

Abstract
Key points
The ventromedial hypothalamus (VMH) and the central melanocortin system both play vital roles in regulating energy balance by modulating energy intake and utilization.
Recent evidence suggests that activation of the VMH alters skeletal muscle metabolism.
We show that intra‐VMH melanocortin receptor activation increases energy expenditure and physical activity, switches fuel utilization to fats, and lowers work efficiency such that excess calories are dissipated by skeletal muscle as heat.
We also show that intra‐VMH melanocortin receptor activation increases sympathetic nervous system outflow to skeletal muscle.
Intra‐VMH melanocortin receptor activation also induced significant changes in the expression of mediators of energy expenditure in muscle.
These results support the role of melanocortin receptors in the VMH in the modulation of skeletal muscle metabolism.
Abstract
The ventromedial hypothalamus (VMH) and the brain melanocortin system both play vital roles in increasing energy expenditure (EE) and physical activity, decreasing appetite and modulating sympathetic nervous system (SNS) outflow. Because of recent evidence showing that VMH activation modulates skeletal muscle metabolism, we propose the existence of an axis between the VMH and skeletal muscle, modulated by brain melanocortins, modelled on the brain control of brown adipose tissue. Activation of melanocortin receptors in the VMH of rats using a non‐specific agonist melanotan II (MTII), compared to vehicle, increased oxygen consumption and EE and decreased the respiratory exchange ratio. Intra‐VMH MTII enhanced activity‐related EE even when activity levels were held constant. MTII treatment increased gastrocnemius muscle heat dissipation during controlled activity, as well as in the home cage. Compared to vehicle‐treated rats, rats with intra‐VMH melanocortin receptor activation had higher skeletal muscle norepinephrine turnover, indicating an increased SNS drive to muscle. Lastly, intra‐VMH MTII induced mRNA expression of muscle energetic mediators, whereas short‐term changes at the protein level were primarily limited to phosphorylation events. These results support the hypothesis that melanocortin peptides act in the VMH to increase EE by lowering the economy of activity via the enhanced expression of mediators of EE in the periphery including skeletal muscle. The data are consistent with the role of melanocortins in the VMH in the modulation of skeletal muscle metabolism.
Key points
The ventromedial hypothalamus (VMH) and the central melanocortin system both play vital roles in regulating energy balance by modulating energy intake and utilization.
Recent evidence suggests that activation of the VMH alters skeletal muscle metabolism.
We show that intra‐VMH melanocortin receptor activation increases energy expenditure and physical activity, switches fuel utilization to fats, and lowers work efficiency such that excess calories are dissipated by skeletal muscle as heat.
We also show that intra‐VMH melanocortin receptor activation increases sympathetic nervous system outflow to skeletal muscle.
Intra‐VMH melanocortin receptor activation also induced significant changes in the expression of mediators of energy expenditure in muscle.
These results support the role of melanocortin receptors in the VMH in the modulation of skeletal muscle metabolism.
Abbreviations
- ACC
acetyl‐CoA carboxylase
- AMPK
AMP‐activated protein kinase
- aMPT
α‐methyl‐p‐tyrosine
- BAT
brown adipose tissue
- CD36/FAT
fatty acid translocase
- CPT1
carnitine palmitoyltransferase 1
- EDL
extensor digitorum longus
- EE
energy expenditure
- EWAT
epididymal white adipose tissue
- gastroc
gastrocnemius
- GWAT
gluteal white adipose tissue
- IWAT
inguinal white adipose tissue
- Kir6.1
ATP‐dependent potassium channel subunit 6.1
- Kir6.2
ATP‐dependent potassium channel subunit 6.2
- MED1
mediator of RNA polymerase II transcription subunit 1
- MTII
melanotan II
- MWAT
mesenteric white adipose tissue
- NE
norepinephrine
- NETO
norepinephrine turnover
- pACC
phosphorylated acetyl‐CoA carboxylase
- pAMPK
phosphorylated AMP‐activated protein kinase
- PGC‐1α
peroxisome proliferator‐activated receptor γ coactivator 1α
- PPARα
peroxisome proliferator‐activated receptor α
- PPARγ
peroxisome proliferator‐activated receptor γ
- PPARδ
peroxisome proliferator‐activated receptor δ
- quad
quadriceps
- RER
respiratory exchange ratio
- RWAT
retroperitoneal white adipose tissue
- SERCA1
sarco/endoplasmic reticulum Ca2+‐ATPase 1
- SERCA2
sarco/endoplasmic reticulum Ca2+‐ATPase 2
- SNS
sympathetic nervous system
- UCP1
uncoupling protein 1
- UCP2
uncoupling protein 2
- UCP3
uncoupling protein 3
- VMH
ventromedial hypothalamus
oxygen consumption
- WAT
white adipose tissue
- ZT
zeitgeber time
Introduction
With the increase in obesity rates, interest in the brain regulation of energy balance has intensified, including how hypothalamic regions regulate autonomic nervous system control of metabolism. For example, peptide systems including brain melanocortins modulate sympathetic nervous system (SNS) outflow to peripheral tissues, including brown adipose tissue (BAT), enhancing energy expenditure (EE) (Labbé et al. 2015). Although interest has focused on BAT and ‘browning’ of white adipose tissue (WAT) (Contreras et al. 2015), hypothalamic nuclei are also involved in the modulation of energetics of other peripheral tissues, including skeletal muscle. Recent studies show that these hypothalamic nuclei modulate skeletal muscle metabolism (Toda et al. 2009; Miyaki et al. 2011; Toda et al. 2013; Schneeberger et al. 2014). Of these, the ventromedial hypothalamus (VMH) is part of a pathway regulating energy balance via its actions on peripheral glucose and lipid allocation, modulating the respiratory exchange ratio (RER), as well as high‐fat diet‐induced thermogenesis via the modulation of SNS (Borg et al. 1999; Miyaki et al. 2011; Stephens et al. 2011).
The brain melanocortin system is an important regulator of energy balance, decreasing appetite at the same time as increasing EE and physical activity (Garfield et al. 2009; Shukla et al. 2012). Activation of central melanocortin receptors also impacts glucose uptake and allocation, BAT thermogenesis and WAT lipolysis via the SNS (Shrestha et al. 2010; Rossi et al. 2011; Vaughan et al. 2011; Xu et al. 2011; Yi et al. 2011; Sohn et al. 2013). Altogether, this strongly implicates melanocortin peptides and receptors in the regulation of peripheral metabolism through sympathetic drive, with melanocortin receptors acting at multiple sites along this pathway (Vaughan et al. 2011; Sohn et al. 2013). Growing evidence also implicates SNS modulation of multiple peripheral systems outside of those most commonly considered, such as BAT and WAT; this includes skeletal muscle. Peripheral signals regarding the energy status of the individual alter the functioning of the hypothalamus, including the VMH, which in turn co‐ordinates energy partitioning in muscle, probably via a sympathetic relay (Belgardt et al. 2010; Braun et al. 2011). This process is mediated at least in part by brain melanocortin receptors, as shown by the ability of hypothalamic administration of melanotan II (MTII), a mixed melanocortin receptor agonist, to increase muscle glucose uptake (Toda et al. 2009).
Based on evidence that the VMH, melanocortins and SNS outflow are each important mediators of the brain control of muscle metabolism (Tanaka et al. 2007; Shiuchi et al. 2009; Toda et al. 2009; Miyaki et al. 2011), we tested the hypothesis that melanocortin receptor activation in the VMH increases SNS outflow to muscle, modulating thermogenesis and fuel selection. We modelled this brain–SNS–muscle axis on the brain control of BAT (Heeren & Münzberg, 2013; Labbé et al. 2015). The investigations described in the present study examined the impact of intra‐VMH melanocortin receptor activation with MTII on EE, particularly EE during activity, and RER, hypothesizing fuel switching and utilization to fats, as well as increased muscle thermogenesis. Lastly, we also determine whether the melanocortin receptor‐mediated activation in the VMH will increase SNS outflow to skeletal muscle and alter the expression of muscle thermogenic mediators and proteins involved in EE and fuel storage and utilization.
Methods
Ethical approval
All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals [‘National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals’, 2011] and with the approval of the Kent State University Institutional Animal Care and Use Committee. Male Sprague–Dawley rats (n = 73; 8 weeks old; ∼230–240 g on arrival) were obtained from Harlan Labs (Indianapolis, Indiana, USA). Each rat was housed individually under a 12 : 12 h light/dark cycle (lights on at 07.00 h EST). Rats received rodent chow (5P00 MRH 3000; T.R. Last Co. Inc., Cabot, PA, USA) and water ad libitum. Details of surgical procedures including anaesthesia, postoperative care and death are discussed below. Exclusion of an animal from final data was dependent on the study, as discussed below (final total n = 60). The investigators understand the ethical principles under which the journal operates and the present study complies with it.
Stereotaxic surgery and transponder implantation
Once rats were acclimated to the housing conditions, stereotaxic surgeries were performed to chronically implant guide cannulae (Plastics One Inc., Roanoke, VA, USA) aimed at the VMH. Rats were anaesthetized using isoflurane and mounted on a stereotaxic apparatus with atraumatic ear bars to prevent potential damage to rats’ tympanic membrane. The following co‐ordinates obtained from a stereotaxic atlas of the rat brain (Paxinos & Watson, 2010) were used for the VMH: anterior–posterior, –2.5 mm; medial–lateral, +0.5 mm; dorsal–ventral, –6 mm (from dura) and an injection needle with 3 mm projection (final dorsal–ventral, –9 mm from dura). The guide cannulae were attached to the skull using a sterile wound clip and dental cement, as described previously (Shukla et al. 2012). Postoperative pain management was carried out using bruprenorphine at 0.05 mg kg−1 on the day of surgery and 0.025 mg kg−1 every 12 h for next 2 days, checking for signs of any discomfort. After completion of the study, rats whose guide cannulae were within 250 μm of the VMH were used for data analysis.
A group of male rats (n = 10) received transponder implants during the stereotaxic surgery. A short incision was made on both hind legs and near the interscapular region. Sterile temperature transponders IPTT‐300 (Bio Medic Data Systems, Inc., Seaford, DE, USA) were implanted on interscapular BAT and also adjacent to the gastrocnemius (gastroc) muscle group of both hind limbs to measure the heat generated by skeletal muscle during activity. Care was taken to place the transponders aiming to avoid disrupting locomotor function. No such disruption or discomfort was noted upon inspection. Rats were allowed to recover for 1 week before the graded treadmill test was performed. Implant placement was confirmed after death by rapid decapitation.
Body composition
Body composition was measured using an EchoMRI‐700 (Echo Medical Systems, Houston, TX, USA) to determine the fat and lean mass (g) of each rat the day before experiment. This did not interfere with temperature transponder function.
Measurement of EE
After body composition determination, the EE and physical activity of rats was measured using small‐animal indirect calorimetry (four‐chamber Oxymax FAST system; Columbus Instruments, Columbus, OH, USA) with 24–48 h of acclimation in their testing cage at thermoneutral conditions to avoid any novelty‐induced alterations in calorimetric parameters, as reported previously (Gavini et al. 2014). On the day of calorimetry, rats were weighed and injected with either a non‐specific melanocortin receptor agonist MTII (Phoenix Pharmaceuticals, Mountain View, CA, USA) (20 pmol per 200 nl) or vehicle (artificial cerebrospinal fluid; 200 nl) over a period of 30 s, with the injection needle held in place for another 30 s to avoid back‐flush. The rats (n = 12) were then placed in the chambers without food but with free access to water, and the chamber was sealed. The calorimeter was calibrated using primary gas standards. Air was pumped into the chamber at 1.9–3.1 litres min–1, depending on the weight of the rat, and chamber air was sampled at 0.4 litres min–1. Measurement of gas exchange took place every 30 s throughout the 4 h period, except for a 3.5 min room‐air reference and settle period after each 60 sample interval. Using infrared beam‐break counts in the x‐ and z‐ axes, physical activity data were collected every 10 s, uninterrupted throughout the 4 h period; the first 15 min of data were not included in the analysis. Gas exchange data (,, RER, EE in kcal h–1) were averaged, and physical activity data were expressed as mean beam breaks min–1. Four days after the first injection, the experiment was repeated with rats now receiving the counterbalanced treatment (day 1 MTII rats receiving vehicle on day 5 and vice versa). Each rat acted as its own control, thereby nullifying the effects of body composition on calorimetric parameters (Tschöp et al. 2012).
As reported previously (Novak et al. 2009; Gavini et al. 2014), physical‐activity EE was indirectly assessed by measuring gas exchange once every 10 s during a treadmill activity test. At least 1 day after a 15 min treadmill acclimation period, rats (n = 12) were placed in the treadmill after weighing them and injecting with either MTII (20 pmol per 200 nl) or vehicle (artificial cerebrospinal fluid) and allowed to acclimate without food for 2 h. After the 2 h resting period, the treadmill was started at 7 m min−1 for 30 min, during which time physical‐activity EE data were collected (no access to food or water); the measurement was repeated after 4 days with rats receiving the counterbalanced injection.
All EE data from both studies were analysed using t tests or repeated‐measures ANOVA for changes in EE over time. Because all rats received both injections, we compared EE between MTII and vehicle treatments using a two‐tailed paired t test. A paired two‐tailed t test was used to compare treadmill gas exchange values between the groups.
Muscle thermogenesis vs. time of day
Adult male Sprague–Dawley rats (n = 11) were housed under a 12 : 12 h light/dark cycle [lights on 07.00 h EST; i.e. 07.00 h is Zeitgeber time (ZT) 0]. To determine whether activity‐induced muscle thermogenesis is influenced by time of day, gastroc temperature was assessed using a IPTT DAS‐7007 s reader during the treadmill walking procedure four times during the day: ZT0, ZT6, ZT12 and ZT18. Data were analysed using a 4 × 9 repeated‐measures ANOVA, with the four times of day (ZTs) as one independent variable and the level of treadmill intensity as the other independent variable. Temperature of BAT was measured only before and after the treadmill test, and these data were analysed using a 4 × 2 repeated‐measures ANOVA.
Home‐cage temperature
To determine whether melanocortin receptor activation in the VMH has any effect on the thermogenics of skeletal muscle, we measured skeletal muscle heat dissipation for 4 h after intra‐VMH MTII or vehicle microinjection, with activation of BAT thermogenesis used as a positive control (Brito et al. 2007). Rats (n = 10) were weighed and injected with either MTII or vehicle, placed in their home cage, and temperature data from the transponder implants on the gastroc and BAT were collected every 15 min over 4 h. Again, all rats received both treatments, and data were analysed using two‐tailed paired t tests and repeated‐measures ANOVA to analyse the time course.
Graded treadmill test
To exclude changes in physical activity in the elevated muscle thermogenesis seen after intra‐VMH MTII, skeletal muscle heat dissipation was assessed during controlled physical activity. The rats (n = 10) were acclimated to the treadmill for 10 min in the days prior to the test as well as immediately before the test. The rats received a microinjection of either MTII or vehicle 1.5 h prior to the test. Gastroc temperatures in each leg were recorded at baseline (before injecting and immediately before start of the test) and at set intervals during a 35 min, five‐level graded treadmill test, as reported previously (Gavini et al. 2014). The measurement was repeated after 4 days, with rats receiving the counterbalanced treatment. Data were analysed using a two‐tailed paired t test or repeated‐measures ANOVA to examine time course.
Norepinephrine turnover (NETO)
NETO was used to assess sympathetic drive to peripheral tissues including liver, heart, BAT and skeletal muscle [including quadriceps (quad), lateral and medial gastroc, extensor digitorum longus (EDL) and soleus], as well as different WAT depots: mesenteric WAT (MWAT), gluteal WAT (GWAT), retroperitoneal WAT (RWAT), inguinal WAT (IWAT) and epididymal WAT (EWAT). Rats (n = 24, eight per group) were individually housed under a 12 : 12 h light/dark cycle and acclimated to daily handling for 1 week after stereotaxic surgery. The level of sympathetic drive to peripheral tissues was determined by measuring NETO using α‐methyl‐p‐tyrosine (aMPT) (Shi et al. 2004; Brito et al. 2007; Vaughan et al. 2014). The rats were divided into three groups (aMPT/MTII, aMPT/vehicle, control). On the day of the study, rats assigned to receive aMPT were given injections (125 mg aMPT/kg body weight; 25 mg ml−1) and an additional dose at same concentration 2 h later. Then, 30 min after the first aMPT injection, rats received intra‐VMH microinjection of either MTII or vehicle, depending on their group. All rats were killed by rapid decapitation between 12.00 h and 15.00 h, 4 h after their first aMPT injection. Tissues were rapidly dissected and snap‐frozen in liquid nitrogen.
Tissue was thawed and homogenized in a solution containing dihydroxybenzylamine (internal standard) in 0.2 m perchloric acid with 1 mg ml−1 ascorbic acid. Following centrifugation for 15 min at 7500 g at 4°C, catecholamines were extracted from the homogenate with alumina and were eluted into the perchloric acid/ascorbic acid. Catecholamines were assayed using an HPLC system with electrochemical detection (Coulochem III), MDTM mobile phase (ESA Inc., Chelmsford, MA, USA) and a reverse phase MD 150 × 3.2 column. NETO in peripheral tissues was calculated using the formula (Shi et al. 2004; Gavini et al. 2014; Vaughan et al. 2014):
where k is the constant rate of NE (norepinephrine) efflux (also known as fractional turnover rate), [NE]0 is the initial NE concentration from the 0‐hour group (control), [NE]4 is the final NE concentration from the 4 h group (aMPT), and K = NETO.
Differences in NETO between intra‐VMH MTII microinjected and vehicle‐microinjected rat tissues were calculated with respect to control‐group rats and compared using separate one‐tailed t tests for each tissue.
Nadolol treatment
We determined the role of β‐adrenergic signalling in modulating muscle thermogenesis using the mixed β adrenergic (β1 and β2) antagonist nadolol, which acts peripherally but not centrally (Johnson et al. 2008). Nadolol (8 mg kg−1 i.p.) or vehicle was injected 30 min prior to the treadmill walking test, with each rat receiving both treatments in counterbalanced order separated by 4 days; gastroc temperature was measured as described above, and BAT temperature was measured before and immediately after treadmill walking. Data (n = 6) were analysed using a 2 × 9 repeated‐measures ANOVA, with drug treatment (nadolol vs. vehicle) and time on treadmill as the independent variables.
Gene expression
Skeletal muscle (gastroc and quad), liver, WAT and BAT were collected from rats 4 h after intra‐VMH microinjection of either MTII or vehicle (death by rapid decapitation) (n = 8 per group). Tissue samples were homogenized and total mRNA was extracted using a RiboPure kit (Ambion, Austin, TX, USA) in accordance with the manufacturer's instructions. The purity of the mRNA was measured using a NANODROP (ND‐1000; NanoDrop, Wilmington, DE, USA) with an A 260/A 280 ratio in the range 1.8–2.1. This mRNA was used to prepare cDNA using an Applied Biosystems kit (Foster City, CA, USA) and thermal cycling at 25°C for 10 min, 48°C for 30 min, 95°C for 5 min and holding at 4°C. The cDNA was used for quantifying the expression of uncoupling proteins 1, 2 and 3 (UCP1, UCP2, UCP3), ATP‐dependent potassium channel (K+ ATP subunits Kir6.1, Kir6.2), mediator of RNA polymerase II transcription subunit 1 (MED1), peroxisome proliferator‐activated receptors α, δ and γ (PPARα, PPARδ, PPARγ), peroxisome proliferator‐activated receptor γ coactivator 1α (PGC‐1α), sarco/endoplasmic reticulum Ca2+‐ATPase 1, 2 (SERCA), β2‐, β3‐adrenergic receptors and glyceraldehyde 3‐phosphate dehydrogenase (used as a control). Relative expression was calculated using the comparative Ct method (ΔCt). Data are expressed as the percentage expression using samples from vehicle‐treated rats as the reference value (defined as 100%) and groups were compared using two‐tailed t tests.
Western blots
Tissues samples from rats (same group of rats used for gene expression) were homogenized with ice‐cold RIPA buffer (Thermo Scientific, Waltham, MA, USA) containing a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). The supernatant from the homogenization and subsequent centrifugation was used for the analysis. Equal quantities of supernatant and sample buffer (150 mm tris‐HCl pH 6.8, Trizma‐base for pH, 6% SDS, 30% glycerol, 0.03% pyronin‐Y, dithiothreitol) were mixed and tubes were heated at 90°C for 3 min. Samples containing equal quantity of protein were loaded onto a gradient gel (4–15%; Bio Rad, Hercules, CA, USA) and electrophoresed using SDS running buffer (0.384 m glycine, 0.05 m Trizma base, 0.1% SDS) at constant voltage (150 V) for 30 min. The gel was blotted onto a poly(vinylidene difluoride) membrane using a semi‐wet blotting apparatus and transfer buffer (49.6 mm Trizma base, 384 mm glycine, 17.5% methanol, 0.01% SDS) at constant current (400 mA) (Otter et al. 1987). The blot was incubated overnight in a blocking solution of 5% milk (Blotto blocking buffer) in 1 × PBST (phosphate‐buffered saline; 84 mm sodium hydrogen phosphate, 16 mm sodium dihydrogen phosphate, 100 mm sodium chloride, Tween 20) and then rinsed using 1 × PBST. Primary antibodies were diluted in blocking solution and incubated with the blot overnight in accordance with the manufacturer's instructions (Abcam). Secondary antibodies were diluted in blocking solution and incubated for 1 h at room temperature. After washing, the blots were developed using a chemiluminiscence detector using an Amersham kit (GE Healthcare, Little Chalfont, UK). The expression levels relative to actin were plotted as a percentage of the reference value (samples from vehicle‐treated rats as 100%) and groups were compared using two‐tailed t tests.
Results
Intra‐VMH MTII microinjection increases EE
As shown in Fig. 1, compared to vehicle microinjection, rats treated with intra‐VMH MTII have significantly higher EE (Fig. 1 A), higher oxygen consumption () (Fig. 1 B) and lower RER (Fig. 1 C). All rats (n = 10) received microinjections of both vehicle and MTII, thereby acting as their own control, eliminating confounding effects of body composition on calorimetric parameters (Tschöp et al. 2012). There was no significant difference in body weight between vehicle vs. MTII treatment groups (333.46 ± 8.01 vs. 326.78 ± 7.2 g). Compared to vehicle, MTII injections significantly increased both horizontal (Fig. 1 D) and ambulatory activity (1.63 ± 0.16 for vehicle vs. 2.57 ± 0.25 for MTII).
Figure 1. Activation of VMH melanocortin receptors increases EE, even in the absence of altered physical activity .
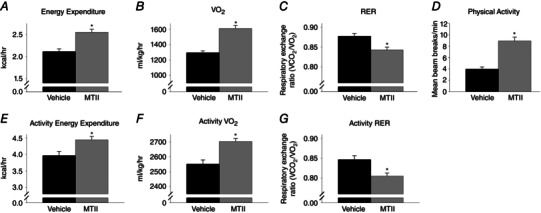
Compared to vehicle microinjection, intra‐VMH microinjection of the mixed melanocortin receptor agonist MTII significantly increased (A) EE, (B) and (D) physical activity levels, and also (C) decreased the RER, implicating increased lipid oxidation. When activity levels and workload were equalized using a treadmill walking protocol, the ability of MTII to (E) elevate EE, (F) increase and (G) lower RER remained, indicating decreased locomotor efficiency. *Significantly different from vehicle, P < 0.05 (n = 10).
Intra‐VMH MTII microinjection increases EE during controlled activity
To determine whether the increase in these calorimetric parameters described above was secondary to increased physical activity after intra‐VMH microinjection of MTII, a controlled treadmill activity study was performed. As shown in Fig. 1, compared to vehicle microinjection, intra‐VMH microinjection of MTII significantly increased EE (Fig. 1 E) and (Fig. 1 F), and lowered RER (Fig. 1 G), during controlled treadmill walking activity (n = 10). Therefore, the elevated EE and calorimetric parameters in intra‐VMH MTII‐injected rats persisted in the absence of differential change in physical activity. Because each rat acted as its own control, and considering the relatively constant body weight, lean mass and fat mass between the two treatments (body weight 330.18 ± 10.32 vs. 329.83 ± 9.65 g), the differential effect of workload during activity was negligible. In summary, MTII increased EE when physical activity and workload were held constant.
High activity energy expenditure after intra‐VMH MTII microinjection is accompanied by skeletal muscle energy dissipation as heat
The results described above showing decreased skeletal muscle work efficiency, as well as studies pointing to the VMH and melanocortin receptors inducing changes in skeletal muscle fuel use and allocation (Tanaka et al. 2007; Shiuchi et al. 2009; Toda et al. 2009; Miyaki et al. 2011), implicate altered skeletal muscle energetics, although the fate of the energy from the enhanced caloric expenditure has not yet been identified. To test this, we microinjected these rats with MTII or vehicle and measured subsequent gastroc and BAT temperatures under resting conditions, as well as during graded treadmill activity. Compared to vehicle, intra‐VMH MTII treatment significantly increased gastroc muscle temperatures in the home cages between 90 and 240 min after microinjection (Fig. 2 B) (n = 8). Baseline gastroc temperature was 36.2 ± 0.3°C before vehicle and 36.2 ± 0.2°C before MTII microinjection, rising to 36.5 ± 0.2°C between 180 and 210 min after MTII injection (gastroc temperature was 36.1 ± 0.3°C at 180–210 min after vehicle injection). During resting, compared to vehicle treatment, intra‐VMH MTII induced a significantly higher rise in BAT temperatures between 30 min and 105 min post‐injection (Fig. 2 A). Baseline BAT temperature was 37.7 ± 0.2°C for vehicle and 37.7 ± 0.2°C for MTII. Peak BAT temperature was 38.1 ± 0.2°C at 90 min after MTII microinjection; BAT temperature was 37.6 ± 0.2°C at 90 min after vehicle microinjection. The relatively elevated temperatures measured by interscapular relative to muscle transponders in this and other experiments (Figs 2 A and 3 B), as well as the differential effects of MTII and treadmill activity (Figs 2 A and 3 B), confirm the measurement by transponders of BAT rather than underlying muscle.
Figure 2. Intra‐VMH microinjections of the melanocortin receptor agonist MTII enhanced thermogenesis .
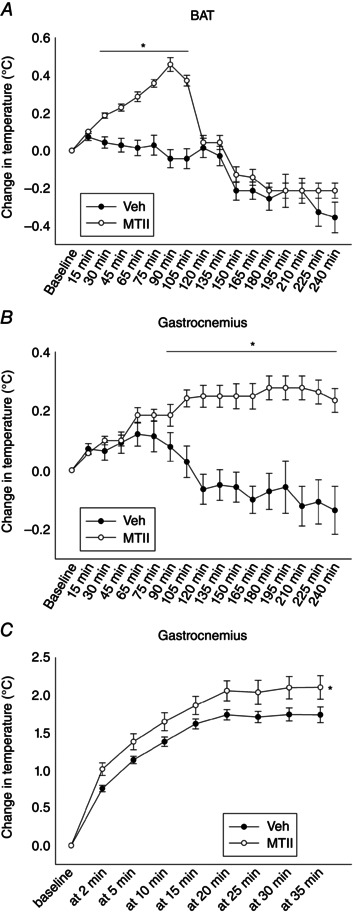
A, change in BAT thermogenesis from baseline levels was significantly higher between 30 and 105 min after MTII microinjection compared to vehicle. B, gastroc muscle thermogenesis was significantly elevated above baseline levels starting 90 min after microinjection and continuing until the end of the 240 min measurement. C, when gastroc temperature was measured after activity levels and workload were equalized using a treadmill, the activity‐associated change in gastroc temperature was significantly higher after intra‐VMH MTII compared to vehicle. *Significantly different from vehicle, P < 0.05 (n = 8).
Figure 3. Time of day, indicated by ZT (lights on ZT0), influenced baseline gastroc muscle and BAT thermogenesis .
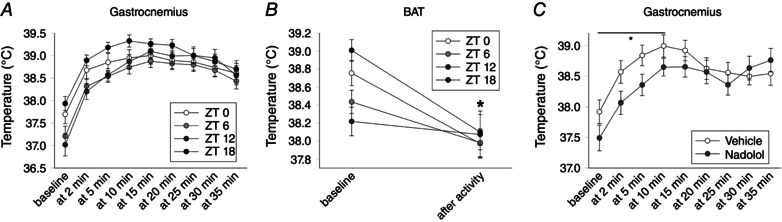
A, baseline gastroc temperature and activity‐related gastroc heat dissipation was highest at ZT18, with the most ZT‐associated differences seen between baseline during the first 10 min of treadmill walking. B, BAT thermogenesis showed a similar pattern of baseline temperature, with treadmill walking activity inducing a significant suppression in temperature (* P < 0.05). C, mixed β‐adrenergic antagonist nadolol significantly suppressed baseline and activity‐related gastroc heat dissipation during 10 min of treadmill walking (* P < 0.05) (n = 8).
To rule out potential changes in physical activity in the MTII‐induced muscle temperature changes, gastroc temperature was also measured during a graded treadmill walking test. Again, all rats received both sets of injections and no significant changes in body composition were observed between treatments. MTII treatment stimulated significantly higher gastroc temperatures, and the maximal rise in temperature was significantly higher after MTII treatment compared to vehicle (Fig. 2 C), demonstrating increased muscle thermogenesis during activity, where caloric expenditure stimulated by MTII is dissipated as heat. Baseline temperature before treadmill walking was 36.6 ± 0.1°C after both vehicle and MTII microinjections, rising to a final temperature of 38.7 ± 0.1°C after MTII injection and 38.3 ± 0.1°C after vehicle injection.
Time of day modulates baseline muscle and BAT thermogenesis
As shown in Fig. 3 A, mean (right and left gastroc) muscle temperature showed a significant interaction between time of day (ZT) and treadmill intensity, as well as significant main effects; individual gastroc temperatures (i.e. right and left) showed the same effects (final n = 8; three rats excluded because of an inability to read temperatures at one or more time points). In short, baseline pre‐treadmill temperatures varied across ZTs, where ZT18 > ZT0 > ZT6, with ZT12 < all time points, except ZT6. These differences in gastroc temperature persisted during 5–10 min of treadmill walking, and gastroc temperature at ZT18 remained higher than other time points during 10–15 min, although there were no differences between any time points at the two highest walking intensities. Change in gastroc temperature from baseline also showed significant main effects and interactions (except for the right leg, for which there was a trend for a main effect of ZT, P = 0.062). The maximal increase in temperature was directly related to baseline temperatures, with the rats and time points with the lower baseline temperatures also showing highest maximal change (r = –0.74); accordingly, temperature change was consistently highest at ZT12. Time of day also significantly affected BAT temperature (significant main effects and interaction). Baseline BAT temperature was highest at ZT18 and decreased through ZT0 and ZT6, reaching its nadir at ZT12. Treadmill activity decreased BAT temperature at every time point, except ZT12.
Intra‐VMH MTII microinjection elevates sympathetic drive to metabolic tissues
As illustrated in Fig. 4, compared to vehicle, intra‐VMH MTII‐microinjected rats had higher NETO in all tissues analysed, indicating higher sympathetic drive to these tissues; this included skeletal muscle (quad, lateral and medial gastroc, EDL, and soleus) (Fig. 4), WAT depots, BAT, heart and liver (Table 1) (n = 8 per group). This implicates a melanocortin‐regulated pathway in the hypothalamus, particularly the VMH, in the regulation of SNS drive, potentially modulating muscle fuel storage and utilization.
Figure 4. Intra‐VMH microinjection of the mixed melanocortin receptor agonist MTII increased skeletal muscle NETO, an indicator of sympathetic drive .
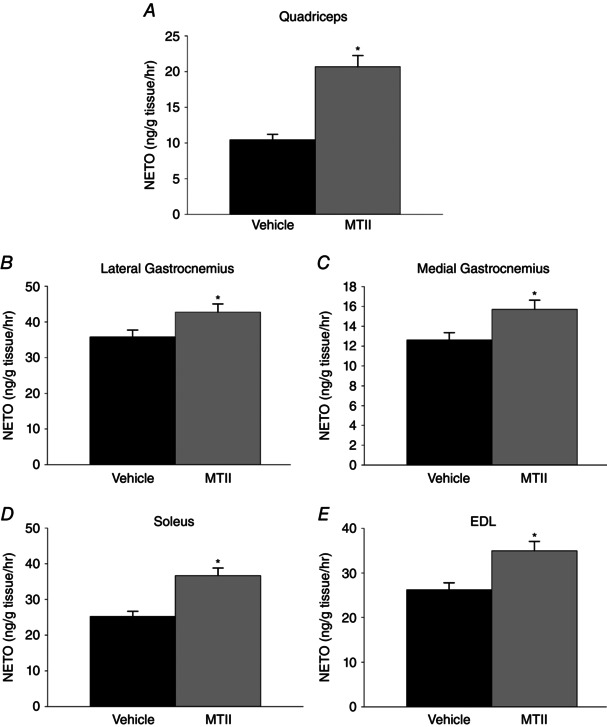
In MTII‐injected rats compared to vehicle‐injected rats, NETO was significantly increased in all muscle groups examined, including the (A) quad, (B) lateral gastroc (C) medial gastroc, (D) soleus and (E) EDL. *Significantly greater than vehicle, P < 0.05 (n = 8 per group).
Table 1.
Intra‐VMH microinjection of the mixed melanocortin receptor agonist MTII increased NETO in peripheral metabolic tissues
| Vehicle | MTII | |
|---|---|---|
| BAT | 103.73 ± 8.34 | 145.37 ± 11.69* |
| EWAT | 0.91 ± 0.06 | 1.18 ± 0.07* |
| RWAT | 25.8 ± 2.21 | 44.1 ± 3.78* |
| MWAT | 29.49 ± 3.18 | 40.77 ± 4.41* |
| IWAT | 17.97 ± 1.14 | 21.99 ± 1.39* |
| GWAT | 7.23 ± 0.62 | 11.82 ± 1.01* |
| Liver | 0.82 ± 0.07 | 1.26 ± 0.11* |
| Heart | 20.61 ± 1.67 | 28.88 ± 2.35* |
*Significantly greater than vehicle, P < 0.05 (n = 8 per group). Data are the mean ± SEM.
β‐adrenergic receptor antagonist decreases muscle heat dissipation
As shown in Fig. 3 C, mean (left and right) gastroc temperature showed a significant interaction between treatment and time on treadmill, as well as a main effect of time on treadmill and a trend toward a main effect of nadolol (P = 0.060). After nadolol injection, mean (left and right) gastroc temperature was significantly lower at baseline during 10 min of treadmill walking, and during 15 min in the left leg only.
Intra‐VMH MTII microinjection elevates expression of mRNA of mediators of energy expenditure, with a trend towards lower expression of energy conserving processes in peripheral metabolic tissues, and some alteration in the protein content
To determine potential sources of calorie use and heat dissipation in the muscle of MTII‐treated rats, we examined the levels of mRNA expression of potential molecular mediators of energy balance: UCP1, UCP2, UCP3, Kir6.1, Kir6.2, MED1, PPARα, PPARδ, PPARγ, PGC‐1α, Ca2+‐ATPase (SERCA), β2‐AR and β3‐AR in skeletal muscle, including quad and gastroc, BAT, WAT and liver. For skeletal muscle, quad of MTII‐microinjected rats showed significantly higher UCP2, UCP3, PPARδ, SERCA1 and SERCA2 mRNA expression (Fig. 5 A), as well as a trend toward higher expression of PPARα, PPARγand β2‐AR (Table 2). Compared to vehicle‐treated rats, gastroc muscle of intra‐VMH MTII rats had significantly higher mRNA expression of UCP2, UCP3, PGC‐1α, PPARα, PPARδ, PPARγ, SERCA2 and β2‐AR (Fig. 5 B). Gastroc muscle also showed significantly lower levels of Kir6.2 (Fig. 5 B), as well as a trend toward lower Kir6.1 and MED1 in MTII‐treated rats (Table 2).
Figure 5. Intra‐VMH microinjection of the mixed melanocortin receptor agonist MTII elevates mRNA expression of mediators of energy expenditure in peripheral metabolic tissues, with some alteration in the protein content and phosphorylation .
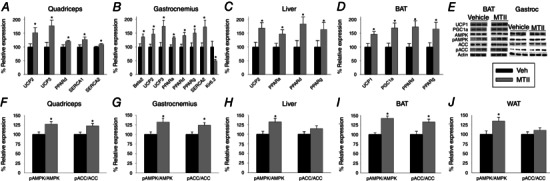
Relative to vehicle‐treated rats, rats with intra‐VMH melanocortin receptor activation had significantly elevated mRNA levels of UCP2 and 3, SERCA2 and PPARδ in the (A) quad and (B) gastroc muscles; quad also had significantly elevated SERCA1, whereas gastroc showed higher β2 adrenergic receptor (Beta 2) and PPARα and δ expression and lower expression of the Kir6.2 subunit of the ATP‐gated K+ channel. C, liver of MTII‐injected rats showed elevated expression UCP2 and PPAR isoforms α, δ, and γ. D, BAT showed elevated expression of UCP1 and PPAR isoforms α, δ and γ in MTII‐injected rats. E, western blots were used to measure protein and phosphoprotein expression levels. In MTII‐treated rats, the pAMPK‐to‐AMPK ratio was significantly elevated in (F) quad, (G) gastroc, (H) liver, (I) BAT and (J) WAT, whereas the pACC/ACC ratio was significantly elevated above vehicle‐treated levels in muscle and BAT only. Mean vehicle‐treated defined as 100%. *Intra‐VMH MTII‐injected rats vs. vehicle‐treated rats, P < 0.05. (n = 7 per group for mRNA, four or five per group for protein).
Table 2.
Tissue expression levels of mRNA in rats 4 h after intra‐VMH microinjections of MTII, relative to vehicle‐treated levels (100%)
| Intra‐VMH | PPAR | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| microinjection | β2AR | β3AR | UCP2 | α | δ | γ | PGC‐1α | SERCA1 | Kir6.1 | Kir6.2 | MED1 | |
| Quad | Veh | 100 ± 6 | – | – | 100 ± 6 | – | 100 ± 4 | 100 ± 5 | – | 100 ± 19 | 100 ± 13 | 100 ± 15 |
| MTII | 110 ± 13 | – | – | 114 ± 3 | – | 106 ± 4 | 113 ± 7 | – | 92 ± 26 | 94 ± 16 | 65 ± 15 | |
| Gastroc | Veh | – | – | – | – | – | – | 100 ± 8 | 100 ± 5 | 100 ± 25 | – | 100 ± 21 |
| MTII | – | – | – | – | – | – | 131 ± 10 | 118 ± 7 | 73 ± 20 | – | 73 ± 14 | |
| BAT | Veh | – | 100 ± 8 | – | 100 ± 13 | – | – | – | – | – | – | – |
| MTII | – | 101 ± 13 | – | 152 ± 26 | – | – | – | – | – | – | – | |
| WAT | Veh | – | 100 ± 6 | 100 ± 19 | 100 ± 15 | 100 ± 15 | 100 ± 11 | 100 ± 11 | – | – | – | – |
| MTII | – | 98 ± 11 | 101 ± 18 | 123 ± 13 | 132 ± 16 | 117 ± 9 | 98 ± 12 | – | – | – | – | |
| Liver | Veh | 100 ± 9 | – | – | – | – | – | 100 ± 7 | – | – | – | – |
| MTII | 112 ± 7 | – | – | – | – | – | 116 ± 8 | – | – | – | – | |
Values calculated relative to glyceraldehyde 3‐phosphate dehydrogenase. β2AR, β2 adrenergic receptor; β3AR, β3 adrenergic receptor; Veh, vehicle (artificial cerebrospinal fluid). Data are the mean ± SEM.
Compared to BAT of vehicle‐treated rats, BAT from rats with intra‐VMH MTII microinjections showed significantly elevated expression levels of UCP1, PGC‐1α, PPARδ and PPARγ, with a trend (1.0 > P > 0.05) toward higher PPARα (Fig. 5 D and Table 2). There were also trends toward increased PPARγ, PPARα and PPARδ in WAT from rats that received intra‐VMH MTII (Table 2). In liver, intra‐VMH MTII‐treated rats showed significantly higher mRNA expression of UCP2, PPARα and PPARδ, as well as a trend for PPARγ (Fig. 5 C and Table 2).
Protein expression levels of some mediators of EE were altered by intra‐VMH MTII, although protein levels did not consistently change in accordance with the effects of MTII treatment on mRNA expression within 4 h of treatment. Compared to vehicle, quad of MTII‐treated rats showed a significant increase in phosphorylated AMP‐activated protein kinase (pAMPK)/AMP‐activated protein kinase (AMPK) and phosphorylated acetyl‐CoA carboxylase (pACC)/acetyl‐CoA carboxylase (ACC) (Fig. 5 F), as well as a trend for increased expression of fatty acid translocase (CD36/FAT), PPARα, PGC‐1α, PPARδ, UCP3, UCP2, PPARγ and SERCA1 (Table 3). Compared to vehicle‐treated rats, gastroc muscle of intra‐VMH MTII‐microinjected rats showed significantly higher expression of PGC‐1α, PPARα, pAMPK/AMPK and pACC/ACC proteins (Fig. 5 G and Table 3). Gastroc muscle also showed trends toward higher expression of several proteins including SERCA1, SERCA2, β2‐AR, PPARγ, PPARδ, UCP2, UCP3, carnitine palmitoyltransferase 1 (CPT1) and CD36/FAT in MTII‐treated rats (Table 3).
Table 3.
Intra‐VMH induction of protein expression in energetic pathways as determined using western blot analysis, relative to actin, with expression in vehicle‐treated rats as 100%
| PPAR | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β2AR | β3AR | UCP1 | UCP2 | UCP3 | CPT1 | CD36/FAT | FAS | α | δ | γ | PGC‐1α | SERCA1 | SERCA2 | Kir6.1 | Kir6.2 | MED1 | ||
| Quad | Veh | 100 ± 9 | – | – | 100 ± 7 | 100 ± 7 | 100 ± 4 | 100 ± 10 | 100 ± 6 | 100 ± 7 | 100 ± 7 | 100 ± 7 | 100 ± 6 | 100 ± 7 | 100 ± 7 | 100 ± 5 | 100 ± 8 | 100 ± 5 |
| MTII | 134 ± 7 | – | – | 115 ± 7 | 117 ± 9 | 101 ± 5 | 118 ± 8 | 96 ± 6 | 122 ± 8 | 116 ± 7 | 120 ± 8 | 118 ± 10 | 117 ± 6 | 118 ± 9 | 98 ± 7 | 103 ± 8 | 92 ± 7 | |
| Gastroc | Veh | 100 ± 8 | – | – | 100 ± 8 | 100 ± 8 | 100 ± 7 | 100 ± 8 | 100 ± 5 | 100 ± 6 | 100 ± 6 | 100 ± 10 | 100 ± 4 | 100 ± 8 | 100 ± 6 | 100 ± 6 | 100 ± 6 | 100 ± 5 |
| MTII | 118 ± 8 | – | – | 114 ± 5 | 116 ± 8 | 115 ± 7 | 120 ± 9 | 94 ± 6 | 130 ± 6* | 118 ± 7 | 119 ± 6 | 124 ± 5* | 120 ± 9 | 119 ± 6 | 98 ± 8 | 93 ± 6 | 94 ± 6 | |
| BAT | Veh | – | 100 ± 6 | 100 ± 3 | – | – | 100 ± 5 | 100 ± 13 | 100 ± 9 | 100 ± 9 | 100 ± 5 | 100 ± 3 | 100 ± 4 | – | – | – | – | – |
| MTII | – | 109 ± 8 | 129 ± 4* | – | – | 111 ± 5 | 124 ± 9* | 99 ± 11 | 121 ± 10 | 110 ± 5 | 105 ± 7 | 132 ± 7 | – | – | – | – | – | |
| WAT | Veh | – | 100 ± 8 | – | 100 ± 8 | – | – | 100 ± 12 | 100 ± 7 | 100 ± 3 | 100 ± 7 | 100 ± 10 | 100 ± 7 | – | – | – | – | – |
| MTII | – | 106 ± 5 | – | 103 ± 7 | – | – | 99 ± 12 | 95 ± 7 | 103 ± 4 | 107 ± 8 | 113 ± 8 | 104 ± 6 | – | – | – | – | – | |
| Liver | Veh | 100 ± 9 | – | – | 100 ± 5 | – | 100 ± 5 | 100 ± 11 | 100 ± 8 | 100 ± 7 | 100 ± 10 | 100 ± 4 | 100 ± 10 | – | – | – | – | – |
| MTII | 111 ± 8 | – | – | 111 ± 6 | – | 108 ± 5 | 96 ± 7 | 93 ± 6 | 11 ± 6 | 111 ± 6 | 111 ± 9 | 110 ± 5 | – | – | – | – | – | |
*Significantly different from vehicle‐treated rats, P < 0.05. β2AR, β2 adrenergic receptor; β3AR, β3 adrenergic receptor; FAS, fatty acid synthase; Veh, vehicle (artificial cerebrospinal fluid). Data are the mean ± SEM.
Compared to vehicle, BAT of intra‐VMH MTII‐treated rats showed significantly higher protein expression of UCP1, PGC‐1α, pAMPK/AMPK and pACC/ACC (Fig. 5 I and Table 3), as well as trends toward increased expression levels of PPARγ, PPARα, β3 AR, PPARδ, CPT1 and CD36/FAT (Table 3). In WAT, no significant differences were observed, except for the expression of pAMPK/AMPK, which was significantly elevated in MTII‐treated rats (Fig. 5 J). Similar to this was the liver of MTII‐microinjected rats, which showed significantly higher pAMPK/AMPK (Fig. 5 H), as well as a trend for increased expression of PPARγ, UCP2, β2‐AR and PGC‐1α (Table 3).
Discussion
Hypothalamic nuclei and neuropeptides including melanocortins modulate SNS outflow to peripheral systems, impacting peripheral metabolism and thermogenesis (Vaughan et al. 2011; Sohn et al. 2013). The investigations reported in the present study demonstrate that intra‐VMH melanocortin receptor activation increases EE, including the EE of activity by reducing locomotor efficiency. Because muscle temperature is also increased by intra‐VMH MTII, this implies that the additional calories expended are dissipated as heat energy. The thermogenesis and reduced energetic efficiency may be attributed to MTII‐induced increases in molecular pathways, including UCPs, PPARs, PGC‐1α, SERCAs and AMPK activation. Lastly, the increased NETO to skeletal muscle implicates intra‐VMH MTII‐induced increases in SNS outflow to muscle as a potential driver of the muscle thermogenesis, similar to SNS‐induced energetic changes in BAT and WAT (Shrestha et al. 2010; Rossi et al. 2011; Stephens et al. 2011; Vaughan et al. 2011; Xu et al. 2011; Yi et al. 2011; Sohn et al. 2013; Lee et al. 2014). Altogether, these studies support a hypothetical pathway through which melanocortin peptides act in the VMH to alter skeletal myocyte energetic pathways, reducing muscle efficiency and increasing EE and dissipating calories as heat energy. Considering the contribution of skeletal muscle to body mass, this induction of thermogenesis has the potential to exert a physiologically relevant impact on total energy expenditure.
Hypothalamic nuclei respond to changes in energy status by regulating the expression of specific homeostasis‐related neuropeptides and neurotransmitters. Although the VMH used to be described as ‘the satiety centre’ (Hetherington & Ranson, 1940; Anand & Brobeck, 1951; Elmquist et al. 1999; King, 2006), it is now clear that ‘VMH obesity’ is not completely secondary to hyperphagia (King, 2006). Today, the VMH is conceptualized as a part of a pathway regulating energy balance through its actions on peripheral glucose and lipid allocation, modulating RER and high‐fat diet‐induced thermogenesis (Akira & Takashi 1981; Toda et al. 2009; Kim et al. 2011; Miyaki et al. 2011). The VMH also shows melanocortin receptor binding (Harrold et al. 1999) and our results demonstrating that VMH melanocortin receptor activation enhances activity‐EE and muscle thermogenesis are consistent with other studies demonstrating the importance of the VMH in the functional modulation of skeletal muscle energetic processes (Tanaka et al. 2007; Shiuchi et al. 2009; Toda et al. 2009; Miyaki et al. 2011). Although the specific identity of the neurons important in this pathway has yet to be determined, VMH steroidogenic factor‐1 neurons have been implicated in BAT thermogenesis and muscle glucose uptake (Orozco‐Solis et al. 2016; Toda et al. 2016) and may modulate the SNS through projections to pre‐autonomic brain regions (Lindberg et al. 2013). The investigations reported in the present study confirm that this brain‐muscle pathway can be targeted to increase activity‐EE.
Compared to vehicle, MTII microinjection increased total EE in freely moving rats, accompanied by increased physical activity and , and a decrease in RER, indicating altered lipid mobilization and utilization (Fig. 1 A–D). The metabolic effects of intra‐VMH MTII were seen in the absence of altered body weight. Moreover, these effects persisted when physical activity was held constant by walking rats on a treadmill (Fig. 1 E–G), demonstrating that the enhanced EE was not dependent on increased physical activity. Although demonstration of melanocortin receptor antagonist‐induced inhibition of this effect would provide further support, these results are consistent with the known role of brain melanocortin peptides in energy balance and SNS outflow (Shrestha et al. 2010; Vaughan et al. 2011; Sohn et al. 2013; Labbé et al. 2015). Genetic alterations in this system are known to result in obesity in humans (Loos et al. 2005; Yako et al. 2012) and animals (Chen et al. 2000; Butler et al. 2001; Mul van Boxtel, et al. 2012), yet these same genetic perturbations are protective of obesity‐induced hypertension (Hall et al. 2010). Brain melanocortins promote negative energy balance through actions on behaviours including food intake (Garfield et al. 2009) and physical activity (Shukla et al. 2012), as well as on SNS outflow (Sohn et al. 2013). The investigations reported in the present study are consistent with recent work demonstrating a central regulation of skeletal muscle metabolism, where peripheral signals relating to energy status alter energy partitioning in muscle, possibly via a sympathetic relay, with melanocortin signalling comprising a critical component (Shiuchi et al. 2009; Toda et al. 2009; Belgardt et al. 2010; Braun et al. 2011). The decreased RER seen during activity in MTII‐injected rats, implicating switching of fuel utilization to fats, is also in line with the role of central melanocortins on peripheral lipid metabolism (Nogueiras et al. 2007).
Based on these data, knowledge that both BAT and skeletal muscle share common progenitors, and the capacity of muscle to mimic energy usage and thermodynamics of BAT (Zurlo et al. 1990), we propose the existence of a pathway between VMH and skeletal muscle, modelled on melanocortin‐regulated VMH control of BAT. Central melanocortin controls peripheral glucose and lipid metabolism and thermogenesis in BAT (Obici et al. 2001; Voss‐Andreae et al. 2006; Nogueiras et al. 2007), demonstrating neuroendocrine control over peripheral cell metabolism, probably via regulation of the SNS (Rahmouni et al. 2003). Hypothalamic peptides probably modulate BAT and muscle through parallel but distinct pathways. For example, although melanocortin can activate both pathways (Toda et al. 2009), the dorsomedial hypothalamus appears to be critical in the regulation of BAT thermogenesis (Tupone et al. 2014), whereas inhibition of the VMH but not the paraventricular or lateral hypothalamus attenuated fatty acid oxidation during endurance exercise (Miyaki et al. 2011). It is possible that the targeted microinjections performed in the present study affected additional brain regions in addition to the VMH; however, the preponderance of evidence highlights the need for signalling in the VMH, rather than adjacent hypothalamic regions, with respect to the modulation of muscle energetics (Toda et al. 2009; Miyaki et al. 2011). Lastly, the activity‐related EE and thermogenesis were probably not influenced by BAT because, first, treadmill activity decreases BAT thermogenesis (Fig. 3 B) and, second, increased BAT thermogenesis was observed only for a short duration (for ∼1.5 h after melanocortin receptor activation) (Fig. 2 A), indicating that alterations at the level of skeletal muscle have a greater probability of playing a role in the observed changes in calorimetric parameters seen 2–4 h after treatment.
Central melanocortin regulation of EE and peripheral metabolism is associated with altered SNS activity to systems including BAT, liver and skeletal muscle (Obici et al. 2001; Rahmouni et al. 2003; Voss‐Andreae et al. 2006; Nogueiras et al. 2007). To determine whether the SNS was activated by intra‐VMH melanocortin receptor stimulation, we measured the level of sympathetic drive to the peripheral tissues by assessing NETO, where endogenous tissue levels of NE decline at a rate proportional to the initial NE concentrations after aMPT administration (Shi et al. 2004; Vaughan et al. 2014). Intra‐VMH MTII‐treated rats had significantly higher NETO, indicating higher sympathetic drive in the tissues analysed (Fig. 4 and Table 1). This supports the overall hypothesis that MTII‐induced melanocortin receptor activation in the VMH and subsequent alteration in EE, thermogenesis, and fuel utilization in muscle is driven by the SNS. Increased SNS outflow to WAT and liver (Table 1) would support the increased energy needs of muscle by increasing lipolysis. Although functional studies have shown the VMH to be a regulator of peripheral metabolism, evidence of a probable neuronal pathway was only recently identified. Steroidogenic factor‐1 neurons send direct projections to the rostral ventrolateral medulla (Lindberg et al. 2013), which is a critical site in the regulation of SNS outflow (Burke et al. 2011; Korim et al. 2011).
The non‐specific β‐adrenergic receptor antagonist nadolol significantly decreased muscle heat dissipation during low‐intensity treadmill activity (Fig. 3 C), suggesting sympathetic modulation of locomotor efficiency, as reflected in the flexibility of muscle heat generation. Although this is consistent with the evidence supporting the roles of SNS and β2 in the control of muscle energetics (Maurya et al. 2015), inhibition of SNS neural signalling is needed to confirm that increased neural SNS activity underlies the muscle thermogenic effects of MTII demonstrated in the present study. Unlike baseline temperatures, muscle heat production at higher‐intensity activity was not significantly affected by β adrenergic blockade (Fig. 3 C). This is consistent with the results of clinical studies where human muscle work efficiency is most labile at lower rather than higher workloads in people who maintain high or reduced body weights (Rosembaum et al. 2003; Rosenbaum et al. 2005; Goldsmith et al. 2010). Although baseline muscle and BAT temperatures varied by time of day (Fig. 3 A and B), similar to body temperature (Abreu‐Vieira et al. 2015), and probably secondary to the activity state of the rat, maximal activity thermogenesis was the same regardless of the time of day. Therefore, the time of day when rats were measured was kept consistent in studies measuring the effects of MTII. Baseline BAT temperatures were higher than that of muscle and varied similarly across the day (data not shown) and treadmill activity suppressed BAT thermogenesis to baseline levels or below, consistent with mathematical models where thermogenic systems and heat dissipation are altered to compensate for activity‐related heat production (Yoo et al. 2015).
Increased sympathetic drive to the skeletal muscle may impact the molecular pathways involved in EE and in energy conservation, playing an important role in determining the propensity of an an individual for obesity. In the present study, we show that there is a significant increase in the expression of several of these mediators of EE at the level of both mRNA and protein (Fig. 5 and Tables 2 and 3) in metabolically active tissues, including muscle. Compared to vehicle, intra‐VMH melanocortin receptor activation also induced an increase in UCP1 in BAT, as well as UCP2 and UCP3 in oxidative gastroc muscle (Fig. 5 B and D). UCPs have several hypothesized functions, including thermogenesis, protection from ROS, mediation of insulin secretion, neuroprotection and export of fatty acids (Fisler & Warden, 2006). The increase in UCP after intra‐VMH melanocortin receptor activation and downstream SNS may account for some of the changes in the calorimetric parameters seen in the present study, such as increased EE, partially through increased BAT and skeletal muscle thermogenesis, and may also be involved in fatty acid transport, thereby affecting RER (Fig. 1 C and G). A longer delay after MTII injection may reveal significant changes to proteins where trends are indicated (Table 3) for example in SERCA1 and SERCA2, regulators of cytosolic calcium levels and muscle thermogenesis (Periasamy & Kalyanasundaram, 2007), and CD36/FAT involved in the transport of fatty acids to heart and skeletal muscle for oxidation in the mitochondria (Bonen et al. 2006). Compared to vehicle, intra‐VMH melanocortin receptor activation induced a higher expression of PPARs and coactivator PGC‐1α, which are involved in the regulation of metabolic pathways including lipid metabolism and mitochondrial biogenesis (Evans et al. 2004; Finck & Kelly, 2006). In metabolically active tissues such as muscle, liver and BAT, activation of PPARs leads to increased β‐oxidation of fatty acids by switching muscle fibres to be oxidative, as well as to adaptive thermogenesis (Wang et al. 2003; Wang et al. 2004). The expression and activity of these PPAR and coactivators such as PGC‐1α depends on the energy demands of the cell, thereby affecting substrate uptake and utilization (Finck & Kelly, 2006). We found an increase in PPARs and PGC‐1α in BAT and skeletal muscle after intra‐VMH melanocortin receptor activation, which probably contributed to the increased fatty acid oxidation as indicated by the low RER (Fig. 1 C and G). This is consistent with previous studies on PGC‐1α altered tissue fuel selection (Finck & Kelly, 2006).
Although only trends were seen in most proteins discussed above (Table 3), intra‐VMH melanocortin receptor activation increased the expression of pAMPK and pACC in most of the tissues analysed (Fig. 5 F and J). In skeletal muscle, the degree of activation of AMPK is dependent on the metabolic stress caused by contraction. Under basal conditions, leptin or adiponectin increases the expression of pAMPK in skeletal muscle and liver by increasing sympathetic drive to these tissues (Minokoshi et al. 2002; Tomas et al. 2002; Yamauchi et al. 2002). This increases EE in muscle by increasing fatty acid oxidation and up‐regulating mitochondrial biogenesis (Minokoshi et al. 2002). pAMPK also inhibits ACC by phosphorylation (pACC), thereby inhibiting fatty acid synthesis (Corton et al. 1995; Henin et al. 1995). MTII‐induced activation of pAMPK would increase pACC, lowering the malonyl‐CoA concentration. This in turn relieves the myocyte of inhibition for the uptake of fatty acids into mitochondria via the carnitine carrier system, thus stimulating fatty acid oxidation and also up‐regulating mitochondrial biogenesis (Merrill et al. 1997; Winder et al. 2000).
The data reported in the present study demonstrate that melanocortins act in the VMH to activate SNS outflow, increasing sympathetic drive to skeletal muscle, and modulating fuel allocation and use, potentially through the differential activation of molecular mediators of energy homeostasis. One or more of these molecular pathways probably increases EE and lowers fuel economy during activity, dissipating wasted calories as heat at the same time as increasing free fatty acid uptake for use as fuel. Even a small change in thermogenesis or EE at the level of skeletal muscle, which makes up to 40% of total body mass (Zurlo F et al. 1990) and is major consumer of fuels (glucose and fatty acids) during acute physiological demand (Maurya et al. 2015), can have a major impact on energy homeostasis. Altogether, these results support the role of melanocortin receptors in the VMH in the regulation of peripheral metabolism, particularly in tissues such as BAT and skeletal muscle, via increased sympathetic drive. Taken together, this suggests that the VMH is important for the regulation of peripheral metabolism, and that tissues such as skeletal muscle are a good target system for changing EE in the treatment of individuals who are overweight and obese.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed in the laboratory of CMN at Kent state University. GCK, CMN and WCJ were involved in the conception and design of the experiments, data collection, assembly, analysis and interpretation of data. GCK, CMN and WCJ drafted the manuscript. GCK and CMN revised the manuscript. All authors agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by National Institutes of Health (NIH) grants NIH R01NS055859 and NIH R15DK097644, as well as American Heart Association grant 12GRNT12050566 to CMN.
Acknowledgements
We thank Lydia Heemstra for organizing personnel and support and the staff of the animal care and housing at Kent State University for their support in taking care of the animals.
Linked articles This article is highlighted by a Perspective by Dunn & Mittendorfer. To read this Perspective, visit http://dx.doi.org/10.1113/JP272701.
This is an Editor's Choice article from the 15 September 2016 issue.
References
- Abreu‐Vieira G, Xiao C, Gavrilova O & Reitman ML (2015). Integration of body temperature into the analysis of energy expenditure in the mouse. Mol Metab 4, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Akira & Shimazu Takashi (1981). Hypothalamic regulation of lipid metabolism in the rat: effect of hypothalamic stimulation on lipolysis. J Auton Nerv Syst 4, 195–205. [DOI] [PubMed] [Google Scholar]
- Anand BK & Brobeck JR (1951). Localization of a “feeding center” in the hypothalamus of the rat. Proc Soc Exp Biol Med 77, 323–324. [DOI] [PubMed] [Google Scholar]
- Belgardt BF & Bruning JC (2010). CNS leptin and insulin action in the control of energy homeostasis. Ann NY Acad Sci 1212, 97–113. [DOI] [PubMed] [Google Scholar]
- Bonen A, Tandon NN, Glatz JF, Luiken JJ & Heigenhauser GJ (2006). The fatty acid transporter FAT/CD36 is upregulated in subcutaneous and visceral adipose tissues in human obesity and type 2 diabetes. Int J Obes (Lond) 30, 877–883. [DOI] [PubMed] [Google Scholar]
- Borg MA, Borg WP, Tamborlane WV, Brines ML, Shulman GI & Sherwin RS (1999). Chronic hypoglycemia and diabetes impair counterregulation induced by localized 2‐ deoxy‐glucose perfusion of the ventromedial hypothalamus in rats. Diabetes 48, 584–587. [DOI] [PubMed] [Google Scholar]
- Braun TP & Marks DL (2011). Hypothalamic regulation of muscle metabolism. Curr Opin Clin Nutr Metab Care 14, 237–242. [DOI] [PubMed] [Google Scholar]
- Brito MN, Brito NA, Baro DJ, Song CK & Bartness TJ (2007). Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology 148, 5339–5347. [DOI] [PubMed] [Google Scholar]
- Burke PG, Neale J, Korim WS, McMullan S & Goodchild AK (2011). Patterning of somatosympathetic reflexes reveals nonuniform organization of presympathetic drive from C1 and non‐C1 RVLM neurons. Am J Physiol Regul Integr Comp Physiol 301, R1112–R1122. [DOI] [PubMed] [Google Scholar]
- Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M & Cone RD (2001). Melanocortin‐4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci 4, 605–611. [DOI] [PubMed] [Google Scholar]
- Butler AA (2006). The melanocortin system and energy balance. Peptides 27, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchi S, Stutzmann F, Cavalcanti‐Proenca C, Durand E, Pouta A, Hartikainen AL, Marre M, Vol S, Tammelin T, Laitinen J, Gonzalez‐Izquierdo A, Blakemore AI, Elliott P, Meyre D, Balkau B, Jarvelin MR & Froguel P (2009). Combined effects of MC4R and FTO common genetic variants on obesity in European general populations. J Mol Med 87, 537–546. [DOI] [PubMed] [Google Scholar]
- Chagnon YC, Chen WJ, Perusse L, Chagnon M, Nadeau A, Wilkison WO & Bouchard C (1997). Linkage and association studies between the melanocortin receptors 4 and 5 genes and obesity‐related phenotypes in the Quebec Family Study. Mol Med 3, 663–673. [PMC free article] [PubMed] [Google Scholar]
- Chari M, Lam CKL & Lam TKT (2010). Hypothalamic Fatty Acid Sensing in the Normal and Disease States, Chapter 20. NCBI Bookshelf. Bookshelf ID: NBK53535 PMID: 21452468. [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal‐Truter S, MacIntyre DE, Chen HY & Van der Ploeg LH (2000). Inactivation of the mouse melanocortin‐3 receptor results in increased fatmass and reduced lean body mass. Nat Genet 26, 97–102. [DOI] [PubMed] [Google Scholar]
- Contreras C, Gonzalez F, Fernø J, Diéguez C, Rahmouni K, Nogueiras R & López M (2015). The brain and brown fat. Ann Med 47, 150–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA & Hardie DG (1995). 5‐aminoimidazole‐4‐carboxamide ribonucleoside. A specific method for activating AMP‐activated protein kinase in intact cells? Eur J Biochem 229, 558–565. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF & Saper CB (1999). From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 22, 221–232. [DOI] [PubMed] [Google Scholar]
- Evans RM, Barish GD & Wang YX (2004). PPARs and the complex journey to obesity. Nat Med 10, 355–361. [DOI] [PubMed] [Google Scholar]
- Finck BN & Kelly DP (2006). PGC‐1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisler JS & Warden CH (2006). Uncoupling proteins, dietary fat and the metabolic syndrome. Nutr Metab (Lond) 3, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield AS, Lam DD, Marston OJ, Przydzial MJ & Heisler LK (2009). Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol Metab 20, 203–15. [DOI] [PubMed] [Google Scholar]
- Gavini CK, Mukherjee S, Shukla C, Britton SL, Koch, LG , Shi H & Novak CM (2014). Leanness and heightened non‐resting energy expenditure: role of skeletal muscle activity thermogenesis. Am J Physiol Endocrinol Metab 306, E635–E647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith R, Joanisse DR, Gallagher D, Pavlovich K, Shamoon E, Leibel RL & Rosenbaum M (2010). Effects of experimental weight perturbation on skeletal muscle work efficiency, fuel utilization, and biochemistry in human subjects. Am J Physiol Regul Integr Comp Physiol 298, R79–R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill HJ (2006). Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity (Silver Spring) 5(Suppl), 216S–221S. [DOI] [PubMed] [Google Scholar]
- Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G & Stec DE (2010). Obesity‐induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 285, 17271–17276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrold JA, Widdowson PS & Williams G (1999). Altered energy balance causes selective changes in melanocortin‐4(MC4‐R), but not melanocortin‐3 (MC3‐R), receptors in specific hypothalamic regions: further evidence that activation of MC4‐R is a physiological inhibitor of feeding. Diabetes 48, 267–271. [DOI] [PubMed] [Google Scholar]
- Heeren J & Münzberg H (2013). Novel aspects of brown adipose tissue biology. Endocrinol Metab Clin 42, 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henin N, Vincent MF, Gruber HE & Van den Berghe G (1995). Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP‐activated protein kinase. FASEB J 9, 541–546. [DOI] [PubMed] [Google Scholar]
- Hetherington AW & Ranson SW (1940). Hypothalamic lesions and adiposity in the rat. Anat Rec 78, 149–172. [Google Scholar]
- Huszar D, Lynch CA, Fairchild‐Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P & Lee F (1997). Targeted disruption of the melanocortin‐4 receptor results in obesity in mice. Cell 88, 131–141. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Cortez V, Kennedy SL, Foley TE, Hanson H 3rd & Fleshner M (2008). Role of central beta‐adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav Immun 22, 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Zhao L, Donato J Jr, Kohno D, Xu Y, Elias CF, Lee C, Parker KL & Elmquist JK (2011). Steroidogenic factor 1 directs programs regulating diet‐induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci USA 108, 10673–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BM (2006). The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Beh 87, 221–244. [DOI] [PubMed] [Google Scholar]
- Korim WS, McMullan S, Cravo SL & Pilowsky PM (2011). Asymmetrical changes in lumbar sympathetic nerve activity following stimulation of the sciatic nerve in rat. Brain Res 1391, 60–70. [DOI] [PubMed] [Google Scholar]
- Labbé SM, Caron A, Lanfray D, Monge‐Rofarello B, Bartness TJ & Richard D (2015). Hypothalamic control of brown adipose tissue thermogenesis. Front Syst Neurosci 9, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Jung YS & Choi D (2014). Recent advance in brown adipose physiology and its therapeutic potential. Exp Mol Med 46, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg D, Chen P & Li C (2013). Conditional viral tracing reveals that steroidogenic factor 1‐positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J Comp Neurol 521, 3167–3190. [DOI] [PubMed] [Google Scholar]
- Loos RJ, Rankinen T, Tremblay A, Perusse L, Chagnon Y & Bouchard C (2005). Melanocortin‐4 receptor gene and physical activity in the Quebec Family Study. Int J Obes (Lond) 29, 420–428. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR & Jaswal JS (2010). Targeting intermediary metabolism in the hypothalamus as a mechanism to regulate appetite. Pharmacol Rev 62, 237–264. [DOI] [PubMed] [Google Scholar]
- Maurya SK, Bal NC, Sopariwala DH, Pant M, Rowland LA, Shaikh SA & Periasamy M (2015). Sarcolipin is a key determinant of the basal metabolic rate, and its overexpression enhances energy expenditure and resistance against diet‐induced obesity. J Biol Chem 290, 10840–10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG & Winder WW (1997). AICA riboside increases AMP‐activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273, E1107–E1112. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D & Kahn BB (2002). Leptin stimulates fatty‐acid oxidation by activating AMP‐activated protein kinase. Nature 415, 339–343. [DOI] [PubMed] [Google Scholar]
- Miyaki T, Fujikawa T, Kitaoka R, Hirano N, Matsumura S, Fushiki T & Inoue K (2011). Noradrenergic projections to the ventromedial hypothalamus regulate fat metabolism during endurance exercise. Neuroscience. 190, 239–250. [DOI] [PubMed] [Google Scholar]
- Mul JD, van Boxtel R, Bergen DJ, Brans MA, Brakkee JH, Toonen PW, Garner KM, Adan RA & Cuppen E (2012). Melanocortin receptor 4 deficiency affects body weight regulation, grooming behavior, and substrate preference in the rat. Obesity 20, 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueiras R, Wiedmer P, Perez‐Tilve D, Veyrat‐Durebex C, Keogh JM, Sutton GM, Pfluger PT, Castaneda TR, Neschen S, Hofmann SM, Howles PN, Morgan DA, Benoit SC, Szanto I, Schrott B, Schurmann A, Joost HG, Hammond C, Hui DY, Woods SC, Rahmouni K, Butler AA, Farooqi IS, O'Rahilly S, Rohner‐Jeanrenaud F & Tschop MH (2007). The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest 117, 3475–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak CM, Escande C, Gerber SM, Chini EN, Zhang M, Britton SL, Koch LG & Levine JA (2009). Endurance capacity, not body size, determines physical activity levels: role of skeletal muscle PEPCK. PLoS ONE 4, e5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obici S, Feng Z, Tan J, Liu L, Karkanias G & Rossetti L (2001). Central melanocortin receptors regulate insulin action. J Clin Invest 108, 1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco‐Solis R, Aguilar‐Arnal L, Murakami M, Peruquetti R, Ramadori G, Coppari R & Sassone‐Corsi P (2016). The circadian clock in the ventromedial hypothalamus controls cyclic energy expenditure. Cell Metab 23, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otter T, King SM & Witman GB (1987). A two‐step procedure for efficient electrotransfer of both high‐molecular‐weight (>400,000) and low‐molecular‐weight (<20,000) proteins. Anal Biochem 162, 370–377. [DOI] [PubMed] [Google Scholar]
- Paxinos G & Watson C (2006). The Rat Brain in Stereotaxic Coordinates, 6th edn. Academic Press, San Diego, CA. [Google Scholar]
- Periasamy M & Kalyanasundaram A (2007). SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35, 430–442. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Haynes WG, Morgan DA & Mark AL (2003). Role of melanocortin‐4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci 23, 5998–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razquin C, Marti A & Martinez JA (2011). Evidences on three relevant obesogenes: MC4R, FTO and PPARgamma. Approaches for personalized nutrition. Mol Nutr Food Res 55, 136–149. [DOI] [PubMed] [Google Scholar]
- Rosenbaum M, Vandenborne K, Goldsmith R, Simoneau JA, Heymsfield S, Joanisse DR, Hirsch J, Murphy E, Matthews D, Segal KR & Leibel RL (2003). Effects of weight change on skeletal muscle work efficiency in human subjects. Am J Physiol Regul Integr Comp Physiol 285, R183–R192. [DOI] [PubMed] [Google Scholar]
- Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, Gallagher D, Mayer L, Murphy E & Leibel RL ( 2005. ) Low‐dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest 115, 3579–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB & Elmquist JK (2011). Melanocortin‐4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini F, Maffei M, Pelosini C, Salvetti G, Scartabelli G & Pinchera A (2009). Melanocortin‐4 receptor mutations in obesity. Adv Clin Chem 48, 95–109. (PMID: 19803416). [PubMed] [Google Scholar]
- Schneeberger M, Gomis R & Claret M (2014). Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J Endocrinol 220, T25–T46. [DOI] [PubMed] [Google Scholar]
- Shi H, Bowers RR & Bartness TJ (2004). Norepinephrine turnover in brown and white adipose tissue after partial lipectomy. Physiol Behav 81, 535–42. [DOI] [PubMed] [Google Scholar]
- Shiuchi T, Haque MS, Okamoto S, Inoue T, Kageyama H, Lee S, Toda C, Suzuki A, Bachman ES, Kim YB, Sakurai T, Yanagisawa M, Shioda S, Imoto K & Minokoshi Y (2009). Hypothalamic orexin stimulates feeding‐associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab 10, 466–480. [DOI] [PubMed] [Google Scholar]
- Shrestha YB, Vaughan CH, Smith BJ, Jr. , Song CK, Baro DJ & Bartness TJ (2010). Central melanocortin stimulation increases phosphorylated perilipin A and hormone‐sensitive lipase in adipose tissues. Am J Physiol Regul Integr Comp Physiol 299, R140–R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla C, Britton SL, Koch LG & Novak CM (2012). Region‐specific differences in brain melanocortin receptors in rats of the lean phenotype. Neuroreport 23, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, Balthasar N, Williams KW & Elmquist JK (2013). Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons. Cell 152, 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Ludgate M & Rees DA (2011). Brown fat and obesity: the next big thing? Clin Endocrinol (Oxf) 74, 661–670. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Masuzaki H, Yasue S, Ebihara K, Shiuchi T, Ishii T, Arai N, Hirata M, Yamamoto H, Hayashi T, Hosoda K, Minokoshi Y & Nakao K (2007). Central melanocortin signaling restores skeletal muscle AMP‐activated protein kinase phosphorylation in mice fed a high‐fat diet. Cell Metab 5, 395–402. [DOI] [PubMed] [Google Scholar]
- Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW & Diano S (2016). UCP2 regulates mitochondrial fission and ventromedial nucleus control of glcose responsiveness. Cell 164, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda C, Shiuchi T, Kageyama H, Okamoto S, Coutinho EA, Sato T, Okamatsu‐Ogura Y, Yokota S, Takagi K, Tang L, Saito K, Shioda S & Minokoshi Y (2013). Extracellular signal‐regulated kinase in the ventromedial hypothalamus mediates leptin‐induced glucose uptake in red‐type skeletal muscle. Diabetes 62, 2295–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda C, Shiuchi T, Lee S, Yamato‐Esaki M, Fujino Y, Suzuki A, Okamoto S & Minokoshi Y (2009). Distinct effects of leptin and a melanocortin receptor agonist injected into medial hypothalamic nuclei on glucose uptake in peripheral tissues. Diabetes 58, 2757–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas E, Tsao TS, Saha AK, Murrey HE, Cc Zhang Cc, Itani SI, Lodish HF & Ruderman NB (2002). Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl‐CoA carboxylase inhibition and AMP‐activated protein kinase activation. Proc Natl Acad Sci USA 99, 16309–16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp MH, Speakman JR, Arch JR, Auwerx J, Brüning JC, Chan L, Eckel RH, Farese RV Jr, Galgani JE, Hambly C, Herman MA, Horvath TL, Kahn BB, Kozma SC, Maratos‐Flier E, Müller TD, Münzberg H, Pfluger PT, Plum L, Reitman ML, Rahmouni K, Shulman GI, Thomas G, Kahn CR & Ravussin E (2012). A guide to analysis of mouse energy metabolism. Nat Methods 9, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupone D, Madden CJ & Morrison SF (2014). Autonomic regulation of brown adipose tissue thermogenesis in health and disease: potential clinical applications for altering BAT thermogenesis. Front Neurosci 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CH, Shrestha YB & Bartness TJ (2011). Characterization of a novel melanocortin receptor‐containing node in the SNS outflow circuitry to brown adipose tissue involved in thermogenesis. Brain Res 1411, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CH, Zarebidaki E, Ehlen JC & Bartness TJ (2014). Analysis and measurement of the sympathetic and sensory innervation of white and brown adipose tissue. Methods Enzymol 537, 199–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss‐Andreae A, Murphy JG, Ellacott KL, Stuart RC, Nillni EA, Cone RD & Fan W (2006). Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology 148, 1550–1560. [DOI] [PubMed] [Google Scholar]
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H & Evans RM (2003). Peroxisome‐proliferator‐activated receptor delta activates fat metabolism to prevent obesity. Cell 113, 159–170. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga‐Ocampo CR, Ham J, Kang H & Evans RM (2004). Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2, e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M & Holloszy JO (2000). Activation of AMP‐activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol (1985) 88, 2219–2226. [DOI] [PubMed] [Google Scholar]
- Xu Y, Elmquist JK & Fukuda M (2011). Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann NY Acad Sci 1243, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yako YY, Hassan MS, Erasmus RT, van der Merwe L, Janse van Rensburg S & Matsha TE (2012). Associations of MC3R polymorphisms with physical activity in South African adolescents. J Phys Act Health 10, 813–825. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB & Kadowaki T (2002). Adiponectin stimulates glucose utilization and fatty‐acid oxidation by activating AMP‐activated protein kinase. Nat Med 8, 1288–1295. [DOI] [PubMed] [Google Scholar]
- Yi CX, Kalsbeek A & Tschop MH (2011). Autonomic MC sets the metabolic tone. Cell Metab. 13, 121–123. [DOI] [PubMed] [Google Scholar]
- Yoo Y, LaPradd M, Kline H, Zaretskaia MV, Behrouzvaziri A, Rusyniak DE, Molkov YI & Zaretsky DV (2015). Exercise activates compensatory thermoregulatory reaction in rats: a modeling study. J Appl Physiol 119, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurlo F, Larson K, Bogardus C & Ravussin E (1990). Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest 86, 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]