

Abstract
Key points
The present study describes the cerebral oxidative and non‐oxidative metabolism in man during a prolonged apnoea (ranging from 3 min 36 s to 7 min 26 s) that generates extremely low levels of blood oxygen and high levels of carbon dioxide.
The cerebral oxidative metabolism, measured from the product of cerebral blood flow and the radial artery‐jugular venous oxygen content difference, was reduced by ∼29% at the termination of apnoea, although there was no change in the non‐oxidative metabolism.
A subset study with mild and severe hypercapnic breathing at the same level of hypoxia suggests that hypercapnia can partly explain the cerebral metabolic reduction near the apnoea breakpoint.
A hypercapnia‐induced oxygen‐conserving response may protect the brain against severe oxygen deprivation associated with prolonged apnoea.
Abstract
Prolonged apnoea in humans is reflected in progressive hypoxaemia and hypercapnia. In the present study, we explore the cerebral metabolic responses under extreme hypoxia and hypercapnia associated with prolonged apnoea. We hypothesized that the cerebral metabolic rate for oxygen (CMRO2) will be reduced near the termination of apnoea, attributed in part to the hypercapnia. Fourteen elite apnoea‐divers performed a maximal apnoea (range 3 min 36 s to 7 min 26 s) under dry laboratory conditions. In a subset study with the same divers, the impact of hypercapnia on cerebral metabolism was determined using varying levels of hypercapnic breathing, against the background of similar hypoxia. In both studies, the CMRO2 was calculated from the product of cerebral blood flow (ultrasound) and the radial artery‐internal jugular venous oxygen content difference. Non‐oxidative cerebral metabolism was calculated from the ratio of oxygen and carbohydrate (lactate and glucose) metabolism. The CMRO2 was reduced by ∼29% (P < 0.01, Cohen's d = 1.18) near the termination of apnoea compared to baseline, although non‐oxidative metabolism remained unaltered. In the subset study, in similar backgrounds of hypoxia (arterial O2 tension: ∼38.4 mmHg), severe hypercapnia (arterial CO2 tension: ∼58.7 mmHg), but not mild‐hypercapnia (arterial CO2 tension: ∼46.3 mmHg), depressed the CMRO2 (∼17%, P = 0.04, Cohen's d = 0.87). Similarly to the apnoea, there was no change in the non‐oxidative metabolism. These data indicate that hypercapnia can partly explain the reduction in CMRO2 near the apnoea breakpoint. This hypercapnic‐induced oxygen conservation may protect the brain against severe hypoxaemia associated with prolonged apnoea.
Key points
The present study describes the cerebral oxidative and non‐oxidative metabolism in man during a prolonged apnoea (ranging from 3 min 36 s to 7 min 26 s) that generates extremely low levels of blood oxygen and high levels of carbon dioxide.
The cerebral oxidative metabolism, measured from the product of cerebral blood flow and the radial artery‐jugular venous oxygen content difference, was reduced by ∼29% at the termination of apnoea, although there was no change in the non‐oxidative metabolism.
A subset study with mild and severe hypercapnic breathing at the same level of hypoxia suggests that hypercapnia can partly explain the cerebral metabolic reduction near the apnoea breakpoint.
A hypercapnia‐induced oxygen‐conserving response may protect the brain against severe oxygen deprivation associated with prolonged apnoea.
Abbreviations
- CO
cardiac output
- CDO2
cerebral delivery of oxygen
- CMRO2
cerebral metabolic rate of oxygen
- gCBF
global cerebral blood flow
- Glu
glucose
- Glu Ext
glucose extraction
- HR
heart rate
- ICA
internal carotid artery
- Lac
lactate
- Lac Ext
lactate extraction
- MAP
mean arterial pressure
- MCA
middle cerebral artery
- MCAv
middle cerebral artery blood velocity
- mild‐HH
mild‐hypercapnic hypoxia
- OCI
oxidative carbohydrate index
- OGI
oxidative glucose index
- O2 Ext
oxygen extraction fraction
oxygen saturation
partial pressure of carbon dioxide
partial pressure of oxygen
partial pressure of end‐tidal carbon dioxide
partial pressure of end‐tidal oxygen
- PCA
posterior cerebral artery
- PCAv
posterior cerebral artery blood veleocity
- QICA
blood flow of the right internal carotid artery
- QVA
blood flow of the left vertebral artery
- Severe‐HH
severe‐hypercapnic hypoxia
- SV
stroke volume
- v
venous
- VA
vertebral artery
Introduction
Although it accounts for only 2% of total body mass, the human brain utilizes a disproportionate 20% of the body's basal oxygen consumption. The relatively high cerebral metabolic rate of oxygen (CMRO2) is required to support a high rate of ATP production and neuronal activity (Brown and Ransom, 2007). A constant oxygen supply is therefore obligatory for the maintenance of normal brain function. With respect to cerebral energy demands for oxygen, the current world record apnoea duration in humans of 11 min 35 s is truly remarkable.
In terms of oxygen conservation, decreasing the cerebral oxidative metabolism and increasing non‐oxidative metabolism is of teleological benefit. Not surprisingly, a reduction in the CMRO2 during deep hypothermia is readily utilized in medicine (e.g. during bypass surgery; Fukui and Takanashi, 2016) and can partly explain extreme survival following prolonged anoxic (>15 min) cold‐water immersion (Young et al. 1980; Antretter et al. 1994). It appears, however, that moderate hypoxia alone does not provide the stimulus of oxygen conservation. For example, no appreciable changes in the CMRO2 have been reported during isocapnic or poikilocapnic hypoxic breathing ( of ∼40–55 mmHg) (Kety and Schmidt, 1948; Cohen et al. 1967; Bailey et al. 2009; Overgaard et al. 2012; Ainslie et al. 2014). Indeed, using recent magnetic imaging techniques, some studies report significant elevations in CMRO2 by 5–10% with acute poikilocapnic hypoxia (Xu et al. 2012; Vestergaard et al. 2015). Recent and available data further indicate no increases in cerebral non‐oxidative metabolism (estimated via the oxidative carbohydrate index; OCI) with isocapnic hypoxic breathing (Ainslie et al. 2014).
An increase in CMRO2 with acute hypoxia shown by some studies (Vestergaard et al. 2015), but not all (Ainslie et al. 2014), may relate to the background or pH. In one study (Ainslie et al. 2014), subjects were kept at eucapnia, whereas, in another study (Vestergaard et al. 2015), the hypoxia‐induced increase in ventilation reduced by an average of 10 mmHg (absolute 31 mmHg). A large body of both human and animal data indicates that changing may impact CMRO2, with hypocapnia increasing, and hypercapnia decreasing it; for a synopsis, see Yablonskiy (2011). Indeed, synaptic transmission (Dulla et al. 2005; Thesen et al. 2012) and phosphofructokinase activity (Folbergrova et al. 1975) are each dependent on pH. Because a prolonged breath‐hold yields both extreme levels of hypoxia and hypercapnia (Willie et al. 2014; Bain et al. 2015 a), it stands to reason that, in contrast to hypoxia alone, the CMRO2 may be reduced during apnoea. Additionally, in contrast to hypoxic breathing (Ainslie et al. 2014), a greater increase in sympathetic nerve activity during apnoea, despite the same chemoreflex stimuli (Steinback et al. 2010 a), may promote a shift towards non‐oxidative metabolic pathways. For example, infusion of high dose adrenaline (0.08 μg kg−1 min−1 for 15 min) in humans significantly increases the percentage of non‐oxidative cerebral metabolism (Seifert et al. 2009) and adrenaline appears to underlie the increased non‐oxidative metabolism during exercise (Larsen et al. 2008). A shift towards non‐oxidative cerebral metabolism, and a decrease in the CMRO2, may in turn help explain how some elite breath‐hold divers may hold their breath for over 10 min.
Despite obvious implications in neuropathology, describing the cerebral metabolic profile under severe apneic stress in humans has remained a methodological and ethical challenge. Elite breath‐hold divers provide a unique population to test the limits of hypoxaemia tolerance beyond what is possible in otherwise healthy humans. As such, the primary purpose of the present study was to examine the oxidative and non‐oxidative cerebral metabolism during a prolonged dry‐land apnoea in elite human breath‐hold competitors. In a subset study, we gathered mechanistic information by quantifying the impact of hypercapnia/pH on cerebral metabolism against the background of hypoxia. We examined the hypotheses that (1) compared to a resting baseline, CMRO2 will be reduced and indices of cerebral non‐oxidative metabolism will be increased during a prolonged apnoea and (2) the reductions in CMRO2 will partly be a function of a hypercapnic‐mechanism.
Methods
Participants
Fourteen competitive and elite breath‐hold divers (two females; age 29.5 ± 7.3 years; body mass index 23.5 ± 2.5 kg m–2) were recruited from the Croatian national apnoea team, and provided their written informed consent for participation. All participants were actively competing, and had been practicing competitive breath hold diving for between 1.5 and 14.0 years (mean 5.2 ± 3.7 years). The majority of subjects were competitive in the discipline of dynamic apnoea (maximal swimming distance underwater). Some were also involved in various depth disciplines (with regular exposure to high pressure). Seven of the subjects were world‐class free diving competitors, having been placed in the top ten within the last 3 years in international competition in at least one event. Three subjects had recently set new official world records. All subjects were normotensive and free from cardiovascular and respiratory disease. The ethical committees of the University of Split School of Medicine, the University of British Columbia and the University of South Wales approved the experimental procedures.
Experimental design
Experimentation was completed on a single day, following strict adherence to pre‐testing protocol, including abstinence from vigorous exercise and alcohol at least 48 h, as well as from caffeine at least 12 h, before arriving at the laboratory. We did not obtain information on previous caffeine use. All testing was performed at the University of Split, School of Medicine, Department of Integrative Physiology. Upon arrival, a medical history and standard anthropometric and pulmonary functioning metrics were assessed. After, a 20‐gauge arterial catheter (Arrow, Markham, Ontario, Canada) was placed in the right radial artery, and a central venous catheter (PediaSat Oximetry Catheter; Edwards Lifesciences, Irvine, CA, USA) was placed in the right internal jugular vein and directed cephalad to the jugular bulb. Using the identical technique and performed by the same anaesthesiologist, correct placement of the jugular bulb catheter has been confirmed by lateral skull X‐ray (Ainslie et al. 2014). Arterial and jugular cannulations were completed under ultrasound guidance with local anaesthesia (1% lidocaine) and under sterile conditions. Following cannulation, the catheters were attached to an in‐line waste‐less sampling system and a pressure transducer located at the height of the right atrium (VAMP System; Edwards Lifesciences) and a TruWave transducer (Edwards Lifesciences). Subjects were then instrumented with the remaining measurements (see measurements).
Apnoea
Baseline measurements were acquired following a minimum of 30 min of supine rest, and prior to preparatory apnoeas. After baseline measurements were acquired, each participant completed two preparatory (non‐experimental) apnoeas. The first preparatory apnoea was performed after a normal end‐expiration until seven involuntary breathing movements were attained. Two minutes later, the second preparatory apnoea was performed at total lung capacity until ten involuntary breathing movements were attained. These preparatory apnoeas were performed to generate the longest possible time for the experimental maximal apnoea, chosen by the national Croatian apnoea team coach (Ivan Drvis) based on a best‐suited standardized preparatory phase for all subjects. Following the preparatory phase, subjects rested for 6 min before commencing the maximal apnoea. Subjects were allowed to lung pack (glossopharyngeal insufflation) prior to the experimental apnoea, based on individual preference to attain the longest apnoea possible. Of note, the individual magnitude of glossopharyngeal insufflation will have profound effects on the initial haemodynamic changes at the apnoea onset (Batinic et al. 2011). The magnitude of glossopharyngeal insufflation was not measured. Data were collected throughout the maximal breath‐hold until breathing was resumed. Arterial and jugular venous blood draws were attained prior to the preparatory apnoeas (baseline), every 30 s throughout the maximal apnoea, and immediately upon termination (100% of apnoea).
Subset study: end‐tidal forcing
Following the maximal apnoea, participants’ rested supine for a minimum of 30 min before starting the hypercapnic‐hypoxic breathing subset trials. The mild‐hypercapnic hypoxic (mild‐HH) and severe‐hypercapnic hypoxia (severe‐HH) breathing was performed using a custom built end‐tidal (PET) forcing system. Subjects were equipped with a mouthpiece and nose clip, and were instructed to breath normally. Gases were sampled at the mouthpiece and analysed by a calibrated gas analyser (ML206; ADInstruments, Colorado Springs, CO, USA). Respiratory flows were measured by pneumotachography (HR 800L; Hans Rudolph, Shawnee, KS, USA). Custom written software (Labview, Austin, TX, USA) determined the breath‐by‐breath tidal volumes and end‐tidal partial pressures of oxygen and carbon dioxide ( and ). The end‐tidal forcing system prospectively delivered inspired gases to clamp and at desired input levels. Independently controlled solenoid valves delivered the desired volumes of O2, CO2, and N2 as determined by an error reduction algorithm incorporating , and inspiratory and expiratory tidal volume from the last breath. Levels of desired for both mild‐HH and severe‐HH were individualized to the average measured during the last 90 s of the maximal breath‐hold. Levels of during the mild‐HH trial were individualized to the achieved at ∼50% of the maximal apnoea. Levels of during the severe‐HH trial were targeted for +10 mmHg from the mild‐HH. Each condition (mild‐HH and severe‐HH) lasted 5 min, and the severe‐HH trial was performed immediately following mild‐HH, at the same time as continually maintaining hypoxia. Arterial and jugular venous blood draws were taken after 4 min 30 s in each stage.
Measurements
Blood gases, oximetry and metabolites
Measurements of arterial and jugular venous PO2, PCO2, O2 saturation (), glucose (Glu) and lactate (La) were analysed immediately following each draw using a commercially available cassette based analyser (ABL90 Flex; Radiometer, Copenhagen, Denmark). Each measure required <2 ml of blood. Arterial (radial) and jugular venous draws were taken at the same time.
Catecholamines
Blood samples (7 ml) were collected into tubes containing K‐EDTA and centrifuged at 600g for 10 min at 4°C. Plasma (2 ml sample volume) was transferred into cryovial tubes and immediately snap‐frozen under liquid nitrogen (N2, Cryopak CP100, Taylor‐Wharton, Theodore, AL, USA) and then stored at –80°C prior to analysis. Analysis was performed within 1 month of receipt of the sample. All chemicals including reagents and standards were of the highest available purity from Sigma‐Aldrich (Poole, UK). Plasma concentrations of adrenaline, and noradrenaline were measured by reverse‐phase ion pair high performance liquid chromatography (Gilson ASTED.XL; Anachem Ltd, Luton, UK) with electrochemical detection (ESA Coulochem II; ESA Analytical, East Grinstead, UK) (Bouloux et al. 1985). The coefficient of variation was 5% for noradrenaline at 4.8 nmol l−1 and 8% for adrenaline at 1.0 nmol l−1.
Cardiovascular
Heart rate (HR) was obtained from the R‐R intervals measured from a three‐lead ECG. Beat‐to‐beat arterial blood pressure was measured by finger photoplethysmography (Finometer PRO; Finapress Medical Systems, Amsterdam, Netherlands) normalized to manual cuff measurements of the brachial artery. Intra‐radial arterial pressure and jugular venous pressure were also recorded; however, as a result of the frequency of blood sampling, measurements were available for ∼5‐ to 10‐s bins around each blood draw. Online calculations of stroke volume were obtained using the three‐element nonlinear arterial model of the arterial blood pressure waveform (Finometer). Cardiac output was then derived from the product of stroke volume and HR.
Cerebrovascular
The cerebral blood velocity of the right middle cerebral artery (MCAv) and left posterior cerebral artery (PCAv) was measured using a 2‐MHz pulsed transcranial Doppler ultrasound system (Spencer Technologies, Seattle, WA, USA). A specialized headband fixation device (model M600 bilateral head frame; Spencer Technologies) was used to secure the probes in position. Signal quality was optimized using standardized search techniques that produce test–retest reliability of ∼3% and 2% for MCAv and PCAv, respectively. MCAv was insonated through the left temporal window, at a depth 1 cm distal to the MCA–anterior cerebral artery bifurcation. PCAv was insonated at the P1 segment through the right temporal window.
Volumetric blood flow of the right internal carotid artery (QICA) and left vertebral artery (QVA) was concomitantly measured using duplex vascular ultrasound (Terason 3000; Teratech, Burlington, MA, USA). The right ICA was insonated ∼2cm from the carotid bifurcation, whereas the left VA was insonated at the C5‐C6 or C5‐C4 space depending on individual anatomy. Care was taken to standardize the insonation location between measurements within subjects. The steering angle was fixed to 60° among all trials, and the sample volume was placed in the centre of the vessel adjusted to cover the entire vascular lumen. All files were collected in AVI format for offline analysis at 30 Hz using custom designed software (Woodman et al. 2001). Simultaneous measurements of luminal diameter and velocity over a minimum of 10 cardiac cycles were used to calculate flow.
Calculations
Under the assumption of symmetrical blood flow of contralateral ICA and VA arteries, global CBF (gCBF) was calculated as: gCBf (ml min–1) = (QICA × 2) + (QVA × 2).
As a result of the onset of involuntary breathing movements during the latter ∼60% of the breath‐hold (struggle phase), which encompasses large movements of the chest wall and movement of the sternocleidomastoid muscles, we were unable to attain reliable QICA and QVA values in all subjects for the entire breath‐hold. However, we were able to reliably track ICA and VA diameter up to the breath‐hold termination. As such, where simultaneous measurements of ICA or VA velocity with diameter were missing, gCBF was derived from incorporating changes in MCAv (indicative of ICA velocity) and PCAv (indicative of VA velocity) with correction for diameter. That is, the percentage change in MCAv or PCAv was estimated to equate with ICA or VA velocity changes (r 2 = 0.7, where simultaneous measurements are available). The inclusion of changes in diameter measurements ensured that gCBF was not underestimated using the metrics of ∆MCAv and ∆PCAv only (Coverdale et al. 2014).
Contents of arterial (CaO2) and venous (CvO2) oxygen were calculated as:
where 1.36 is the affinity for oxygen to haemoglobin for a given arterial saturation and 0.003 is the percentage of oxygen dissolved in the blood. Values are expressed as ml of O2 per 100 ml of blood (ml dl–1).
Cerebral delivery of oxygen (CDO2) was calculated as:
The cerebral metabolic rate of oxygen (CMRO2) was calculated as:
Cerebral oxygen, glucose and lactate extraction fraction were calculated from the arterial‐venous content difference divided by the arterial value, and then multiplied by 100.
The OCI, which provides an estimation of oxidative vs. non‐oxidative metabolism (Ainslie et al. 2014), was calculated from the equation below. The oxidative glucose index (OGI) was calculated the same way but with omission of the addition of the lactate arterial–venous difference. In short, a reduction of 6 reflects the presence of non‐oxidative metabolism given that the oxidization of glucose by oxygen requires one oxygen molecule per six carbon atoms found in glucose, and carboxylation of lactate to pyruvate acid yields only one molecule of pyruvate, compared to the two derived from the breakdown of glucose. This was then converted to a percent OCI by dividing by 6 and then multiplying by 100.
Statistical analysis
Data are the mean ± SD. Baseline measurements were acquired during quite rest prior to the preparatory apnoeas (∼15 min before the maximal apnoea) and 1 min before the end‐tidal forcing trials. Baseline measurements were averaged over 1 min around the baseline blood draw. Mean values for MAP (Finometer corrected with intra‐arterial) HR and gCBF during the apnoea were averaged over 20 s around the blood draws.
Statistical analysis for both apnoea and the end‐tidal forcing was performed using one‐way repeated measures ANOVA. When a main effect was observed, pre‐planned paired comparisons were performed to baseline only using two‐tailed Student t tests. A Bonferroni adjustment was applied to correct for multiple comparisons (six stages of the breath hold and three stages of end‐tidal forcing). When significant, Cohen's d was calculated for effect size of the primary outcome variable (CMRO2).
Results
Apnoea
Maximal apnoea times ranged from 3 min 36 s to 7 min 26 s with an average of 5 min 14 s.
Blood gases, oximetry and metabolites
Arterial and jugular venous blood gas, oximetry, pH and metabolite data are provided in Table 1. To illustrate the near normalization of arterial with venous blood at the end of the apnoea, arterial and jugular venous PO2 from baseline to 100% of the apnoea are shown in Fig. 1. As expected, there was a main effect for all blood gas variables (all P < 0.05). Significant post hoc comparisons to baseline are denoted in Table 1.
Table 1.
Arterial (radial) and cerebral venous (internal jugular bulb) measurements of the , , haematocrit (Hct), haemoglobin (tHb), Glu, Lac and pH at baseline and during 20% increments of the maximal apnoea
| (mmHg) | (%) | Hct (%) | tHb (mmol L–1) | Glu (mmol L–1) | Lac (mmol L–1) | pH | |
|---|---|---|---|---|---|---|---|
| Baseline | |||||||
| Arterial | 40.9 ± 3.3 | 97.8 ± 0.9 | 44.4 ± 3.1 | 14.5 ± 1.0 | 5.6 ± 0.8 | 0.8 ± 0.3 | 7.41 ± 0.02 |
| Venous | 51.8 ± 3.6 | 62.1 ± 5.1 | 44.1 ± 2.6 | 14.4 ± 0.9 | 4.9 ± 0.8 | 0.8 ± 0.3 | 7.36 ± 0.02 |
| 20% | |||||||
| Arterial | 32.4 ± 5.1* | 99.0 ± 0.4* | 44.3 ± 3.2 | 14.4 ± 1.0 | 5.3 ± 0.6 | 0.9 ± 0.2 | 7.48 ± 0.04* |
| Venous | 47.6 ± 3.5* | 50.5 ± 8.1* | 45.3 ± 3.4 | 14.8 ± 1.1 | 4.6 ± 0.6 | 0.9 ± 0.2 | 7.40 ± 0.02* |
| 40% | |||||||
| Arterial | 39.6 ± 4.3 | 96.9 ± 1.9 | 44.8 ± 4.2 | 14.6 ± 1.4 | 5.3 ± 0.6 | 0.9 ± 0.2 | 7.42 ± 0.02 |
| Venous | 50.0 ± 3.1 | 61.2 ± 6.5 | 45.5 ± 3.6 | 14.9 ± 1.2 | 4.8 ± 0.6 | 0.9 ± 0.3 | 7.38 ± 0.02 |
| 60% | |||||||
| Arterial | 46.1 ± 5.0* | 89.2 ± 5.1* | 45.4 ± 4.4 | 14.8 ± 1.5 | 5.3 ± 0.7 | 1.0 ± 0.3 | 7.38 ± 0.03* |
| Venous | 52.5 ± 3.4 | 63.7 ± 6.0 | 45.7 ± 3.8 | 14.9 ± 1.2 | 4.9 ± 0.6 | 1.0 ± 0.3 | 7.36 ± 0.02 |
| 80% | |||||||
| Arterial | 49.7 ± 4.7* | 80.6 ± 7.8* | 45.5 ± 3.7* | 14.8 ± 1.2* | 5.5 ± 0.6 | 1.0 ± 0.3 | 7.36 ± 0.02* |
| Venous | 54.9 ± 3.7 | 59.5 ± 6.0 | 45.7 ± 3.2* | 14.9 ± 1.0* | 5.1 ± 0.6 | 1.0 ± 0.3 | 7.34 ± 0.02* |
| 100% | |||||||
| Arterial | 53.4 ± 4.8* | 60.9 ± 14.0* | 45.8 ± 3.4* | 14.9 ± 1.1* | 5.4 ± 0.7 | 1.1 ± 0.3* | 7.34 ± 0.02* |
| Venous | 57.8 ± 4.7* | 45.6 ± 13.0* | 46.0 ± 3.7* | 15.0 ± 1.2* | 5.1 ± 0.6 | 1.2 ± 0.4* | 7.32 ± 0.02* |
*Significant difference from baseline.
Figure 1. Absolute arterial and internal jugular venous artial pressure of oxygen .
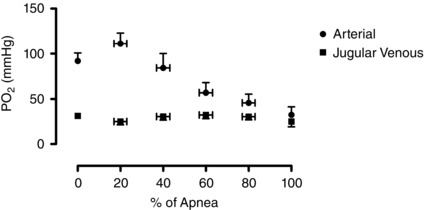
Mean ± SD of absolute arterial () and internal jugular venous () partial pressure of oxygen throughout the duration of the maximal apnoea.
Haemodynamics
Heart rate (HR), mean arterial pressure (MAP), stroke volume (SV), cardiac output (CO) and global cerebral blood flow (gCBF) are provided in Table 2. There was a main effect of for all variables (all P < 0.05). Despite a reduction in CO from baseline, the MAP was elevated throughout the majority of the apnoea (all P < 0.05), indicating an elevated total peripheral resistance. The reductions in indices of SV from 20–80% of the apnoea (all P < 0.05) are attributable to the glossopharyngeal insufflation prior to the start of the apnoea. As expected, gCBF was reduced at the onset of the apnoea, and then was progressively elevated by ∼70% at 100% of the apnoea. All significant post hoc comparisons to baseline are denoted in Table 2.
Table 2.
HR, SV, CO, MAP and gCBF at baseline and during 20% increments of the maximal apnoea
| HR (beats min–1) | SV (ml) | CO (mmol L–1) | MAP (mmHg) | gCBF (ml min–1) | |
|---|---|---|---|---|---|
| Baseline | 64±11 | 108 ± 21 | 6.78 ± 1.65 | 88±8 | 610 ± 153 |
| 20% | 80±17* | 58 ± 16* | 4.52 ± 1.47* | 92±10 | 469 ± 140* |
| 40% | 77±16* | 62 ± 13* | 4.54 ± 1.03* | 99±11* | 613 ± 196 |
| 60% | 66±16 | 81 ± 15* | 5.11 ± 1.46* | 115±21* | 828 ± 267* |
| 80% | 59±14 | 95 ± 18 | 5.41 ± 1.52* | 131±21* | 991 ± 325* |
| 100% | 56±15 | 96 ± 33 | 5.17 ± 1.91* | 139±23* | 1041 ± 318* |
*Significant difference from baseline.
Cerebral metabolic dynamics and oxygen delivery
Cerebral delivery of oxygen (CDO2), oxygen extraction (O2 Ext), lactate extraction (Lac Ext), glucose extraction (Glu Ext), OCI and CMRO2 are provided in Table 3. Individual CMRO2 at baseline and at 100% of the apnoea are shown in Fig. 2. There was a main effect of CDO2 (P < 0.01), O2 Ext (P < 0.01), Glu Ext (P < 0.01), OCI (P = 0.02) and CMRO2 (P < 0.01). Compared to baseline, the CMRO2 was reduced by ∼29% at 100% of the apnoea (P < 0.01, d = 1.18) but similar at all other time points (all P < 0.05). The O2 Ext was increased at 20% of the apnoea, and decreased at the latter half of the apnoea (all P < 0.05). The CDO2 fell below baseline values at 20% of the apnoea (P < 0.01), above baseline at 60% and 80% of the apnoea (both P < 0.01) and was similar to baseline at 100% of the apnoea. All significant post hoc comparisons to baseline are denoted in Table 3. Although there was a main effect of OCI, no post hoc comparisons were different. Values for OGI were identical to the OCI. There was no correlation between the absolute or change in or with the change in CMRO2 from baseline to 100% of the apnoea.
Table 3.
CDO2, O2 Ext, Lac Ext, Glu Ext, OCI and CMRO2 at baseline and during 20% increments of the maximal apnoea
| CDO2 (ml min–1) | O2 Ext (%) | Lac Ext (%) | Glu Ext (%) | OCI (%) | CMRO2 (ml min–1) | |
|---|---|---|---|---|---|---|
| Baseline | 122 ± 30 | 37 ± 6 | −2 ± 11 | 12 ± 3 | 83 ± 13 | 45 ± 11 |
| 20% | 92 ± 24* | 47 ± 10* | −3 ± 17 | 12 ± 6 | 127 ± 75 | 43 ± 10 |
| 40% | 120 ± 34 | 36 ± 7 | −3 ± 20 | 9 ± 6 | 125 ± 61 | 41 ± 10 |
| 60% | 152 ± 48* | 28 ± 7* | 0 ± 15 | 7 ± 4* | 139 ± 94 | 41 ± 12 |
| 80% | 163 ± 50* | 26 ± 4* | −3 ± 17 | 7 ± 2* | 92 ± 37 | 41 ± 11 |
| 100% | 128 ± 36 | 25 ± 9* | −2 ± 13 | 5 ± 3* | 92 ± 57 | 32 ± 12* |
*Significant difference from baseline.
Figure 2. Individual data of the absolute CMRO2 before the apnoea (baseline) and at apnoea termination (100% apnoea) .
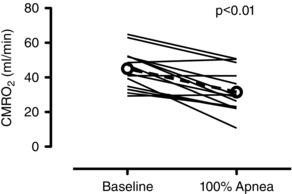
Dashed line with open circles denotes mean data. Compared to baseline, the CMRO2 was reduced by ∼29% at 100% of the apnoea (P < 0.01, d = 1.18).
Catecholamines
The radial arterial plasma adrenaline and noradrenaline increased by ∼380% (670 ± 380 to 2012 ± 1020 pmol l−1) and ∼483% (1893 ± 673 to 8260 ± 4375 pmol l−1) from baseline to 100% of the apnoea, respectively (both P < 0.01). Similarly, the jugular venous plasma adrenaline and noradrenaline increased by ∼258% (584 ± 258 to 1762 ± 801 pmol l−1) and ∼385% (1860 ± 577 to 8559 ± 3707 pmol l−1) (both P < 0.01). The arterial–venous adrenaline and noradrenaline differences were not different from baseline to 100% of the apnoea (both P < 0.05).
Subset study: variable hypercapnic breathing with similar hypoxia
Three participants were unable to complete the end‐tidal forcing trial as a result of ventilatory volumes during the severe‐hypercapnic hypoxia (severe‐HH) that exceeded the capacity of the end‐tidal forcing system (i.e. >100 L min−1). The sample size for the hypoxic breathing trial was in turn reduced from 14 to 11 subjects.
Blood gases, oximetry and metabolites
Arterial and jugular venous blood gas, oximetry, pH and metabolite data are provided in Table 4. By design, there was an increase in and a reduction in , SaO2 and pH in both mild‐HH and severe‐HH from baseline. There was no main effect of glucose or lactate (both P < 0.05).
Table 4.
Arterial (radial) and cerebral venous (internal jugular bulb) measurements of and , , haematocrit (Hct), haemoglobin (tHb), Glu, Lac and pH at baseline and during similar hypoxic breathing with mild‐HH and severe‐HH
| (mmHg) | (mmHg) | (%) | Hct (%) | tHb (mmol L–1) | Glu (mmol L–1) | Lac (mmol L–1) | pH | |
|---|---|---|---|---|---|---|---|---|
| Baseline | ||||||||
| Arterial | 91.6 ± 13.7 | 38.6 ± 5.1 | 98 ± 1 | 43.9 ± 3.7 | 14.3 ± 1.2 | 5.4 ± 0.5 | 0.9 ± 0.3 | 7.43 ± 0.05 |
| Venous | 29.5 ± 4.1 | 50.1 ± 4.2 | 60 ± 5 | 44.6 ± 4.1 | 14.6 ± 1.3 | 4.9 ± 0.6 | 1.0 ± 0.3 | 7.37 ± 0.04 |
| Mild‐HH | ||||||||
| Arterial | 38.9 ± 7.8* | 46.3 ± 3.2* | 75 ± 8* | 44.7 ± 3.8* | 14.6 ± 1.2* | 5.3 ± 0.5 | 0.9 ± 0.3 | 7.38 ± 0.03* |
| Venous | 28.0 ± 3.4 | 52.6 ± 2.6* | 54 ± 7* | 44.6 ± 3.4 | 14.6 ± 1.1 | 5.0 ± 0.5 | 0.9 ± 0.3 | 7.35 ± 0.02* |
| Severe‐HH | ||||||||
| Arterial | 38.0 ± 8.9* | 58.7 ± 4.3* | 68 ± 12* | 45.3 ± 3.4* | 14.8 ± 1.1* | 5.3 ± 0.4 | 0.8 ± 0.3 | 7.30 ± 0.02* |
| Venous | 30.8 ± 5.5 | 62.3 ± 4.1* | 55 ± 11* | 45.2 ± 3.6 | 14.8 ± 1.2 | 5.2 ± 0.4 | 0.9 ± 0.3 | 7.29 ± 0.02* |
*Significant difference from baseline.
Haemodynamics
Absolute HR, MAP, SV, CO and gCBF are provided in Table 5. Except for SV, all measurements in both conditions were elevated from baseline (all P < 0.05).
Table 5.
HR, SV, CO, MAP and gCBF during baseline and hypoxic breathing with mild‐HH and severe‐HH
| HR (mmHg) | SV (ml) | CO (L min–1) | MAP (mmHg) | gCBF (ml min–1) | |
|---|---|---|---|---|---|
| Baseline | 69 ± 18 | 102 ± 18 | 7 ± 1 | 89 ± 6 | 593 ± 151 |
| Mild‐HH | 88 ± 14* | 104 ± 17 | 9 ± 2* | 102 ± 11* | 983 ± 204* |
| Severe‐HH | 94 ± 16* | 107 ± 15 | 10 ± 3* | 113 ± 18* | 1340 ± 343* |
*Significant difference from baseline.
Cerebral metabolism and oxygen delivery
Absolute CDO2, O2 Ext, Glu Ext, Lac Ext, CMRO2 and OCI are provided in Table 6. Individual CMRO2 are shown in Fig. 3. There was a main effect of CMRO2 (P < 0.01). Compared to baseline, post hoc comparisons revealed no change in CMRO2 during mild‐HH (P > 0.05), although there was an ∼17% reduction in the severe‐HH condition (P = 0.04, d = 0.87). The OCI was unchanged from baseline in both conditions. Values for OGI were identical to the OCI. As expected in both mild‐HH and severe‐HH, the O2 Ext was reduced from baseline, and the CDO2 was elevated (all P < 0.05).
Table 6.
QICA, QVA, flow velocity in the MCA and PCA, total CDO2 and total CMRO2 during baseline and hypoxic breathing with mild‐HH and severe‐HH
| CDO2 (ml min–1) | O2 Ext (%) | Lac Ext (%) | Glu Ext (%) | OCI (%) | CMRO2 (ml min–1) | |
|---|---|---|---|---|---|---|
| Baseline | 117 ± 32 | 38 ± 7 | –5 ± 6 | 10 ± 5 | 88 ± 16 | 44 ± 13 |
| Mild‐HH | 149 ± 32* | 28 ± 5* | 2 ± 8 | 7 ± 3 | 80 ± 18 | 41 ± 11 |
| Severe‐HH | 186 ± 53* | 19 ± 4* | –5 ± 15 | 3 ± 3* | 80 ± 26 | 34 ± 11* |
*Significant difference from baseline.
Figure 3. Individual data of the absolute CMRO2 at baseline, during mild‐HH and severe‐HH .
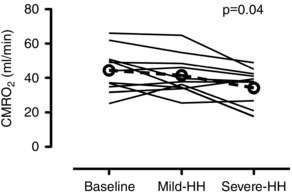
Hypoxia was on average a partial pressure of arterial oxygen of ∼38.5 mmHg for both mild‐HH and severe‐HH. Hypercapnia was on average set at an arterial partial pressure of carbon dioxide of ∼46.3 mmHg and ∼58.7 mmHg in the mild‐HH and severe‐HH conditions respectively. Dashed line with open circles denotes mean data. There was a ∼17% reduction in the severe‐HH condition (P = 0.04, d = 0.87) compared to baseline.
Discussion
The present study reports the first known measurements of cerebral metabolism in humans during a prolonged dry apnoea lasting on average greater than 5 min. A marked reduction in CMRO2 was observed immediately prior to the termination of the apnoea (∼29%, d = 1.18) compared to baseline measurements. By contrast, there was no change at any time point in the indirect estimation of non‐oxidative metabolism, despite an ∼380% increase in arterial adrenaline concentrations from baseline to the end of apnoea. With manipulation of end‐tidal gases, mild hypercapnic hypoxia had little influence on CMRO2; however, the same level of hypoxia but with severe hypercapnia reduced the CMRO2 by ∼17% (d = 0.87). We interpret these findings to indicate that the increased levels of hypercapnia and reduced pH partly explain the reduction in CMRO2 near the termination of apnoea.
Impact of hypercapnia on the CMRO2
The notion of a reduced CMRO2 resulting from hypercapnia is not novel but has remained debatable (Yablonskiy, 2011). With obvious implications in general medicine and anaesthesia (Waaben et al. 1989), the impact of on CMRO2 deserves attention. Unfortunately, the results obtained in human studies with similar MRI‐based experimental designs and in the absence of anaesthesia have remained inconsistent. For example, Jain et al. (2011) report no change in CMRO2 with an ∼8 mmHg increase in end‐tidal CO2; Chen et al. (2010) report an ∼7% reduction in CMRO2 with an ∼9 mmHg increase in end‐tidal CO2; and Xu et al. (2011) report an ∼13% reduction in CMRO2 with an ∼7 mmHg increase in end‐tidal CO2. Related to this, an increase in CMRO2 has been shown during high altitude (Smith et al. 2013), which is suspected to be a consequence of the hyperventilatory‐induced hypocapnia and therefore brain alkalosis. This finding is corroborated by the finding that an increase in CMRO2 following 6 h of normobaric hypoxia is removed with acetazolamide (Wang et al. 2015).
Using arterial and cerebral venous sampling, the present study provides evidence for reductions in CMRO2 with hypercapnia, at least in the presence of hypoxia. The pH‐dependent activity of phosphofructokinase (the enzyme responsible for the phosphorylation of fructose 6‐phosphate in glycolysis) provides mechanistic support for reductions in CMRO2 with hypercapnia. Indeed, an accumulation of glucose 6‐phosphate and fructose 6‐phosphate is shown in rats exposed to acute hypercapnia, which cannot be explained by glycogen breakdown (Folbergrova et al. 1975). Additionally, hypercapnia has consistently been shown to depress cortical activity, which is suspected to result from acidotic‐induced increases in extracellular adenosine (Dulla et al. 2005; Zappe et al. 2008; Thesen et al. 2012). Ultimately, the bulk of literature now collectively supports a modulation of CMRO2. The end‐tidal forcing data in the present study may indicate that this modulation is dose‐dependent, whereby a threshold change in is required.
Non‐oxidative metabolism and adrenaline
During apnoea, cessation of ventilation, chemosensory stimuli and baroreflex inhibition significantly increase muscle sympathetic nerve activity (burst frequency, magnitude and recruitment) (Heusser et al. 2010; Steinback et al. 2010 b) and therefore catecholamine release. Ascribed to the known effect of catecholamines on muscle metabolism (Tank and Lee Wong, 2015), an increase in whole‐body anaerobic metabolism is often held integral to the mammalian dive reflex (Blix and Folkow, 2011). Whether the cerebral tissue significantly partakes in the non‐oxidative metabolism is unclear, although it is generally accepted that, similar to in the muscle, adrenaline (but not noradrenaline) increases the cerebral non‐oxidative metabolism (Seifert et al. 2009). The ∼380% increase in arterial adrenaline from baseline to the termination of apnoea therefore offers potential for an increased cerebral non‐oxidative metabolism (Seifert et al. 2009). However, there was no reduction in the indirect estimation of non‐oxidative carbohydrate use (OCI) at any time point during the apnoea compared to baseline. Indeed, from 20% to 60% of the apnoea, non‐oxidative metabolism trended above 100%. An oxidative carbohydrate index above 100% may indicate the presence of carbon oxidation from sources other than arterial glucose or lactate. Based on animal studies, these data may in turn indicate an allosteric glycogen mobilization at the astrocytes (Saez et al. 2014).
Under the same metabolic paradigm by which adrenaline increases non‐oxidative metabolism, adrenaline should also increase the oxidative metabolism (Tank and Lee Wong, 2015). For example, i.v. adrenaline infusion (∼ 8 μg ml–1 min–1), with concurrent increases in MAP to ensure amine passage across the blood–brain barrier, significantly increases the CMRO2 in humans (King et al. 1952). However, when the adrenaline concentrations result in glycogenolytic and glycolytic flux being increased and impacting the CMRO2, circulating blood glucose would also be elevated (Seifert et al. 2009; Tank and Lee Wong, 2015). Therefore, the slightly reduced arterial glucose, reduced CMRO2 and unchanged oxidative carbohydrate index collectively indicate that the ∼380% increase in adrenaline was insufficient to elicit noticeable metabolic changes. This is perhaps not surprising given that, when non‐oxidative metabolism does increase, plasma adrenaline may be increased >10‐fold (e.g. during exhaustive exercise) (Pott et al. 1996; Tank and Lee Wong, 2015). Of note, anaerobic‐tolerant animals adapt to hypoxia by slowing the oxidative metabolism, rather than increasing the non‐oxidative metabolism (Hochachka et al. 1996). Our findings reveal that a similar metabolic outcome is initiated in humans during prolonged apnoea.
Other potential modulating factors of the cerebral metabolic response
Although we emphasize the impact of on CMRO2, other factors, including oxygen‐conserving reflexes attending the dive reflex, cannot be dismissed. Indeed, it is conceivable that the CMRO2 may be reduced to a greater extent had the apnoea been performed in water, with activation of the trigeminal nerve and therefore a stronger oxygen‐conserving/dive reflex (Schaller et al. 2009; Lemaitre et al. 2015). However, it appears that the activation of the trigeminal nerve has a greater responsibility for blood redistribution (from the periphery to the brain) and bradycardia, without directly influencing the CMRO2 (Reis et al. 1997; Schaller et al. 2009; Lemaitre et al. 2015). Reductions in CMRO2 associated with oxygen‐conserving reflexes may relate more to mechanisms observed in rapid ischaemic preconditioning (Gidday, 2006; Schaller et al. 2009). In such a case, neural inhibitory factors (e.g. adenosine and ATP sensitive potassium channels) are probably responsible, as reported in the anoxic tolerant turtle (Perez‐Pinzon et al. 1993). It is difficult to speculate on these potential oxygen‐conserving mechanisms in the present study; however, given that the average level of acidosis was greater in the severe‐hypercapnic hypoxia trial than it was at the termination of apnoea (arterial pH: 7.30 vs. 7.34, respectively), oxygen‐conserving reflexes independent of acidosis probably also contributed to the reduction in CMRO2 during apnoea. It should also be emphasized that the metabolic changes associated with described in the present study are against the background of hypoxia. Nevertheless, although acidosis decreased the oxygen saturation for the same during severe‐hypercapnic hypoxia compared to mild‐hypercapnic hypoxia (via the Bohr effect; Table 5), this probably did not influence the CMRO2. Indeed, using the same technique, we have demonstrated that similar eucapnic oxygen saturations (∼70%) of 15 min in duration do not alter the CMRO2 (Ainslie et al. 2014). If anything, the further oxygen desaturation would have increased the CMRO2, rather than decrease it (Xu et al. 2012; Vestergaard et al. 2015). Finally, the gCBF was higher in the severe‐HH condition compared to the mild‐HH condition. Therefore, it may be suggested that reduced temperature (via increased tissue to blood heat transfer; Bain et al. 2015 b) or a potential oxygen diffusion limitation can partly explain the reduction in CMRO2. A diffusion limitation, however, is not probable given that cerebral O2 extraction is well maintained even during exercise under both normoxic and hypoxic conditions (Smith et al. 2014). A diffusion limitation is also not probable based on the mathematical cerebral blood flow/metabolism relationship described by Gjedde (2005).
Surprisingly, at the termination of apnoea, neither the , nor the was independently correlated with the reduction in CMRO2. This may suggest individual variability in the CMRO2 response to extreme blood gas changes, an interacting facet of hypoxia and hypercapnia, or the impact of other poorly defined brain oxygen‐conserving reflexes in humans (Schaller et al. 2009).
Conclusions
In summary, the present study reports a clear reduction in CMRO2 immediately prior to the termination of a prolonged apnoea, which is attributable in part to hypercapnia. By contrast, there appears to be no change in non‐oxidative metabolism, despite a three‐ to four‐fold increase in arterial adrenaline concentrations. To our knowledge, this is the first study to examine the cerebral metabolism in healthy humans during prolonged apnoea, yielding some of the most extreme levels of hypoxia and hypercapnia reported to date. It is suggested that hypercapnia promotes brain oxygen‐conservation, and therefore provides a tenable protective mechanism against severe hypoxia relating to apnoea.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
ARB, PNA, ZD, ID and DBM conceived and designed the research. All authors performed the experiments. ARB analysed the data. All authors interpreted the results of the experiments. ARB prepared the artwork. ARB drafted the manuscript. All authors edited and revised the manuscript and approved the final version of the manuscript submitted for publication. All authors agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was funded through a Canadian Research Chair and NSERC Discovery grant held by Professor P. N. Ainslie. Drs Z. Dujic, o. F. Barak and A. R. Ainslie were also funded through the Croatian Science Foundation (IP‐2014‐09‐1937). Mr A. R. Bain was funded through a postgraduate NSERC scholarship.
Acknowledgements
We thank Dr Hitesh Gokani (Epsom and St Helier University Hospitals NHS Trust, UK) for his technical input. We would like to especially acknowledge the apnoea divers from the Croatia National Apnoea team for their participation.
References
- Ainslie PN, Shaw AD, Smith KJ, Willie CK, Ikeda K, Graham J & Macleod DB (2014). Stability of cerebral metabolism and substrate availability in humans during hypoxia and hyperoxia. Clin Sci (Lond) 126, 661–670. [DOI] [PubMed] [Google Scholar]
- Antretter H, Dapunt OE & Mueller LC (1994). Survival after prolonged hypothermia. N Engl J Med 330, 219. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Taudorf S, Berg RM, Lundby C, McEneny J, Young IS, Evans KA, James PE, Shore A, Hullin DA, McCord JM, Pedersen BK & Moller K (2009). Increased cerebral output of free radicals during hypoxia: implications for acute mountain sickness? Am J Physiol Regul Integr Comp Physiol 297, R1283–R1292. [DOI] [PubMed] [Google Scholar]
- Bain AR, Dujic Z, Hoiland RL, Barak OF, Madden D, Drvis I, Stembridge M, MacLeod DB, MacLeod DM & Ainslie PN (2015. a). Peripheral chemoreflex inhibition with low‐dose dopamine: new insight into mechanisms of extreme apnea. Am J Physiol Regul Integr Comp Physiol 309, R1162–R1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain AR, Nybo L & Ainslie PN (2015. b). Cerebral vascular control and metabolism in heat stress. Compr Physiol 5, 1–36. [DOI] [PubMed] [Google Scholar]
- Batinic T, Utz W, Breskovic T, Jordan J, Schulz‐Menger J, Jankovic S, Dujic Z & Tank J (2011). Cardiac magnetic resonance imaging during pulmonary hyperinflation in apnea divers. Med Sci Sports Exerc 43, 2095–2101. [DOI] [PubMed] [Google Scholar]
- Blix AS & Folkow B (2011). Cardiovascular adjustments to diving in mammals and birds. Compr Physiol, 917–945. DOI: 10.1002/cphy.cp020325. [Google Scholar]
- Bouloux P, Perrett D & Besser GM (1985). Methodological considerations in the determination of plasma catecholamines by high‐performance liquid chromatography with electrochemical detection. Ann Clin Biochem 22, 194–203. [DOI] [PubMed] [Google Scholar]
- Brown AM & Ransom BR (2007). Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Chen JJ & Pike GB (2010). Global cerebral oxidative metabolism during hypercapnia and hypocapnia in humans: implications for BOLD fMRI. J Cereb Blood Flow Metab 30, 1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PJ, Alexander SC, Smith TC, Reivich M & Wollman H (1967). Effects of hypoxia and normocarbia on cerebral blood flow and metabolism in conscious man. J Appl Physiol 23, 183–189. [DOI] [PubMed] [Google Scholar]
- Coverdale NS, Gati JS, Opalevych O, Perrotta A & Shoemaker JK (2014). Cerebral blood flow velocity underestimates cerebral blood flow during modest hypercapnia and hypocapnia. J Appl Physiol (1985) 117, 1090–1096. [DOI] [PubMed] [Google Scholar]
- Dulla CG, Dobelis P, Pearson T, Frenguelli BG, Staley KJ & Masino SA (2005). Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron 48, 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folbergrova J, Norberg K, Quistorff B & Siesjo BK (1975). Carbohydrate and amino acid metabolism in rat cerebral cortex in moderate and extreme hypercapnia. J Neurochem 25, 457–462. [DOI] [PubMed] [Google Scholar]
- Fukui T & Takanashi S (2016). Moderate to deep hypothermia in patients undergoing thoracoabdominal aortic repair. Ann Vasc Surg 31, 39–45. [DOI] [PubMed] [Google Scholar]
- Gidday JM (2006). Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 7, 437–448. [DOI] [PubMed] [Google Scholar]
- Gjedde A (2005). The pathways of oxygen in brain. I. Delivery and metabolism of oxygen. Adv Exp Med Biol 566, 269–275. [DOI] [PubMed] [Google Scholar]
- Heusser K, Dzamonja G, Breskovic T, Steinback CD, Diedrich A, Tank J, Jordan J & Dujic Z (2010). Sympathetic and cardiovascular responses to glossopharyngeal insufflation in trained apnea divers. J Appl Physiol (1985) 109, 1728–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ & Land SC (1996). Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 93, 9493–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V, Langham MC, Floyd TF, Jain G, Magland JF & Wehrli FW (2011). Rapid magnetic resonance measurement of global cerebral metabolic rate of oxygen consumption in humans during rest and hypercapnia. J Cereb Blood Flow Metab 31, 1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kety SS & Schmidt CF (1948). The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest 27, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BD, Sokoloff L & Wechsler RL (1952). The effects of l‐epinephrine and l‐norepinephrine upon cerebral circulation and metabolism in man. J Clin Invest 31, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen TS, Rasmussen P, Overgaard M, Secher NH & Nielsen HB (2008). Non‐selective beta‐adrenergic blockade prevents reduction of the cerebral metabolic ratio during exhaustive exercise in humans. J Physiol 586, 2807–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre F, Chowdhury T & Schaller B (2015). The trigeminocardiac reflex – a comparison with the diving reflex in humans. Arch Med Sci 11, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard M, Rasmussen P, Bohm AM, Seifert T, Brassard P, Zaar M, Homann P, Evans KA, Nielsen HB & Secher NH (2012). Hypoxia and exercise provoke both lactate release and lactate oxidation by the human brain. FASEB J 26, 3012–3020. [DOI] [PubMed] [Google Scholar]
- Perez‐Pinzon MA, Lutz PL, Sick TJ & Rosenthal M (1993). Adenosine, a ‘retaliatory’ metabolite, promotes anoxia tolerance in turtle brain. J Cereb Blood Flow Metab 13, 728–732. [DOI] [PubMed] [Google Scholar]
- Pott F, Jensen K, Hansen H, Christensen NJ, Lassen NA & Secher NH (1996). Middle cerebral artery blood velocity and plasma catecholamines during exercise. Acta Physiol Scand 158, 349–356. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Golanov EV, Galea E & Feinstein DL (1997). Central neurogenic neuroprotection: central neural systems that protect the brain from hypoxia and ischemia. Ann NY Acad Sci 835, 168–186. [DOI] [PubMed] [Google Scholar]
- Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martinez‐Pons C, Milan M & Guinovart JJ (2014). Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab 34, 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller B, Cornelius JF, Sandu N, Ottaviani G & Perez‐Pinzon MA (2009). Oxygen‐conserving reflexes of the brain: the current molecular knowledge. J Cell Mol Med 13, 644–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert TS, Brassard P, Jorgensen TB, Hamada AJ, Rasmussen P, Quistorff B, Secher NH & Nielsen HB (2009). Cerebral non‐oxidative carbohydrate consumption in humans driven by adrenaline. J Physiol 587, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, MacLeod D, Willie CK, Lewis NC, Hoiland RL, Ikeda K, Tymko MM, Donnelly J, Day TA, MacLeod N, Lucas SJ & Ainslie PN (2014). Influence of high altitude on cerebral blood flow and fuel utilization during exercise and recovery. J Physiol 592, 5507–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZM, Krizay E, Guo J, Shin DD, Scadeng M & Dubowitz DJ (2013). Sustained high‐altitude hypoxia increases cerebral oxygen metabolism. J Appl Physiol (1985) 114, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinback CD, Breskovic T, Frances M, Dujic Z & Shoemaker JK (2010. a). Ventilatory restraint of sympathetic activity during chemoreflex stress. Am J Physiol Regul Integr Comp Physiol 299, R1407–R1414. [DOI] [PubMed] [Google Scholar]
- Steinback CD, Salmanpour A, Breskovic T, Dujic Z & Shoemaker JK (2010. b). Sympathetic neural activation: an ordered affair. J Physiol 588, 4825–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank AW & Lee Wong D (2015). Peripheral and central effects of circulating catecholamines. Compr Physiol 5, 1–15. [DOI] [PubMed] [Google Scholar]
- Thesen T, Leontiev O, Song T, Dehghani N, Hagler DJ, Jr ., Huang M, Buxton R & Halgren E (2012). Depression of cortical activity in humans by mild hypercapnia. Hum Brain Mapp 33, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard MB, Lindberg U, Aachmann‐Andersen NJ, Lisbjerg K, Christensen SJ, Law I, Rasmussen P, Olsen NV & Larsson HB (2015). Acute hypoxia increases the cerebral metabolic rate – a magnetic resonance imaging study. J Cereb Blood Flow Metab 36, 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaben J, Husum B, Hansen AJ & Gjedde A (1989). Hypocapnia prevents the decrease in regional cerebral metabolism during isoflurane‐induced hypotension. J Neurosurg Anesthesiol 1, 29–34. [DOI] [PubMed] [Google Scholar]
- Wang K, Smith ZM, Buxton RB, Swenson ER & Dubowitz DJ (2015). Acetazolamide during acute hypoxia improves tissue oxygenation in the human brain. J Appl Physiol (1985) 119, 1494–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie CK, Ainslie PN, Drvis I, MacLeod DB, Bain AR, Madden D, Maslov PZ & Dujic Z (2014). Regulation of brain blood flow and oxygen delivery in elite breath‐hold divers. J Cereb Blood Flow Metab 35, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman RJ, Playford DA, Watts GF, Cheetham C, Reed C, Taylor RR, Puddey IB, Beilin LJ, Burke V, Mori TA & Green D (2001). Improved analysis of brachial artery ultrasound using a novel edge‐detection software system. J Appl Physiol (1985) 91, 929–937. [DOI] [PubMed] [Google Scholar]
- Xu F, Liu P, Pascual JM, Xiao G & Lu H (2012). Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation, and oxidative metabolism. J Cereb Blood Flow Metab 32, 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Uh J, Brier MR, Hart J, Jr. , Yezhuvath US, Gu H, Yang Y & Lu H (2011). The influence of carbon dioxide on brain activity and metabolism in conscious humans. J Cereb Blood Flow Metab 31, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonskiy DA (2011). Cerebral metabolic rate in hypercapnia: controversy continues. J Cereb Blood Flow Metab 31, 1502–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RS, Zalneraitis EL & Dooling EC (1980). Neurological outcome in cold water drowning. JAMA 244, 1233–1235. [PubMed] [Google Scholar]
- Zappe AC, Uludag K, Oeltermann A, Ugurbil K & Logothetis NK (2008). The influence of moderate hypercapnia on neural activity in the anesthetized nonhuman primate. Cereb Cortex 18, 2666–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]