP & Sipkema P (1999). Role of cytochrome P‐450 4A in oxygen sensing and NO production in rat cremaster resistance arteries. Am J Physiol 277, H1546–H1552. [DOI] [PubMed] [Google Scholar]
- Kisilevsky M, Mardimae A, Slessarev M, Han J, Fisher J & Hudson C (2008). Retinal arteriolar and middle cerebral artery responses to combined hypercarbic/hyperoxic stimuli. Invest Ophthalmol Vis Sci 49, 5503–5509. [DOI] [PubMed] [Google Scholar]
- Klitzman B, Damon DN, Gorczynski RJ & Duling BR (1982). Augmented tissue oxygen supply during striated muscle contraction in the hamster. Relative contributions of capillary recruitment, functional dilation, and reduced tissue PO2. Circ Res 51, 711–721. [DOI] [PubMed] [Google Scholar]
- Kontos HA & Wei EP (1985). Oxygen‐dependent mechanisms in cerebral autoregulation. Ann Biomed Eng 13, 329–334. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Raper AJ, Rosenblum WI, Navari RM & Patterson JL Jr (1978). Role of tissue hypoxia in local regulation of cerebral microcirculation. Am J Physiol 234, H582–H591. [DOI] [PubMed] [Google Scholar]
- Kroetz DL & Xu F (2005). Regulation and inhibition of arachidonic acid ω‐hydroxylases and 20‐HETE formation. Annu Rev Pharmacol Toxicol 45, 413–438. [DOI] [PubMed] [Google Scholar]
- Kunert MP, Friesma J, Falck JR & Lombard JH (2009). CYP450 4A inhibition attenuates O2 induced arteriolar constriction in chronic but not acute Goldblatt hypertension. Microvasc Res 78, 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert MP, Roman RJ, Alonso‐Galicia M, Falck JR & Lombard JH (2001. a). Cytochrome P‐450 omega‐hydroxylase: a potential O2 sensor in rat arterioles and skeletal muscle cells. Am J Physiol Heart Circ Physiol 280, H1840–H1845. [DOI] [PubMed] [Google Scholar]
- Kunert MP, Roman RJ, Falck JR & Lombard JH (2001. b). Differential effect of cytochrome P‐450 omega‐hydroxylase inhibition on O2‐induced constriction of arterioles in SHR with early and established hypertension. Microcirculation 8, 435–443. [DOI] [PubMed] [Google Scholar]
- Lamb FS & Barna TJ (1998). Chloride ion currents contribute functionally to norepinephrine‐induced vascular contraction. Am J Physiol 275, H151–H160. [DOI] [PubMed] [Google Scholar]
- Lands WE, Sauter J & Stone GW (1978). Oxygen requirement for prostaglandin biosynthesis. Prostaglandins Med 1, 117–120. [DOI] [PubMed] [Google Scholar]
- Lange A, Gebremedhin D, Narayanan J & Harder D (1997). 20‐Hydroxyeicosatetraenoic acid‐induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J Biol Chem 272, 27345–27352. [DOI] [PubMed] [Google Scholar]
- Large WA & Wang Q (1996). Characteristics and physiological role of the Ca2+‐activated Cl− conductance in smooth muscle. Am J Physiol 271, C435–C454. [DOI] [PubMed] [Google Scholar]
- Lash JM & Bohlen HG (1987). Perivascular and tissue PO2 in contracting rat spinotrapezius muscle. Am J Physiol 252, H1192–H1202. [DOI] [PubMed] [Google Scholar]
- Lindbom L, Tuma RF & Arfors KE (1980). Influence of oxygen on perfused capillary density and capillary red cell velocity in rabbit skeletal muscle. Microvasc Res 19, 197–208. [DOI] [PubMed] [Google Scholar]
- Lombard JH & Duling BR (1977). Relative importance of tissue oxygenation and vascular smooth muscle hypoxia in determining arteriolar responses to occlusion in the hamster cheek pouch. Circ Res 41, 546–551. [DOI] [PubMed] [Google Scholar]
- Lombard JH & Duling BR (1981). Multiple mechanisms of reactive hyperemia in arterioles of the hamster cheek pouch. Am J Physiol 241, H748–H755. [DOI] [PubMed] [Google Scholar]
- Lombard JH, Frisbee JC, Roman RJ & Falck JR (2004). Evaluation of cytochrome P450‐4A ω‐hydroxylase and 20‐hydroxyeicosatetraenoic acid as an O2 sensing mechanism in the microcirculation In Methods in Enzymology, ed. Chandan KS. & Gregg LS, pp. 140–165. Academic Press. [DOI] [PubMed] [Google Scholar]
- Lombard JH, Hess ME & Stekiel WJ (1986). Enhanced response of arterioles to oxygen during development of hypertension in SHR. Am J Physiol 250, H761–H764. [DOI] [PubMed] [Google Scholar]
- Lombard JH, Kunert MP, Roman RJ, Falck JR, Harder DR & Jackson WF (1999). Cytochrome P‐450 ω‐hydroxylase senses O2 in hamster muscle, but not cheek pouch epithelium, microcirculation. Am J Physiol 276, H503–H508. [DOI] [PubMed] [Google Scholar]
- Lund N, Jorfeldt L & Lewis DH (1980. a). Skeletal muscle oxygen pressure fields in healthy human volunteers. A study of the normal state and the effects of different arterial oxygen pressures. Acta Anaesthesiol Scand 24, 272–278. [DOI] [PubMed] [Google Scholar]
- Lund N, Odman S & Lewis DH (1980. b). Skeletal muscle oxygen pressure fields in rats. A study of the normal state and the effects of local anesthetics, local trauma and hemorrhage. Acta Anaesthesiol Scand 24, 155–160. [DOI] [PubMed] [Google Scholar]
- McCullough WT, Collins DM & Ellsworth ML (1997). Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol 272, H1886–H1891. [DOI] [PubMed] [Google Scholar]
- Marshall JM (1994). Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev 74, 543–594. [DOI] [PubMed] [Google Scholar]
- Marshall JM (2000). Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiol Scand 168, 561–573. [DOI] [PubMed] [Google Scholar]
- Marshall JM & Ray CJ (2012). Contribution of non‐endothelium‐dependent substances to exercise hyperaemia: are they O2 dependent? J Physiol 590, 6307–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LD, Barnes SD & Wetzel RC (1992). Acute hypoxia alters eicosanoid production of perfused pulmonary artery endothelial cells in culture. Prostaglandins 43, 371–382. [DOI] [PubMed] [Google Scholar]
- Marvar PJ, Falck JR & Boegehold MA (2007). High dietary salt reduces the contribution of 20‐HETE to arteriolar oxygen responsiveness in skeletal muscle. Am J Physiol Heart Circ Physiol 292, H1507–H1515. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS, Bender PR, Brooks GA, Butterfield GE, Groves BM, Sutton JR, Wolfel EE & Reeves JT (1991). Arterial catecholamine responses during exercise with acute and chronic high‐altitude exposure. Am J Physiol 261, E419–E424. [DOI] [PubMed] [Google Scholar]
- Messina EJ, Sun D, Koller A, Wolin MS & Kaley G (1992). Role of endothelium‐derived prostaglandins in hypoxia‐elicited arteriolar dilation in rat skeletal muscle. Circ Res 71, 790–796. [DOI] [PubMed] [Google Scholar]
- Messina EJ, Sun D, Koller A, Wolin MS & Kaley G (1994). Increases in oxygen tension evoke arteriolar constriction by inhibiting endothelial prostaglandin synthesis. Microvasc Res 48, 151–160. [DOI] [PubMed] [Google Scholar]
- Min SA, Stapleton MP & Tabrizchi R (1999). Influence of chloride ions on α1‐adrenoceptor mediated contraction and Ca2+ influx in rat caudal artery. Life Sci 64, 1631–1641. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Brinkley DW, Chang KM & Roth JP (2007). Molecular oxygen dependent steps in fatty acid oxidation by cyclooxygenase‐1. Biochemistry 46, 3975–3989. [DOI] [PubMed] [Google Scholar]
- Nase GP, Tuttle J & Bohlen HG (2003). Reduced perivascular PO2 increases nitric oxide release from endothelial cells. Am J Physiol Heart Circ Physiol 285, H507–H515. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF & Standen NB (1990). Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol 259, C3–C18. [DOI] [PubMed] [Google Scholar]
- Ngo AT, Jensen LJ, Riemann M, Holstein‐Rathlou NH & Torp‐Pedersen C (2010). Oxygen sensing and conducted vasomotor responses in mouse cremaster arterioles in situ. Pflugers Arch 460, 41–53. [DOI] [PubMed] [Google Scholar]
- Ngo AT, Riemann M, Holstein‐Rathlou NH, Torp‐Pedersen C & Jensen LJ (2013). Significance of KATP channels, L‐type Ca2+ channels and CYP450‐4A enzymes in oxygen sensing in mouse cremaster muscle arterioles in vivo. BMC Physiol 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata H, Gebremedhin D, Narayanan J, Harder DR & Koehler RC (2010). Onset of pulmonary ventilation in fetal sheep produces pial arteriolar constriction dependent on cytochrome p450 omega‐hydroxylase activity. J Appl Physiol (1985) 109, 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohwada A, Kira S & Yamashita T (1990). Influence of hypoxia on 5‐lipoxygenase pathway in rat alveolar macrophages. Prostaglandins Leukot Essent Fatty Acids 39, 69–73. [DOI] [PubMed] [Google Scholar]
- Pacaud P, Loirand G, Baron A, Mironneau C & Mironneau J (1991). Ca2+ channel activation and membrane depolarization mediated by Cl− channels in response to noradrenaline in vascular myocytes. Br J Pharmacol 104, 1000–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NAM (1986). Influence of hypoxia on histamine and leukotriene release from dispersed porcine lung cells. J Appl Physiol (1985) 61, 1790–1795. [DOI] [PubMed] [Google Scholar]
- Peters‐Golden M (1998). Cell biology of the 5‐lipoxygenase pathway. Am J Respir Crit Care Med 157, S227–S231. [PubMed] [Google Scholar]
- Peters‐Golden M & Brock TG (2001). Intracellular compartmentalization of leukotriene synthesis: unexpected nuclear secrets. FEBS Lett 487, 323–326. [DOI] [PubMed] [Google Scholar]
- Pittman RN & Duling BR (1973). Oxygen sensitivity of vascular smooth muscle. I. In vitro studies. Microvasc Res 6, 202–211. [DOI] [PubMed] [Google Scholar]
- Pohl U & Busse R (1989). Hypoxia stimulates release of endothelium‐derived relaxant factor. Am J Physiol 256, H1595–H1600. [DOI] [PubMed] [Google Scholar]
- Prewitt RL & Johnson PC (1976). The effect of oxygen on arteriolar red cell velocity and capillary density in the rat cremaster muscle. Microvasc Res 12, 59–70. [DOI] [PubMed] [Google Scholar]
- Pries AR, Heide J, Ley K, Klotz KF & Gaehtgens P (1995). Effect of oxygen tension on regulation of arteriolar diameter in skeletal muscle in situ. Microvasc Res 49, 289–299. [DOI] [PubMed] [Google Scholar]
- Proctor KG, Damon DN & Duling BR (1981). Tissue PO2 and arteriolar responses to metabolic stimuli during maturation of striated muscle. Am J Physiol 241, H325–H331. [DOI] [PubMed] [Google Scholar]
- Proctor KG & Duling BR (1982. a). Adenosine and free‐flow functional hyperemia in striated muscle. Am J Physiol 242, H688–H697. [DOI] [PubMed] [Google Scholar]
- Proctor KG & Duling BR (1982. b). Oxygen‐derived free radicals and local control of striated muscle blood flow. Microvasc Res 24, 77–86. [DOI] [PubMed] [Google Scholar]
- Randriamboavonjy V, Busse R & Fleming I (2003). 20‐HETE‐induced contraction of small coronary arteries depends on the activation of Rho‐kinase. Hypertension 41, 801–806. [DOI] [PubMed] [Google Scholar]
- Reglin B & Pries AR (2014). Metabolic control of microvascular networks: oxygen sensing and beyond. J Vasc Res 51, 376–392. [DOI] [PubMed] [Google Scholar]
- Renkin EM (1984). Control of microcirculation and blood‐tissue exchange In Handbook of Physiology, section 2, The Cardiovascular System, vol. IV, Microcirculation, part 2, ed. Renkin EM. & Michel CC, pp. 627–687. American Physiological Society, Bethesda. [Google Scholar]
- Riemann M, Rai A, Ngo AT, Dziegiel MH, Holstein‐Rathlou NH & Torp‐Pedersen C (2011). Oxygen‐dependent vasomotor responses are conducted upstream in the mouse cremaster microcirculation. J Vasc Res 48, 79–89. [DOI] [PubMed] [Google Scholar]
- Sakai N, Mizuno R, Ono N, Kato H & Ohhashi T (2007). High oxygen tension constricts epineurial arterioles of the rat sciatic nerve via reactive oxygen species. Am J Physiol Heart Circ Physiol 293, H1498–H1507. [DOI] [PubMed] [Google Scholar]
- Salter KJ & Kozlowski RZ (1998). Differential electrophysiological actions of endothelin‐1 on Cl− and K+ currents in myocytes isolated from aorta, basilar and pulmonary artery. J Pharmacol Exp Ther 284, 1122–1131. [PubMed] [Google Scholar]
- Sato A, Sakuma I & Gutterman DD (2003). Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol 285, H2345–H2354. [DOI] [PubMed] [Google Scholar]
- Schjorring OL, Carlsson R & Simonsen U (2015). Pressure myography to study the function and structure of isolated small arteries. Methods Mol Biol 1339, 277–295. [DOI] [PubMed] [Google Scholar]
- Segal SS, Damon DN & Duling BR (1989). Propagation of vasomotor responses coordinates arteriolar resistances. Am J Physiol 256, H832–H837. [DOI] [PubMed] [Google Scholar]
- Segal SS & Duling BR (1986). Communication between feed arteries and microvessels in hamster striated‐muscle – segmental vascular‐responses are functionally coordinated. Circ Res 59, 283–290. [DOI] [PubMed] [Google Scholar]
- Shah S, Allen J, Wood JG & Gonzalez NC (2003). Dissociation between skeletal muscle microvascular PO2 and hypoxia‐induced microvascular inflammation. J Appl Physiol (1985) 94, 2323–2329. [DOI] [PubMed] [Google Scholar]
- Shepherd RK & Duling BR (1995). Use of Ruthenium Red staining to detect mast cell degranulation in vivo. Microcirculation 2, 363–370. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Yokoshiki H, Sperelakis N & Paul RJ (2000). Role of voltage‐dependent and Ca2+‐activated K+ channels on the regulation of isometric force in porcine coronary artery. J Vasc Res 37, 16–25. [DOI] [PubMed] [Google Scholar]
- Sinkler SY & Segal SS (2014). Aging alters reactivity of microvascular resistance networks in mouse gluteus maximus muscle. Am J Physiol Heart Circ Physiol 307, H830–H839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Barbee RW, Ward KR & Pittman RN (2004). Prolonged tissue PO2 reduction after contraction in spinotrapezius muscle of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 287, H401–H407. [DOI] [PubMed] [Google Scholar]
- Smith LM, Golub AS & Pittman RN (2002). Interstitial PO2 determination by phosphorescence quenching microscopy. Microcirculation 9, 389–395. [DOI] [PubMed] [Google Scholar]
- Sparks HV (1980). Effect of local metabolic factors on vascular smooth muscle In Handbook of Physiology, section 2, The Cardiovascular System, vol. II, Microcirculation, part 2, ed. Bohr DF, Somlyo AP. & Sparks HV, pp. 181–309. American Physiological Society, Bethesda. [Google Scholar]
- Stiris TA, Blanco D, Codoceo R, Lasa D, Suguihara C, Bancalari E & Quero J (1996). Effects of dexamethasone on retinal and choroidal blood flow during normoxia and hyperoxia in newborn piglets. Pediatr Res 40, 592–596. [DOI] [PubMed] [Google Scholar]
- Storch U, Blodow S, Gudermann T & Mederos YSM (2015). Cysteinyl leukotriene 1 receptors as novel mechanosensors mediating myogenic tone together with angiotensin II type 1 receptors – Brief report. Arterioscler Thromb Vasc Biol 35, 121–126. [DOI] [PubMed] [Google Scholar]
- Sullivan SM & Johnson PC (1981. a). Effect of oxygen on arteriolar dimensions and blood flow in cat sartorius muscle. Am J Physiol 241, H547–H556. [DOI] [PubMed] [Google Scholar]
- Sullivan SM & Johnson PC (1981. b). Effect of oxygen on blood flow autoregulation in cat sartorius muscle. Am J Physiol 241, H807–H815. [DOI] [PubMed] [Google Scholar]
- Sun CW, Falck JR, Harder DR & Roman RJ (1999). Role of tyrosine kinase and PKC in the vasoconstrictor response to 20‐HETE in renal arterioles. Hypertension 33, 414–418. [DOI] [PubMed] [Google Scholar]
- Tateishi J & Faber JE (1995. a). ATP‐sensitive K+ channels mediate α2D‐adrenergic receptor contraction of arteriolar smooth muscle and reversal of contraction by hypoxia. Circ Res 76, 53–63. [DOI] [PubMed] [Google Scholar]
- Tateishi J & Faber JE (1995. b). Inhibition of arteriole alpha 2‐ but not alpha 1‐adrenoceptor constriction by acidosis and hypoxia in vitro. Am J Physiol 268, H2068–H2076. [DOI] [PubMed] [Google Scholar]
- Teixeira MC, Coelho RR, Leal‐Cardoso JH & Criddle DN (2000). Comparative effects of niflumic acid and nifedipine on 5‐hydroxytryptamine‐ and acetylcholine‐induced contraction of the rat trachea. Eur J Pharmacol 394, 117–122. [DOI] [PubMed] [Google Scholar]
- Thorborg P, Gustafsson U, Sjöberg F, Harrison DK & Lewis DH (1990). Effect of hyperoxemia and ritanserin on skeletal muscle microflow. J Appl Physiol (1985) 68, 1494–1500. [DOI] [PubMed] [Google Scholar]
- Tsai AL & Kulmacz RJ (2010). Prostaglandin H synthase: resolved and unresolved mechanistic issues. Arch Biochem Biophys 493, 103–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma RF, Lindbom L & Arfors KE (1977). The effect of tissue oxygen tension on reactive hyperemia in skeletal muscle. Bibl Anat, 425–428. [PubMed] [Google Scholar]
- Vucetic M, Jensen PK & Jansen EC (2004). Diameter variations of retinal blood vessels during and after treatment with hyperbaric oxygen. Br J Ophthalmol 88, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Schmidt JR, Roman RJ, Anjaiah S, Falck JR & Lombard JH (2009). Modulation of vascular O2 responses by cytochrome 450‐4A ω‐hydroxylase metabolites in Dahl salt‐sensitive rats. Microcirculation 16, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME (1999). Dilation of rat diaphragmatic arterioles by flow and hypoxia: roles of nitric oxide and prostaglandins. J Appl Physiol (1985) 86, 1644–1650. [DOI] [PubMed] [Google Scholar]
- Wei EP, Kontos HA & Beckman JS (1996). Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am J Physiol 271, H1262–H1266. [DOI] [PubMed] [Google Scholar]
- Welsh DG, Jackson WF & Segal SS (1998). Oxygen induces electromechanical coupling in arteriolar smooth muscle cells: a role for L‐type Ca2+ channels. Am J Physiol 274, H2018–H2024. [DOI] [PubMed] [Google Scholar]
- Welsh DG & Segal SS (1998). Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol 274, H178–H186. [DOI] [PubMed] [Google Scholar]
- Westcott EB, Goodwin EL, Segal SS & Jackson WF (2012). Function and expression of ryanodine receptors and inositol 1,4,5‐trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol 590, 1849–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott EB & Jackson WF (2011). Heterogeneous function of ryanodine receptors, but not IP3 receptors, in hamster cremaster muscle feed arteries and arterioles. Am J Physiol Heart Circ Physiol 300, H1616–H1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen WJ, Nair P, Buerk D & Thuning CA (1974). Tissue PO2 in normal and denervated cat skeletal muscle. Am J Physiol 227, 1221–1225. [DOI] [PubMed] [Google Scholar]
- Whorton AR, Simonds DB & Piantadosi CA (1997). Regulation of nitric oxide synthesis by oxygen in vascular endothelial cells. Am J Physiol 272, L1161–L1166. [DOI] [PubMed] [Google Scholar]
- Woods JW, Coffey MJ, Brock TG, Singer II & Peters‐Golden M (1995). 5‐Lipoxygenase is located in the euchromatin of the nucleus in resting human alveolar macrophages and translocates to the nuclear envelope upon cell activation. J Clin Invest 95, 2035–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J & Duling BR (1995). Electromechanical coupling and the conducted vasomotor response. Am J Physiol 269, H2022–H2030. [DOI] [PubMed] [Google Scholar]
- Yuan XJ (1997). Role of calcium‐activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol 272, L959–L968. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Park TS & Gidday JM (1998). Mechanisms of hyperoxia‐induced reductions in retinal blood flow in newborn pig. Exp Eye Res 67, 357–369. [DOI] [PubMed] [Google Scholar]
- Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR & Roman RJ (1996). 20‐HETE is an endogenous inhibitor of the large‐conductance Ca2+‐activated K+ channel in renal arterioles. Am J Physiol 270, R228–R237. [DOI] [PubMed] [Google Scholar]