

Abstract
The production of reactive oxygen/nitrogen species (ROS/RNS) is generally considered to increase during physical exercise. Nevertheless, direct measurements of ROS/RNS often show modest increases in ROS/RNS in muscle fibres even during intensive fatiguing stimulation, and the major source(s) of ROS/RNS during exercise is still being debated. In rested muscle fibres, mild and acute exposure to exogenous ROS/RNS generally increases myofibrillar submaximal force, whereas stronger or prolonged exposure has the opposite effect. Endogenous production of ROS/RNS seems to preferentially decrease submaximal force and positive effects of antioxidants are mainly observed during fatigue induced by submaximal contractions. Fatigued muscle fibres frequently enter a prolonged state of reduced submaximal force, which is caused by a ROS/RNS‐dependent decrease in sarcoplasmic reticulum Ca2+ release and/or myofibrillar Ca2+ sensitivity. Increased ROS/RNS production during exercise can also be beneficial and recent human and animal studies show that antioxidant supplementation can hamper the beneficial effects of endurance training. In conclusion, increased ROS/RNS production have both beneficial and detrimental effects on skeletal muscle function and the outcome depends on a combination of factors: the type of ROS/RNS; the magnitude, duration and location of ROS/RNS production; and the defence systems, including both endogenous and exogenous antioxidants.
Abbreviations
- [Ca2+]i
free cytosolic Ca2+ concentration
- DTT
dithiothreitol
- eNOS
endothelial NO• synthase
- FDB
flexor digitorum brevis
- GSH
reduced glutathione
- iNOS
inducible NO• synthase
- NAC
N‐acetylcysteine
- NOS
NO• synthase
- nNOS
neuronal NO• synthase
- NOX
NADPH oxidase
- PLFFD
prolonged low‐frequency force depression
- RNS
reactive nitrogen species
- roGFP
redox‐sensitive green fluorescent protein
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SOD
superoxide dismutase
- SR
sarcoplasmic reticulum
- t‐BOOH
tert‐butyl hydroperoxide
- Tn
troponin
Introduction
In a classical study, Reid et al. showed that the general antioxidant N‐acetylcysteine (NAC) mitigated the force decline when human subjects performed repeated submaximal contractions (Reid et al. 1994). These results imply that the production of reactive oxygen/nitrogen species (ROS/RNS) increases in skeletal muscle during physical exercise and ameliorating the resulting ‘oxidative stress’ with antioxidants lessens the force decrease. Moreover, several studies show increases in ROS/RNS in conditions with skeletal muscle dysfunction and muscle weakness, such as rheumatoid arthritis (Yamada et al. 2015 a,b), Duchenne muscle dystrophy (Khairallah et al. 2012), malignant hyperthermia (Lanner et al. 2012), and in normal ageing (Andersson et al. 2011). On the other hand, submaximal force in intact and skinned fast‐twitch skeletal muscle fibres has been shown to increase with acute exposure to the ROS hydrogen peroxide (H2O2) (Andrade et al. 1998 a, 2001; Murphy et al. 2008; Mollica et al. 2012). Furthermore, recent human and animal studies show that treatment with antioxidants hampers the beneficial effects of endurance training (Gomez‐Cabrera et al. 2008; Ristow et al. 2009; Paulsen et al. 2014). Thus, increases in ROS/RNS can have both beneficial and detrimental effects on skeletal muscle contractile function and fitness, and the outcome probably depends on a combination of factors: the type of ROS/RNS, the size of ROS/RNS increase, the duration of ROS/RNS elevation (e.g. milliseconds vs. hours), and the localization of ROS/RNS production and accumulation (Droge, 2002; Westerblad & Allen, 2011; Ristow, 2014). In the first part of this review, we will discuss the metabolism and sources of ROS/RNS that are likely to increase during exercise and ways to measure them. In the latter part, we will discuss ROS/RNS effects on muscle fibre contractility during exercise (i.e. during induction of fatigue) and in the subsequent recovery phase.
ROS/RNS in contracting muscle: metabolism, sources and methods to measure
A free radical is an atom, molecule or ion with unpaired valency electron(s), generally making them highly unstable and reactive (Halliwell & Gutteridge, 1984). The dominant ROS in cells are superoxide (O2 •−) and its downstream derivatives, such as H2O2. Similarly, the central RNS in cells are nitric oxide (NO•) along with its downstream derivatives, such as peroxynitrite (ONOO•−). ROS/RNS production in skeletal muscle is generally considered to increase during physical exercise (Powers & Jackson, 2008). However, the direct mechanisms and sources of the increased ROS/RNS production during exercise remain uncertain and they are likely to differ depending on the type of activity, e.g. endurance vs. resistance training or short‐term high‐intensity vs. prolonged low‐intensity exercise. Moreover, direct measurements of ROS/RNS during exercise are relatively rare and the increases detected with conventional fluorescent indicators are often rather modest (e.g. Reid et al. 1992; Pye et al. 2007; Sakellariou et al. 2013; Cheng et al. 2015), which implies that such measurements are methodologically problematic.
Superoxide
O2 •− is generated through either incomplete reduction of oxygen in the mitochondrial electron transport chain or as a specific product of enzymatic reactions. O2 •− is negatively charged and hence relatively impermeable to cell membranes. It has a relatively long half‐life (∼1 μs), which permits diffusion within the muscle cell and allows interaction with several cellular targets (Winkler et al. 1999). Calculations in endothelial cells indicate that the steady‐state cellular concentration of O2 •− is in the pico‐ to nanomolar range (Carballal et al. 2014).
Complexes I and III are the two major sites of O2 •− production in the mitochondria (Cadenas et al. 1977; Turrens & Boveris, 1980; Murphy, 2009; Tahara et al. 2009; Quinlan et al. 2012). Early reports suggested that 2–5% of the total oxygen consumed by mitochondria was reduced to O2 •− (Boveris & Chance, 1973; Loschen et al. 1974). This measure of O2 •− production seems far too high in contracting skeletal muscle fibres, because oxygen consumption increases massively during intense exercise and prolonged exercise would then result in dangerously high ROS levels, severe oxidative stress, and muscle damage (Westerblad & Allen, 2011). Accordingly, more recent data indicate that only ∼0.1–0.2% of the oxygen consumed by the mitochondria forms O2 •− (St‐Pierre et al. 2002; Murphy, 2009; Tahara et al. 2009; Brand, 2010). The extent of mitochondrial O2 •− production in muscle also critically depends on other factors, such as sub‐ vs. supramaximal exercise intensity or sufficient vs. limited O2 delivery. For instance, mitochondrial ROS production appears to be higher during state 4 (basal) than during state 3 (maximal ADP‐stimulated) respiration, and the latter dominates during aerobic exercise (Powers & Jackson, 2008). Thus, only a very small fraction of the oxygen consumed by the mitochondria during exercise is reduced to O2 •− and there is no fixed relation between the rates of mitochondrial oxygen consumption and O2 •− production.
Enzymes that produce O2 •− in skeletal muscle include NADPH oxidase (NOX) (Pal et al. 2013), phospholipase A2 (Nethery et al. 1999), xanthine oxidase (Gomez‐Cabrera et al. 2010) and uncoupled NO• synthase (NOS) (Stuehr et al. 2001). Of these enzymes, NOX has received most recent attention and is proposed to be a major contributor to O2 •− production in skeletal muscle both at rest and during contractile activities (Michaelson et al. 2010; Pal et al. 2013; Sakellariou et al. 2013). A skeletal muscle fibre expresses two NOX isoforms, NOX2 and NOX4, and of these NOX2 has received most attention (Sakellariou et al. 2014). NOX2 has several subunits and is localized in the sarcolemma, either at the surface or in the t‐tubular system (Javeshghani et al. 2002; Hidalgo et al. 2006; Sakellariou et al. 2014). NOX4 is less studied in skeletal muscle. It has been proposed to be localized in the sarcoplasmic reticulum (SR) where it may affect the Ca2+ release channel (ryanodine receptor 1; RyR1) (Sun et al. 2011; Sakellariou et al. 2014).
For decades, mitochondria have been considered as the major site for O2 •− production in contracting skeletal muscle. Accordingly, a recent study using mouse flexor digitorum brevis (FDB) fibres transfected with a novel mitochondrial‐targeted superoxide biosensor (mt‐cpYFP) shows strictly localized mitochondrial O2 •− production during repetitive contractions (Wei et al. 2011). Moreover, pretreatment with a mitochondrial‐targeted antioxidant (SS‐31) decreased ROS production, as measured with the fluorescent ROS indicator MitoSOX Red, in isolated mouse muscle fibres during repeated tetanic contractions (Cheng et al. 2015). Conversely, recent studies propose NOX2 as the main producer of ROS in contracting skeletal muscles, because the contraction‐mediated increase in cytosolic ROS was prevented by pharmacological inhibition or genetic knockdown of NOX2 (Michaelson et al. 2010; Pal et al. 2013; Sakellariou et al. 2013). Thus, there are conflicting results regarding the importance of different sources of O2 •− in contracting muscle and further studies are required to resolve this issue.
Hydrogen peroxide
Dismutation of O2 •−, both spontaneous and catalysed by superoxide dismutase (SOD), constitutes the major source of H2O2 in muscle cells (2O2 •− + 2H+ → H2O2 + O2). Two out of three SOD isoforms are highly abundant (∼10–20 μm) within the skeletal muscle fibres: SOD1 requires copper–zinc as a cofactor and is located in the cytosol and in the mitochondrial intermembrane space; SOD2 uses manganese as a cofactor and is located in the mitochondrial matrix (Powers & Jackson, 2008). Of the total SOD activity in skeletal muscle fibres, ∼15–35% is in the mitochondria and the remaining 65–85% in the cytosol. The highest SOD activities are present in oxidative slow‐twitch muscle fibres and endurance exercise increases SOD activity in both slow‐ and fast‐twitch fibres (Higuchi et al. 1985; Powers et al. 1994; Oh‐ishi et al. 1997; Bruton et al. 2008). H2O2 is cell permeable and relatively stable with a half‐life of seconds to minutes. The concentration of H2O2 in skeletal muscle has been calculated to be ∼10–100 nm at rest and to increase to ∼100–200 nm during contractions (Vasilaki et al. 2006). In comparison to other ROS, H2O2 is a relatively weak oxidizing agent, but in the presence of Fe2+ it can be converted into the highly reactive and cytotoxic hydroxyl radical (OH•) (Halliwell & Gutteridge, 1984; Powers & Jackson, 2008). In skeletal muscle, H2O2 is metabolized to H2O via three major antioxidant enzymatic systems: glutathione peroxidases (H2O2 + 2GSH → 2H2O + GSSG), where GSH is reduced glutathione and GSSG is oxidized glutathione; catalase (2H2O2 → 2H2O + O2); and peroxiredoxins (PRX(reduced) + H2O2 → PRX(oxidized) + 2H2O) (Sakellariou et al. 2014).
Methods to measure ROS
Quantitative measurements of ROS in skeletal muscle are rare and this is to a large extent due to the fact that methods available to measure O2 •− and H2O2 production have severe shortcomings. Many studies of mitochondrial ROS production are performed with in vitro measurements on isolated mitochondria. This approach is problematic because the techniques used to isolate mitochondria from muscle alter mitochondrial structure and function by, for instance, causing fragmentation, loss of soluble matrix enzymes and altered respiratory rates and O2 •−/H2O2 production (Schwerzmann et al. 1989; Picard et al. 2011 a,b). Fluorescent ROS indicators (e.g. dichlorofluorescein, dihydroethidium, MitoSOX Red) commonly used to assess changes in ROS in intact muscle fibres have limitations in that they, for instance, are not specific to a certain ROS (Kalyanaraman et al. 2012). Moreover, increases in the overall fluorescence of these indicators in response to repeated contractions are relatively small (e.g. Reid et al. 1992; Pye et al. 2007; Sakellariou et al. 2013; Cheng et al. 2015) and therefore it is difficult to assess contraction‐induced ROS changes in a quantitative and spatially/temporally confined manner. To deal with the limited specificity of these fluorescent ROS indicators, it has been suggested that they should be combined with HPLC‐based methods (Kalyanaraman et al. 2012), but such methods are cumbersome when following real‐time changes in ROS, e.g. during repeated contractions and in the following recovery period.
Promising new tools for dynamic and site‐specific assessment of ROS have recently been developed and these will hopefully lead to a better understanding of the role(s) of ROS in muscle biology. For instance, genetically engineered fusion of redox‐sensitive green fluorescent protein (roGFP) and the peroxide Orp1 has been used to measure the concentration of H2O2 (Gutscher et al. 2009). Furthermore, Rodney and co‐workers recently used roGFP fused to the regulatory subunit p47phox of NOX2 for real‐time ROS measurements in contracting mouse skeletal muscle fibres (Pal et al. 2013). Furthermore, roGFP‐based ROS indicators are relatively sensitive, reversible, ratiometric (i.e. independent of changes in indicator concentration) and pH insensitive (Hanson et al. 2004; Gutscher et al. 2009; Meyer & Dick, 2010; Pal et al. 2013). Thus, genetically engineered fusion proteins constitute a novel group of ROS indicators that allow more specific detection of one ROS and can be targeted to organelles or proteins (e.g. mitochondria or NOX2).
Nitric oxide
NO• is a versatile biological signalling molecule that is generated via enzymatic reactions of nitric oxide synthase (NOS) and the production increases in muscle fibres during repeated contractions (Pye et al. 2007; Cheng et al. 2015). NO• can also be formed from the inorganic anions nitrate (NO3 −) and nitrite (NO2 −) (Weitzberg et al. 2010). Skeletal muscle constitutively expresses neuronal and endothelial NOS (nNOS and eNOS, respectively), whereas inducible NOS (iNOS) is upregulated in response to acute inflammatory insults. Despite its name, nNOS is expressed at higher levels in human skeletal muscle than in human brain (Nakane et al. 1993). NO• is synthesized by NOS from l‐arginine, NADPH and O2. nNOS and eNOS are activated by increases in the free cytosolic Ca2+ concentration ([Ca2+]i) (Forstermann et al. 1994). The K m of [Ca2+]i for the activation of nNOS is ∼200 nm and [Ca2+]i reaches higher levels during contractions (>1 μm), hence nNOS will become highly active during contractile activities (Bredt & Snyder, 1990). Under normal conditions, nNOS is mostly compartmentalized to submembrane scaffolds that are part of the dystrophin glycoprotein complex (Brenman et al. 1995). In addition, minor fractions of nNOS are detectable in association with the SR and with mitochondria (Buchwalow et al. 2005). In line with this, data from our group and others show that some nNOS co‐localize with the RyR1 in skeletal muscle from mouse and human subjects (Salanova et al. 2008; Yamada et al. 2015 b). Interestingly, the amount of nNOS co‐localized with RyR1 was markedly increased in muscles from mice with collagen‐induced arthritis (Yamada et al. 2015 b).
NO• on its own is relatively stable but it reacts rapidly with numerous cellular targets, which results in a biological half‐life time of ∼0.3 s in skeletal muscle under physiological conditions (Thomas et al. 2001). Nevertheless, NO• is small, uncharged and freely diffusible through membranes and is therefore considered to exert effects over distances even exceeding 100 μm (Thomas et al. 2001). The concentration of NO• has been calculated to be ∼20 nm in resting rat diaphragm muscle fibres (Boczkowski et al. 1999).
Peroxynitrite
ONOO•−, formed when NO• reacts with O2 •−, is a potent oxidizing and nitrating agent able to react with a wide range of biomolecules. The biological half‐life under physiological conditions is ∼10 ms and it is estimated to influence cellular targets within ∼5–20 μm (Radi, 1998; Romero et al. 1999; Szabó et al. 2007; Carballal et al. 2014). Calculations in endothelial cells indicate that the steady‐state level of ONOO•− is ∼1 nm (Carballal et al. 2014).
The rate constant of ONOO•− formation has been estimated to be within the range of (4–16) × 109 m −1 s−1 (Goldstein & Czapski, 1995; Botti et al. 2010), which is higher than the rate of SOD conversion of O2 •− to H2O2 ((1–2) × 109 m −1 s−1) (Klug‐Roth et al. 1973; Hsu et al. 1996). Thus, when NO• is produced at a high rate, it will rapidly react with O2 •− to produce significant amounts of ONOO•− even in the presence of the high physiological concentrations of SOD (∼10–20 μm). In fact, the formation of ONOO•− from NO• and O2 •− can occur about six times faster than the rate at which SOD can convert O2 •− to H2O2 (Beckman & Koppenol, 1996). Peroxiredoxins represent an efficient detoxification system for ONOO•− and CO2 accounts for another fraction of the ONOO•− consumption by forming carbonate radicals and nitrogen dioxide (Carballal et al. 2014). On the other hand, GSH (present at mm concentration in muscles) does not react sufficiently fast in vivo to directly scavenge ONOO•−; instead, glutathione may counteract ONOO•−‐dependent processes via reactions with secondary radicals (Ferrer‐Sueta & Radi, 2009).
Acute effects of ROS/RNS on myofibrillar function in skeletal muscle
The intracellular events leading to contraction of skeletal muscle fibres start with RyR1‐mediated Ca2+ release from SR. Ca2+ then binds to the troponin (Tn) complex consisting of TnC, TnI and TnT. Ca2+ binding to the Tn–Ca2+ complex moves the position of the tropomyosin filaments, hence turning on cross‐bridge cycling and contraction by uncovering active sites of actin for myosin binding (Gordon et al. 2000). In addition, the degree of myofibrillar activation depends on the kinetics of cross‐bridge attachment and detachment to actin, because an actin‐bound cross‐bridge exerts mechanical impact on tropomyosin that facilitates the binding of neighbouring cross‐bridges (Brenner, 1988; Gordon et al. 2000). The result of these interacting processes is the steep, sigmoidal force–[Ca2+]i relationship. Changes in the Tn–Ca2+ interaction and/or cross‐bridge kinetics can affect the steepness of the force–[Ca2+]i relationship as well as the position on the [Ca2+]i axis; for simplicity, we will refer to alterations in the latter parameter as changes in myofibrillar Ca2+ sensitivity. Generally, acute ROS/RNS‐mediated changes in force generation are more marked with activation at submaximal than at maximal frequencies (Lamb & Westerblad, 2011). Submaximal contractions occur on the steep part of the force–[Ca2+]i relationship, which means that even small changes in myofibrillar Ca2+ sensitivity, or SR Ca2+ release, have large effects on the generated force (Fig. 1). In this context it is worth noting that everyday activities generally require low to moderate forces and the firing frequencies of motor units are therefore set to produce submaximal contractions (Marsden et al. 1971; Grimby & Hannerz, 1977)
Figure 1. Changes in myofibrillar Ca2+ sensitivity have a much larger effect on submaximal than on maximal force .
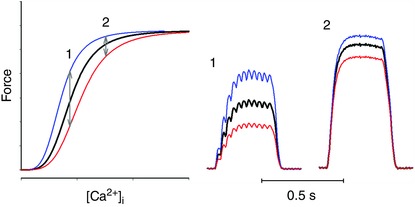
Schematic representation of the effect of increased (blue lines) and decreased (red lines) myofibrillar Ca2+ sensitivity. (1) Stimulation at frequencies giving unfused tetani results in forces on the steep part of the force–[Ca2+]i relationship and changes in sensitivity have a large effect (∼40% in the example) on force output. (2) Conversely, the same changes in sensitivity have little effect (here ∼10%) at higher stimulation frequencies and fused tetani. Similar changes occur with changes in tetanic [Ca2+]i, i.e. larger effects in unfused contractions.
Intact fast‐twitch mouse muscle fibres acutely exposed to the reducing agent dithiothreitol (DTT) showed decreased submaximal (30–60 Hz) force. Acute exposure to H2O2, or its non‐metabolizable analogue tert‐butyl hydroperoxide (t‐BOOH), has the opposite effect, that is, it increased submaximal force (Andrade et al. 1998 a, 2001; Cheng et al. 2015). The effect on [Ca2+]i during contractions was small both with DTT and H2O2/t‐BOOH exposure, hence the changes in force production were explained by altered myofibrillar Ca2+ sensitivity. The force‐potentiating effect of H2O2/t‐BOOH exposure was transient and prolonged (more than 5 min) exposures resulted in a marked decrease in submaximal force, again accompanied by only minor changes in tetanic [Ca2+]i (Andrade et al. 1998 a, 2001; Cheng et al. 2015) (Fig. 2 A). Intriguingly, the depression of submaximal force induced by prolonged exposure to H2O2 was reversed by exposure to DTT and, vice versa, the depression induced by initial exposure to DTT was reversed by H2O2 (Andrade et al. 1998 a) (Fig. 2 B). To sum up, myofibrillar Ca2+ sensitivity is highly susceptible to acute exogenous exposure to oxidizing and reducing agents, the effect is readily reversed by the opposite redox challenge, and rested muscle fibres appear to be in a suboptimally reduced state (Andrade et al. 1998 a; Lamb & Westerblad, 2011; Powers et al. 2011).
Figure 2. Transient and reversible effects of H2O2 on myofibrillar contractile function .
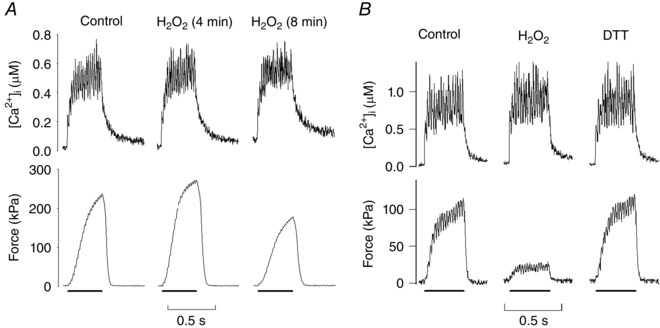
Original [Ca2+]i and force records from submaximal (50 Hz) contractions of single FDB muscle fibres exposed to H2O2 (300 μm) for up to 8 min (A) and H2O2 for 6 min followed by exposure to the reducing agent DTT (1 mm) for 10 min (B). Note that H2O2 causes major force changes whereas [Ca2+]i is little affected, which means that H2O2 mainly acts at the myofibrillar level. The effects on force are time dependent with brief H2O2 exposure resulting in increased submaximal force, which is followed by a progressive force decline (A). Furthermore, the force depression caused by prolonged H2O2 exposure is reversed by reduction with DTT (B). Conversely, prolonged exposure to DTT results in depressed submaximal force that can be reversed by application of H2O2 (not shown). Figure adapted from Andrade et al. (1998).
The effect of H2O2 application on the myofibrillar Ca2+ sensitivity of skinned fast‐twitch fibres differs markedly depending on the presence or absence of myoglobin and glutathione, which are normally present in intact skeletal muscle fibres (Murphy et al. 2008; Lamb & Westerblad, 2011). Thus, application of H2O2 on its own had little effect in skinned rat fast‐twitch fibres, whereas myofibrillar Ca2+ sensitivity was severely decreased in the presence of myoglobin. The proposed mechanism was that H2O2, through the Fenton reaction, interacts with Fe2+ on myoglobin to produce the highly reactive OH• (Murphy et al. 2008). On the other hand, application of H2O2 to skinned fast‐twitch fibres in the presence of myoglobin and glutathione resulted in an initial increase in myofibrillar Ca2+ sensitivity followed by a decrease (Murphy et al. 2008), i.e. a pattern very similar to that observed when intact fast‐twitch fibres are exposed to H2O2 or t‐BOOH (see Fig. 2). Subsequent experiments on skinned fast‐twitch rat and human muscle fibres revealed a likely mechanism for the initial increase in myofibrillar Ca2+ sensitivity: GSH reacts with oxidized cysteine residues on the fast isoform of TnI (TnIf; probably Cys133) and the resulting S‐glutathionylation increases myofibrillar Ca2+ sensitivity (Mollica et al. 2012). Accordingly, human muscle showed a marked increase in TnIf S‐glutathionylation following 40 min of low‐intensity cycling at ∼60% peak oxygen consumption (Mollica et al. 2012), and a recent study shows that repeated stimulation can increase myofibrillar Ca2+ sensitivity by S‐glutathionylation of TnIf in in situ experiments performed on rat gastrocnemius muscles (Watanabe et al. 2015). Notably, no corresponding increase in myofibrillar Ca2+ sensitivity was observed in mammalian slow‐twitch fibres, or in chicken and toad fibres, which have TnI isoforms that lack the equivalent of Cys133 (Mollica et al. 2012).
Intact fast‐twitch fibres showed several changes (albeit relatively minor, ∼5%) in cross‐bridge function in response to acute exposure to either H2O2 or t‐BOOH: decreased maximum shortening velocity, increased maximum force production (i.e. force at saturating [Ca2+]i), and increased rate of force redevelopment after shortening (Andrade et al. 2001). The same treatment resulted in increased myofibrillar Ca2+ sensitivity and the faster force redevelopment might contribute to this increase (Brenner, 1988). The concentration of peroxide used by Andrade et al. (1 μm) would be within or close to the physiological range and hence endogenous increases in H2O2 occurring during, for instance, high‐intensity exercise are likely to affect cross‐bridge function (Andrade et al. 2001).
What about the effects of acute exposure to ROS/RNS other than H2O2 on myofibrillar function? Myofibrillar Ca2+ sensitivity appears not to be affected by O2 •− (Bruton et al. 2008; Murphy et al. 2008). Acute exposure to NO• donors leads to decreased myofibrillar Ca2+ sensitivity (Andrade et al. 1998 b; Dutka et al. 2011). Studies on intact muscle fibres exposed to NO• donors indicate that the NO•‐induced decrease in Ca2+ sensitivity is not caused by altered cross‐bridge kinetics (Morrison et al. 1996; Andrade et al. 1998 b) and therefore it would be due to impaired Ca2+–Tn interaction. ONOO•− is known to decrease both myofibrillar Ca2+ sensitivity and maximum force in slow‐ and fast‐twitch muscles (Dutka et al. 2011).
Effects of ROS/RNS on contractile function during fatigue and recovery
Studies of human exercise show a clear positive effect of decreased ROS/RNS on endurance when fatigue is induced by submaximal contractions, whereas the effect is small or absent with maximal contractions (Reid et al. 1994; Powers et al. 2011). ‘Classical’ fatigue‐causing factors may dominate during the latter type of exercise and potential effects of ROS/RNS would therefore be difficult to discern (Allen et al. 2008). Such ‘classical’ factors that contribute to the force decrease in acute fatigue include accumulation of inorganic phosphate ions due breakdown of creatine phosphate (Dahlstedt et al. 2003), depletion of inter‐ and intramyofibrillar glycogen (Ørtenblad et al. 2013; Nielsen et al. 2014), and impaired action potential propagation (Pedersen et al. 2004; de Paoli et al. 2013). In a general sense, the positive effects of reducing ROS/RNS during fatigue with submaximal contractions fit with the fact that acute ROS/RNS effects are most marked on the steep part of the force–Ca2+ relationship (see above). However, acute exogenous application of H2O2 results in a transient increase in myofibrillar Ca2+ sensitivity (see Fig. 2) and some skinned fibre experiments show increased rather than decreased myofibrillar Ca2+ sensitivity after fatiguing contractions (Gejl et al. 2016; Watanabe et al. 2015). Thus, these results suggest that reducing ROS/RNS during fatigue would impair rather than improve performance. One tentative explanation for this apparent conflict is that exogenously applied and endogenously produced H2O2 have different effects. Another tentative explanation is that the effect of increases in ROS/RNS other than H2O2 dominates; for instance, both NO• and ONOO•− have been shown to decrease myofibrillar Ca2+ sensitivity (Andrade et al. 1998 b; Dutka et al. 2011). Furthermore, deleterious effects of oxidants may overpower any potentiating effects when the physical activity is prolonged and/or highly intense (Lamb & Westerblad, 2011).
In contrast to the situation during the actual induction of acute fatigue, obvious effects of increased ROS/RNS are seen during the subsequent recovery period (Westerblad & Allen, 2011). For instance, isolated mouse soleus fibres did not fatigue prematurely when exposed to severe oxidative stress (fatigued at 43°C in the presence of 10 μm H2O2 or t‐BOOH), but contractures developed and the fibres died ∼10 min after the end of stimulation (Place et al. 2009). Under less extreme conditions, fatigued muscle fibres frequently enter a prolonged state of severely depressed submaximal force, i.e. prolonged low‐frequency force depression (PLFFD) (Allen et al. 2008). At the muscle fibre level, depressed submaximal force can be due to decreased SR Ca2+ release and/or reduced myofibrillar Ca2+ sensitivity. Acutely fatigued fast‐twitch mouse FDB fibres displayed a marked PLFFD that is mainly caused by decreased SR Ca2+ release (Westerblad et al. 1993). Intriguingly, the cause of PLFFD changes towards reduced myofibrillar Ca2+ sensitivity in genetically modified mouse FDB fibres overexpressing the mitochondrial matrix redox enzyme SOD2 (Bruton et al. 2008), in rat FDB fibres that endogenously express more SOD2 (Bruton et al. 2008), and in mouse FDB fibres treated with the mitochondria‐targeted antioxidant SS‐31 or the NOS inhibitor l‐NAME (Cheng et al. 2015). In a recent study we show that PLFFD is accompanied by RyR1 fragmentation in vastus lateralis muscles of recreationally active human subjects after one session of high‐intensity interval training (6 × 30 s all‐out cycling) (Place et al. 2015). Conversely, when elite endurance athletes performed the same exercise, a similar PLFFD was observed but the RyR1 remained intact. The elite endurance athletes had higher levels of the antioxidant enzymes SOD2 and catalase in their muscles than recreationally active subjects. Moreover, a similar RyR1 fragmentation could be induced by high‐intensity stimulation of isolated mouse FDB muscles and this fragmentation was blocked by the antioxidant NAC (Place et al. 2015). Collectively, these results indicate that accumulation of mitochondrially generated O2 •−, or ONOO•−, preferentially affects SR Ca2+ release, probably via redox modifications of RyR1 (Bellinger et al. 2008 a,b; Andersson et al. 2011; Lanner et al. 2012). Conversely, when O2 •− is more efficiently metabolized, the resulting increase in H2O2, or downstream products, preferentially leads to changes in myofibrillar Ca2+ sensitivity. In other words, antioxidants do not prevent PLFFD, but they can change the underlying mechanism from impaired SR Ca2+ release to reduced myofibrillar Ca2+ sensitivity. The question is then: Does this matter? Our answer is: Yes, it probably does.
Impaired SR Ca2+ release caused by redox modifications of RyR1 is associated with increased SR Ca2+ leak at rest, and the resulting increase in resting [Ca2+]i may stimulate mitochondrial biogenesis and thereby improve muscle endurance (Wright et al. 2007; Bruton et al. 2010). On the other hand, major adaptations are unlikely to be triggered when PLFFD is caused by decreased myofibrillar Ca2+ sensitivity. Accordingly, prolonged changes in gene expression were recently shown after one session of high‐intensity interval training that induced PLFFD accompanied by ROS/RNS‐dependent RyR1 fragmentation in recreationally active subjects. Conversely, prolonged changes in mRNA levels were not observed in elite endurance athletes where PLFFD occurred while RyR1 remained intact (Place et al. 2015). Thus, ROS/RNS‐induced changes in RyR1 structure and function provide a mechanism as to why treatment with antioxidants hamper the beneficial effects of endurance training (Gomez‐Cabrera et al. 2008; Ristow et al. 2009; Paulsen et al. 2014).
A recent study from our laboratory highlights the complexity of fatigue‐induced redox effects (Cheng et al. 2015). Neither exposure to DTT nor t‐BOOH had any clear‐cut effect on the impaired SR Ca2+ release during PLFFD. Unexpectedly, application of DTT as well as t‐BOOH resulted in a major, but transient, increase in myofibrillar Ca2+ sensitivity during PLFFD. The effect of DTT during PLFFD was opposite to that in the rested state, where DTT decreased force production, which implies that redox‐sensitive sites were suboptimally reduced under resting conditions, became too oxidized during fatigue, and this was reversed by DTT. The effect of t‐BOOH was more intriguing since the results showed a transiently improved myofibrillar force production both in the unfatigued state and during PLFFD. Thus, some functionally important sites on myofibrillar proteins appeared to remain in a suboptimally reduced state during fatigue and the function of these was then temporarily improved by exogenous peroxide application (Fig. 3). While these intriguing findings illustrate the complexity of redox effects, further studies are clearly required before they can be understood at a more precise mechanistic level.
Figure 3. The prolonged low‐frequency force depression (PLFFD) after fatiguing stimulation is the result of complex ROS/RNS effects on SR Ca2+ release and myofibrillar Ca2+ sensitivity .
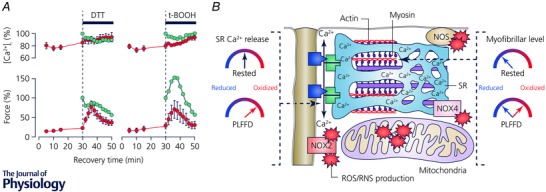
A, [Ca2+]i (upper panel) and force (lower panel; mean data ± SEM) in 30 Hz contractions produced during PLFFD (red circles) initially in standard Tyrode solution, followed by addition of DTT (1 mm) or the non‐metabolizable analogue of H2O2, t‐BOOH (10 μm). The effect of the same exposures on unfatigued fibres are also shown (green circles). B, simplified model of ROS/RNS effects on SR Ca2+ release and myofibrillar Ca2+ sensitivity. Key proteins for SR Ca2+ release are the t‐tubular voltage sensors, the dihydropyridine receptors (blue boxes), and the SR Ca2+ release channels, RyR1 (green boxes). These proteins appear to be in an optimal redox state at rest and become overly oxidized during fatiguing stimulation resulting in decreased [Ca2+]i, which is not affected by application of either t‐BOOH or DTT (see A). In the rested state, myofibrillar proteins are in a suboptimal reduced state. Some myofibrillar proteins become overly oxidized during induction of fatigue and the resulting force decrease is transiently counteracted by application of DTT. Intriguingly, other myofibrillar proteins apparently remain reduced during fatigue since application of the oxidizing agent t‐BOOH temporarily improves force generation, i.e. similar to the effect in the rested state (see A). Figure adapted from Cheng et al. (2015).
Conclusions
In this review we discuss intricate ROS/RNS effects on myofibrillar contractile function. While mechanisms underlying some changes in myofibrillar function are fairly well established, others are more uncertain. For instance, it is fairly well established that S‐glutathionylation of TnIf can cause an initial increase in myofibrillar Ca2+ sensitivity during exogenous H2O2 exposure, whereas the mechanism(s) behind the decreased sensitivity during prolonged exposure remains uncertain. Due to such uncertainties we find it premature to discuss complex changes in contractile function, e.g. mechanisms underlying PLFFD, in terms of one specific ROS/RNS acting on one specific molecular target. This type of knowledge would, for instance, require improved methods to measure changes in a specific ROS/RNS with good temporal and spatial resolution. Moreover, an obvious risk with reductionistic approaches to assess the complex ROS/RNS effects is that simplistic cellular or subcellular experiments are prioritized to get at the molecular mechanism. Our opinion is that such experiments often provide the correct answer to the wrong question.
In studies of ROS/RNS‐induced changes in myofibrillar function, we find it essential to measure both force and [Ca2+]i, since both are likely to be affected. Experiments on enzymatically isolated muscle fibres are technically much easier to perform than experiments on dissected fibres. However, enzymatically dissociated fibres lack tendons and force measurements become cumbersome. Intact whole muscles are also easier to use than dissected single muscle fibres, but [Ca2+]i is then difficult to measure, especially in deeper parts of the muscle. The mechanism of PLFFD shifts from decreased tetanic [Ca2+]i in wild‐type muscles to reduced myofibrillar Ca2+ sensitivity in SOD2‐overexpressing muscles (Bruton et al. 2008). Experiments with enzymatically dissociated fibres would detect decreased tetanic [Ca2+]i during recovery of wild‐type fibres, whereas [Ca2+]i returned to the pre‐fatigue level with SOD2 overexpression. Thus, the conclusion would be that increased SOD2 activity prevents PLFFD. Conversely, experiments with intact whole muscles would detect a similar force depression during recovery of wild‐type and SOD2‐overexpressing muscle, and the conclusion would be that SOD2 overexpression has no effect on PLFFD. Thus, the correct conclusion requires experiments with simultaneous measurements of force and [Ca2+]i in fully intact fibres, or the combination of experiments with enzymatically dissociated fibres and whole muscles.
A wide range of antioxidants, including gp91ds‐tat and SS‐31, could not prevent the loss of force observed after acute fatiguing stimulation of healthy mouse muscle fibres (Cheng et al. 2015). Moreover, improved antioxidant capacity in muscles of endurance athletes did not prevent the development of PLFFD after a session of high‐intensity interval training (Place et al. 2015). On the other hand, prolonged administration of antioxidants (e.g. vitamin C and E) has been shown to hamper the beneficial effects of endurance training in humans and rodents (Gomez‐Cabrera et al. 2008; Ristow et al. 2009; Paulsen et al. 2014). Thus, the overall impact of antioxidant supplementation in association with endurance training is negative: antioxidants do not prevent the exercise‐induced prolonged ROS/RNS‐dependent decline in contractile function, whereas they can hamper the beneficial adaptations that come with endurance exercise.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors acknowledge support from the Swedish Research Council and the Swedish National Centre for Sports Research.
Biographies
Arthur J. Cheng is a researcher at Karolinska Institutet. His research is focused on skeletal muscle fatigue and recovery and the role of ROS/RNS in these processes.

Johanna T. Lanner is assistant professor and leads a research group at Karolinska Institutet. She has discovered novel ways to regulate the ryanodine receptor and her recent research focuses on molecular mechanisms in skeletal muscle weakness.
References
- Allen DG, Lamb GD & Westerblad H (2008). Skeletal muscle fatigue: cellular mechanisms. Physiol Rev 88, 287–332. [DOI] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A & Marks AR (2011). Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG & Westerblad H (1998. a). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol 509, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG & Westerblad H (1998. b). Effect of nitric oxide on single skeletal muscle fibres from the mouse. J Physiol 509, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB & Westerblad H (2001). Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox‐modulation. FASEB J 15, 309–311. [DOI] [PubMed] [Google Scholar]
- Beckman JS & Koppenol WH (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271, C1424–C1437. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Mongillo M & Marks AR (2008. a). Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest 118, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, Lacampagne A & Marks AR (2008. b). Remodeling of ryanodine receptor complex causes ‘leaky’ channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA 105, 2198–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczkowski J, Lisdero CL, Lanone S, Samb A, Carreras MC, Boveris A, Aubier M & Poderoso JJ (1999). Endogenous peroxynitrite mediates mitochondrial dysfunction in rat diaphragm during endotoxemia. FASEB J 13, 1637–1646. [DOI] [PubMed] [Google Scholar]
- Botti H, Möller MN, Steinmann D, Nauser T, Koppenol WH, Denicola A & Radi R (2010). Distance‐dependent diffusion‐controlled reaction of •NO and O2 •− at chemical equilibrium with ONOO− . J Phys Chem B 114, 16584–16593. [DOI] [PubMed] [Google Scholar]
- Boveris A & Chance B (1973). The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD (2010). The sites and topology of mitochondrial superoxide production. Exp Gerontol 45, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS & Snyder SH (1990). Isolation of nitric oxide synthetase, a calmodulin‐requiring enzyme. Proc Natl Acad Sci USA 87, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K & Bredt DS (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752. [DOI] [PubMed] [Google Scholar]
- Brenner B (1988). Effects of Ca2+ on cross‐bridge turnover kinetics in skinned single rabbit psoas fibers: Implications for regulation of muscle contraction. Proc Natl Acad Sci USA 85, 3265–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton JD, Aydin J, Yamada T, Shabalina IG, Ivarsson N, Zhang SJ, Wada M, Tavi P, Nedergaard J, Katz A & Westerblad H (2010). Increased fatigue resistance linked to Ca2+‐stimulated mitochondrial biogenesis in muscle fibres of cold‐acclimated mice. J Physiol 588, 4275–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton JD, Place N, Yamada T, Silva JP, Andrade FH, Dahlstedt AJ, Zhang SJ, Katz A, Larsson NG & Westerblad H (2008). Reactive oxygen species and fatigue‐induced prolonged low‐frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J Physiol 586, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalow IB, Minin EA, Samoilova VE, Boecker W, Wellner M, Schmitz W, Neumann J & Punkt K (2005). Compartmentalization of NO signaling cascade in skeletal muscles. Biochem Biophys Res Commun 330, 615–621. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Boveris A, Ragan CI & Stoppani AO (1977). Production of superoxide radicals and hydrogen peroxide by NADH‐ubiquinone reductase and ubiquinol‐cytochrome c reductase from beef‐heart mitochondria. Arch Biochem Biophys 180, 248–257. [DOI] [PubMed] [Google Scholar]
- Carballal S, Bartesaghi S & Radi R (2014). Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim Biophys Acta 1840, 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AJ, Bruton JD, Lanner JT & Westerblad H (2015). Antioxidant treatments do not improve force recovery after fatiguing stimulation of mouse skeletal muscle fibres. J Physiol 593, 457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstedt AJ, Katz A, Tavi P & Westerblad H (2003). Creatine kinase injection restores contractile function in creatine‐kinase‐deficient mouse skeletal muscle fibres. J Physiol 547, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paoli FV, Broch‐Lips M, Pedersen TH & Nielsen OB (2013). Relationship between membrane Cl− conductance and contractile endurance in isolated rat muscles. J Physiol 591, 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W (2002). Free radicals in the physiological control of cell function. Physiol Rev 82, 47–95. [DOI] [PubMed] [Google Scholar]
- Dutka TL, Mollica JP & Lamb GD (2011). Differential effects of peroxynitrite on contractile protein properties in fast‐ and slow‐twitch skeletal muscle fibers of rat. J Appl Physiol (1985) 110, 705–716. [DOI] [PubMed] [Google Scholar]
- Ferrer‐Sueta G & Radi R (2009). Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol 4, 161–177. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I & Kleinert H (1994). Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 23, 1121–1131. [DOI] [PubMed] [Google Scholar]
- Gejl KD, Hvid LG, Willis SJ, Andersson E, Holmberg HC, Jensen R, Frandsen U, Hansen J, Plomgaard P & Ørtenblad N (2016). Repeated high‐intensity exercise modulates Ca2+ sensitivity of human skeletal muscle fibers. Scand J Med Sci Sports 26, 488–497. [DOI] [PubMed] [Google Scholar]
- Goldstein S & Czapski G (1995). The reaction of NO• with O2 •− and HO2 •−: a pulse radiolysis study. Free Radic Biol Med 19, 505–510. [DOI] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Close GL, Kayani A, McArdle A, Vina J & Jackson MJ (2010). Effect of xanthine oxidase‐generated extracellular superoxide on skeletal muscle force generation. Am J Physiol Regul Integr Comp Physiol 298, R2–R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J & Vina J (2008). Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training‐induced adaptations in endurance performance. Am J Clin Nutr 87, 142–149. [DOI] [PubMed] [Google Scholar]
- Gordon AM, Homsher E & Regnier M (2000). Regulation of contraction in striated muscle. Physiol Rev 80, 853–924. [DOI] [PubMed] [Google Scholar]
- Grimby L & Hannerz J (1977). Firing rate and recruitment order of toe extensor motor units in different modes of voluntary contraction. J Physiol 264, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y & Dick TP (2009). Proximity‐based protein thiol oxidation by H2O2‐scavenging peroxidases. J Biol Chem 284, 31532–31540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B & Gutteridge JM (1984). Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J 219, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY & Remington SJ (2004). Investigating mitochondrial redox potential with redox‐sensitive green fluorescent protein indicators. J Biol Chem 279, 13044–13053. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Sanchez G, Barrientos G & Aracena‐Parks P (2006). A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S‐glutathionylation. J Biol Chem 281, 26473–26482. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Cartier LJ, Chen M & Holloszy JO (1985). Superoxide dismutase and catalase in skeletal muscle: adaptive response to exercise. J Gerontol 40, 281–286. [DOI] [PubMed] [Google Scholar]
- Hsu JL, Hsieh Y, Tu C, O'Connor D, Nick HS & Silverman DN (1996). Catalytic properties of human manganese superoxide dismutase. J Biol Chem 271, 17687–17691. [DOI] [PubMed] [Google Scholar]
- Javeshghani D, Magder SA, Barreiro E, Quinn MT & Hussain SN (2002). Molecular characterization of a superoxide‐generating NAD(P)H oxidase in the ventilatory muscles. Am J Respir Crit Care Med 165, 412–418. [DOI] [PubMed] [Google Scholar]
- Kalyanaraman B, Darley‐Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ 2nd & Ischiropoulos H (2012). Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 52, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen YW, Raiteri R, Lederer WJ, Dorsey SG & Ward CW (2012). Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci Signal 5, ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug‐Roth D, Fridovich I & Rabani J (1973). Pulse radiolytic investigations of superoxide catalyzed disproportionation. Mechanism for bovine superoxide dismutase. J Am Chem Soc 95, 2786–2790. [DOI] [PubMed] [Google Scholar]
- Lamb GD & Westerblad H (2011). Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J Physiol 589, 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Dagnino‐Acosta A, Ainbinder A, Cheng Q, Joshi AD, Chen Z, Yarotskyy V, Oakes JM, Lee CS, Monroe TO, Santillan A, Dong K, Goodyear L, Ismailov II, Rodney GG, Dirksen RT & Hamilton SL (2012). AICAR prevents heat‐induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat Med 18, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschen G, Azzi A, Richter C & Flohe L (1974). Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett 42, 68–72. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Meadows JC & Merton PA (1971). Isolated single motor units in human muscle and their rate of discharge during maximal voluntary effort. J Physiol 217, 12–13P. [PubMed] [Google Scholar]
- Meyer AJ & Dick TP (2010). Fluorescent protein‐based redox probes. Antioxid Redox Signal 13, 621–650. [DOI] [PubMed] [Google Scholar]
- Michaelson LP, Shi G, Ward CW & Rodney GG (2010). Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve 42, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica JP, Dutka TL, Merry TL, Lamboley CR, McConell GK, McKenna MJ, Murphy RM & Lamb GD (2012). S‐Glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast‐twitch muscle fibres of rats and humans. J Physiol 590, 1443–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison RJ, Miller CC 3rd & Reid MB (1996). Nitric oxide effects on shortening velocity and power production in the rat diaphragm. J Appl Physiol (1985) 80, 1065–1069. [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RM, Dutka TL & Lamb GD (2008). Hydroxyl radical and glutathione interactions alter calcium sensitivity and maximum force of the contractile apparatus in rat skeletal muscle fibres. J Physiol 586, 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakane M, Schmidt HH, Pollock JS, Forstermann U & Murad F (1993). Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle. FEBS Lett 316, 175–180. [DOI] [PubMed] [Google Scholar]
- Nethery D, Stofan D, Callahan L, DiMarco A & Supinski G (1999). Formation of reactive oxygen species by the contracting diaphragm is PLA2 dependent. J Appl Physiol (1985) 87, 792–800. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Cheng AJ, Ørtenblad N & Westerblad H (2014). Subcellular distribution of glycogen and decreased tetanic Ca2+ in fatigued single intact mouse muscle fibres. J Physiol 592, 2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh‐ishi S, Kizaki T, Nagasawa J, Izawa T, Komabayashi T, Nagata N, Suzuki K, Taniguchi N & Ohno H (1997). Effects of endurance training on superoxide dismutase activity, content and mRNA expression in rat muscle. Clin Exp Pharmacol Physiol 24, 326–332. [DOI] [PubMed] [Google Scholar]
- Ørtenblad N, Westerblad H & Nielsen J (2013). Muscle glycogen stores and fatigue. J Physiol 591, 4405–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Basu Thakur P, Li S, Minard C & Rodney GG (2013). Real‐time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One 8, e63989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen G, Cumming KT, Holden G, Hallen J, Ronnestad BR, Sveen O, Skaug A, Paur I, Bastani NE, Ostgaard HN, Buer C, Midttun M, Freuchen F, Wiig H, Ulseth ET, Garthe I, Blomhoff R, Benestad HB & Raastad T (2014). Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double‐blind, randomised, controlled trial. J Physiol 592, 1887–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen TH, Nielsen OB, Lamb GD & Stephenson DG (2004). Intracellular acidosis enhances the excitability of working muscle. Science 305, 1144–1147. [DOI] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Gouspillou G & Hepple RT (2011. a). Mitochondria: isolation, structure and function. J Physiol 589, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MM, Romestaing C & Hepple RT (2011. b). Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One 6, e18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place N, Ivarsson N, Venckunas T, Neyroud D, Brazaitis M, Cheng AJ, Ochala J, Kamandulis S, Girard S, Volungevicius G, Pauzas H, Mekideche A, Kayser B, Martinez‐Redondo V, Ruas JL, Bruton J, Truffert A, Lanner JT, Skurvydas A & Westerblad H (2015). Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high‐intensity interval exercise. Proc Natl Acad Sci USA 112, 15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place N, Yamada T, Zhang SJ, Westerblad H & Bruton JD (2009). High temperature does not alter fatigability in intact mouse skeletal muscle fibres. J Physiol 587, 4717–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Criswell D, Lawler J, Ji LL, Martin D, Herb RA & Dudley G (1994). Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am J Physiol 266, R375–R380. [DOI] [PubMed] [Google Scholar]
- Powers SK & Jackson MJ (2008). Exercise‐induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Ji LL, Kavazis AN & Jackson MJ (2011). Reactive oxygen species: impact on skeletal muscle. Compr Physiol 1, 941–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye D, Palomero J, Kabayo T & Jackson MJ (2007). Real‐time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J Physiol 581, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Treberg JR, Perevoshchikova IV, Orr AL & Brand MD (2012). Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic Biol Med 53, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R (1998). Peroxynitrite reactions and diffusion in biology. Chem Res Toxicol 11, 720–721. [DOI] [PubMed] [Google Scholar]
- Reid MB, Haack KE, Franchek KM, Valberg PA, Kobzik L & West MS (1992). Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J Appl Physiol (1985) 73, 1797–1804. [DOI] [PubMed] [Google Scholar]
- Reid MB, Stokic DS, Koch SM, Khawli FA & Leis AA (1994). N‐acetylcysteine inhibits muscle fatigue in humans. J Clin Invest 94, 2468–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M (2014). Unraveling the truth about antioxidants: mitohormesis explains ROS‐induced health benefits. Nat Med 20, 709–711. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR & Blüher M (2009). Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 106, 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero N, Denicola A, Souza JM & Radi R (1999). Diffusion of peroxynitrite in the presence of carbon dioxide. Arch Biochem Biophys 368, 23–30. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, Jackson MJ & Vasilaki A (2014). Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic Res 48, 12–29. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A & Jackson MJ (2013). Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18, 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salanova M, Schiffl G, Rittweger J, Felsenberg D & Blottner D (2008). Ryanodine receptor type‐1 (RyR1) expression and protein S‐nitrosylation pattern in human soleus myofibres following bed rest and exercise countermeasure. Histochem Cell Biol 130, 105–118. [DOI] [PubMed] [Google Scholar]
- Schwerzmann K, Hoppeler H, Kayar SR & Weibel ER (1989). Oxidative capacity of muscle and mitochondria: correlation of physiological, biochemical, and morphometric characteristics. Proc Natl Acad Sci USA 86, 1583–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St‐Pierre J, Buckingham JA, Roebuck SJ & Brand MD (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277, 44784–44790. [DOI] [PubMed] [Google Scholar]
- Stuehr D, Pou S & Rosen GM (2001). Oxygen reduction by nitric‐oxide synthases. J Biol Chem 276, 14533–14536. [DOI] [PubMed] [Google Scholar]
- Sun QA, Hess DT, Nogueira L, Yong S, Bowles DE, Eu J, Laurita KR, Meissner G & Stamler JS (2011). Oxygen‐coupled redox regulation of the skeletal muscle ryanodine receptor‐Ca2+ release channel by NADPH oxidase 4. Proc Natl Acad Sci USA 108, 16098–16103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C, Ischiropoulos H & Radi R (2007). Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6, 662–680. [DOI] [PubMed] [Google Scholar]
- Tahara EB, Navarete FD & Kowaltowski AJ (2009). Tissue‐, substrate‐, and site‐specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med 46, 1283–1297. [DOI] [PubMed] [Google Scholar]
- Thomas DD, Liu X, Kantrow SP & Lancaster JR Jr (2001). The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2 . Proc Natl Acad Sci USA 98, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF & Boveris A (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilaki A, Mansouri A, Van Remmen H, van der Meulen JH, Larkin L, Richardson AG, McArdle A, Faulkner JA & Jackson MJ (2006). Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell 5, 109–117. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Kanzaki K, Kuratani M, Matsunaga S, Yanaka N & Wada M (2015). Contribution of impaired myofibril and ryanodine receptor function to prolonged low‐frequency force depression after in situ stimulation in rat skeletal muscle. J Muscle Res Cell Motil 36, 275–286. [DOI] [PubMed] [Google Scholar]
- Wei L, Salahura G, Boncompagni S, Kasischke KA, Protasi F, Sheu SS & Dirksen RT (2011). Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB J 25, 3068–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzberg E, Hezel M & Lundberg JO (2010). Nitrate‐nitrite‐nitric oxide pathway: implications for anesthesiology and intensive care. Anesthesiology 113, 1460–1475. [DOI] [PubMed] [Google Scholar]
- Westerblad H & Allen DG (2011). Emerging roles of ROS/RNS in muscle function and fatigue. Antioxid Redox Signal 15, 2487–2499. [DOI] [PubMed] [Google Scholar]
- Westerblad H, Duty S & Allen DG (1993). Intracellular calcium concentration during low‐frequency fatigue in isolated single fibers of mouse skeletal muscle. J Appl Physiol (1985) 75, 382–388. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Boulton ME, Gottsch JD & Sternberg P (1999). Oxidative damage and age‐related macular degeneration. Mol Vis 5, 32. [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Han DH, Jones TE & Holloszy JO (2007). Calcium induces increases in peroxisome proliferator‐activated receptor γ coactivator‐1α and mitochondrial biogenesis by a pathway leading to p38 mitogen‐activated protein kinase activation. J Biol Chem 282, 18793–18799. [DOI] [PubMed] [Google Scholar]
- Yamada T, Abe M, Lee J, Tatebayashi D, Himori K, Kanzaki K, Wada M, Bruton JD, Westerblad H & Lanner JT (2015. a). Muscle dysfunction associated with adjuvant induced‐arthritis is prevented by antioxidant treatment. Skelet Muscle 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Fedotovskaya O, Cheng AJ, Cornachione AS, Minozzo FC, Aulin C, Fridén C, Turesson C, Andersson DC, Glenmark B, Lundberg IE, Rassier DE, Westerblad H & Lanner JT (2015. b). Nitrosative modifications of the Ca2+ release complex and actin underlie arthritis‐induced muscle weakness. Ann Rheum Dis 74, 1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]