

Abstract
Key points
Reactive oxygen species (ROS) and nitric oxide (NO) regulate exercise‐induced nuclear factor erythroid 2‐related factor 2 (NFE2L2) expression in skeletal muscle.
NFE2L2 is required for acute exercise‐induced increases in skeletal muscle mitochondrial biogenesis genes, such as nuclear respiratory factor 1 (NRF‐1) and mitochondrial transcription factor A, and anti‐oxidant genes, such as superoxide dismutase (SOD)1, SOD2 and catalase.
Following exercise training mice with impaired NFE2L2 expression have reduced exercise performance, energy expenditure, mitochondrial volume and anti‐oxidant activity.
In muscle cells, ROS and NO can regulate mitochondrial biogenesis via a NFE2L2/NRF‐1‐dependent pathway.
Abstract
Regular exercise induces adaptations to skeletal muscle, which can include mitochondrial biogenesis and enhanced anti‐oxidant reserves. These adaptations and others are at least partly responsible for the improved health of physically active individuals. Reactive oxygen species (ROS) and nitric oxide (NO) are produced during exercise and may mediate the adaptive response to exercise in skeletal muscle. However, the mechanisms through which they act are unclear. In the present study, we aimed to determine the role of the redox‐sensitive transcription factor nuclear factor erythroid‐derived 2‐like 2 (NFE2L2) in acute exercise‐ and training‐induced mitochondrial biogenesis and the anti‐oxidant response. We report that ROS and NO regulate acute exercise‐induced expression of NFE2L2 in mouse skeletal muscle and muscle cells, and that deficiency in NFE2L2 prevents normal acute treadmill exercise‐induced increases in mRNA of the mitochondrial biogenesis markers, nuclear respiratory factor 1 (NRF‐1) and mitochondrial transcription factor A (mtTFA), and the anti‐oxidants superoxide dismutase (SOD) 1 and 2, as well as catalase, in mouse gastrocnemius muscle. Furthermore, after 5 weeks of treadmill exercise training, mice deficient in NFE2L2 had reduced exercise capacity and whole body energy expenditure, as well as skeletal muscle mitochondrial mass and SOD activity, compared to wild‐type littermates. In C2C12 myoblasts, acute treatment with exogenous H2O2 (ROS)‐ and diethylenetriamine/NO adduct (NO donor) induced increases in mtTFA, which was prevented by small interfering RNA and short hairpin RNA knockdown of either NFE2L2 or NRF‐1. Our results suggest that, during exercise, ROS and NO can act via NFE2L2 to functionally regulate skeletal muscle mitochondrial biogenesis and anti‐oxidant defence gene expression.
Key points
Reactive oxygen species (ROS) and nitric oxide (NO) regulate exercise‐induced nuclear factor erythroid 2‐related factor 2 (NFE2L2) expression in skeletal muscle.
NFE2L2 is required for acute exercise‐induced increases in skeletal muscle mitochondrial biogenesis genes, such as nuclear respiratory factor 1 (NRF‐1) and mitochondrial transcription factor A, and anti‐oxidant genes, such as superoxide dismutase (SOD)1, SOD2 and catalase.
Following exercise training mice with impaired NFE2L2 expression have reduced exercise performance, energy expenditure, mitochondrial volume and anti‐oxidant activity.
In muscle cells, ROS and NO can regulate mitochondrial biogenesis via a NFE2L2/NRF‐1‐dependent pathway.
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AMPK
5′ adenosine monophosphate‐activated protein kinase
- COX4
cytochrome c oxidase subunit 4
- Deta/NO
diethylenetriamine/nitric oxide adduct
- GSH
reduced glutathione
- GSR
glutathione reductase
- GSSG
oxidized glutathione
- GST
glutathione S‐transferase
- H2O2
hydrogen peroxide
- KO
knockout
- l‐NAME
NG‐nitro‐l‐arginine
- MAPK
mitogen‐activated protein kinase
- mtTFA
mitochondrial transcription factor A
- NAC
N‐acetylcysteine
- NFE2L2
nuclear factor erythroid 2‐related factor 2
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NO
nitric oxide
- NRF
nuclear respiratory factor
- PGC1α
peroxisome proliferator‐activated receptor γ coactivator 1α
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SOD
superoxide dismutase
- WT
wild‐type
Introduction
Regular exercise reduces the incidence of disease and increases life expectancy (Warburton et al. 2006). Skeletal muscle is particularly responsive to acute exercise stress, and exercise training causes adaptive changes such as increased anti‐oxidant capacity, mitochondrial volume and insulin signalling intermediates, which improve the function of the muscle and protect against the development metabolic disorders (Egan & Zierath, 2013). One of the mechanisms through which exercise may promote adaptive changes in skeletal muscle is by acutely elevating oxidative and nitrosative stress (Gomez‐Cabrera et al. 2015).
During exercise or contraction, skeletal muscle produces reactive oxygen species (ROS) and nitric oxide (NO) (Powers & Jackson, 2008). Both NO and ROS can promote mitochondrial biogenesis (Suzuki et al. 1998; Nisoli et al. 2003; Piantadosi & Suliman, 2006). Preventing increases in ROS during exercise by supplementing with anti‐oxidants has been shown, in some studies (Gomez‐Cabrera et al. 2005; Gomez‐Cabrera et al. 2008; Ristow et al. 2009; Strobel et al. 2011; Wadley et al. 2013; Paulsen et al. 2014 a) but not others (Yfanti et al. 2010; Higashida et al. 2011; Wadley et al. 2013), to attenuate acute exercise and exercise training inducing increases in mitochondrial biogenesis markers and anti‐oxidant responses. However, the mechanisms through which ROS and NO may be acting to co‐ordinate the adaptive response to exercise stress are not well understood.
The transcription factor peroxisome proliferator‐activated receptor γ coactivator 1α (PGC1α) is viewed as the master regulator of exercise‐induced mitochondrial biogenesis and the anti‐oxidant response (Lin et al. 2005). PGC1α is increased after exercise, and coactivates nuclear respiratory factors (NRF)‐1 and ‐2, which activate mitochondrial transcription factor A (mtTFA) to facilitate mitochondrial replication (Bassel‐Duby & Olson, 2006). Indeed, NO and ROS can increase PGC1α, NRF1 and mtTFA expression in skeletal muscle, and anti‐oxidant supplementation can attenuate exercise‐induced increases in PGC1α levels (Nisoli et al. 2003; Gomez‐Cabrera et al. 2005; Ristow et al. 2009). These observations have resulted in the assertion that ROS act via PGC1α to regulate exercise‐induced adaptive responses in skeletal muscle. However, PGC1α is not required for normal exercise‐induced increases in markers of mitochondrial biogenesis and anti‐oxidant expression (Leick et al. 2008), suggesting that alternative mechanisms are also involved in regulating skeletal muscle adaptation to exercise.
Nuclear factor erythroid‐derived 2‐like 2 (NFE2L2; also frequently referred to as Nrf2) is a ROS and NO sensitive transcription factor (Kensler et al. 2007). Exposure of cells to oxidative or nitrosative stress causes NFE2L2 to translocate from the cytoplasm (where it is sequestered by Keap1 for degradation) to the nucleus, where it binds to the anti‐oxidant response element to initiate defences that protect cells against cytotoxic and oxidative damage (Kensler et al. 2007). The primary function of NFE2L2 is to co‐ordinate anti‐oxidant responses to stress and, accordingly, NFE2L2 ablation prevents increases in skeletal muscle anti‐oxidant gene mRNA levels of old (>23 month) mice immediately after the last bout of very intense short‐term (2 weeks) treadmill training (Narasimhan et al. 2014). In addition, NFE2L2 is required for mitochondrial biogenesis induced by carbon monoxide stress in cardiac muscle (Piantadosi et al. 2008) and inflammation in the liver (Piantadosi et al. 2011).
Because NFE2L2 can regulate both mitochondrial biogenesis and the anti‐oxidant response to acute oxidative and nitrosative stress, we determined whether skeletal muscle NFE2L2 is activated by acute exercise and whether NFE2L2 regulates acute and exercise training‐induced increases in mitochondrial biogenesis and the anti‐oxidant response. We report that both NO and ROS contribute to increasing skeletal muscle NFE2L2 mRNA following an acute bout of exercise, and that NFE2L2 expression is required for normal increases in mitochondrial biogenesis markers and the anti‐oxidant response in skeletal muscle following acute exercise and exercise training. Furthermore, our data suggest that both ROS (H2O2) and NO‐induced increases in mitochondrial biogenesis markers (mRNA) are dependent on NFE2L2 and NRF1, whereas only NO‐mediated mitochondrial biogenesis gene expression requires PGC1α.
Methods
Animal ethics
All procedures described in the present study were approved by the Canton of Zurich Veterinary Office, Switzerland (license number ZH245/14), and conducted in accordance with the ETH Zurich and Swiss national guidelines for animal welfare. The investigators understand the ethical principles under which The Journal of Physiology operates and this work complies with the animal ethics checklist.
Murine breeding and housing conditions
Mice were maintained in a temperature‐controlled facility under a 12 : 12 h light/dark cycle and had ad libitum access to a diet unsupplemented with vitamins E and C (S8022‐S005; ssniff Spezialdiäten GmbH, Soest, Germany) and water. NFE2L2−/− (also known as Nrf2−/−) mice have been described previously (Chan et al. 1994) and were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and bred inhouse on a C57Bl/6 background. Male (∼25–30 g, 15–30 weeks old) NFE2L2−/− mice and NEF2L2+/+ littermates, herein referred to as NFE2L2 knockout (KO) and wild‐type (WT), respectively, and C57Bl/6 mice were used for all the experiments. All mice were killed humanly via cervical dislocation.
Exercise protocols and metabolic cages
All exercised mice underwent at least two treadmill (Panlab/Harvard Apparatus, Holliston, MA, USA) familiarization sessions on separate days >5 days prior to the initial exercise test. Familiarization consisted of a 5 min exposure to the treadmill at speeds from 0–9 m min−1, with a 0° incline. A mild electrical stimulus was provided as motivation via a shock‐grid located at end of the treadmill belt. Aerobic capacity was determined using an incremental treadmill test to exhaustion. The treadmill was set at a 10° incline and the speed was increased by 2 m min−1 every 2 min until the mouse spent >5 s on the shock grid without attempting to continue running. To test endurance capacity, the treadmill was set to an incline of 15° and starting speed of 5 m min−1, which was continuously increased (ramped) at a rate of 0.17 m min−1 until the mouse spent >5 s on the shock grid without attempting to continue running.
Exercise training involved 30–60 min of treadmill running at 10–15 m min−1, at a 10° incline, 4–5 days week−1 for 4–6 weeks. Acute exercise protocol consisted of 1 h of treadmill running at 12 m min−1, at a 10° incline. Tissue was collected and snap‐frozen in liquid nitrogen following acute exercise at the time points indicated, and at least 36 h after the final exercise session for exercise‐trained mice.
A PhenoMaster (TSE Systems, Bad Homburg, Germany) open‐circuit calorimetry system was used to measure oxygen consumption and ambulatory activity over 48 h (two light/dark cycles) following an acclimation period of 24–48 h.
Antibodies and reagents
Rabbit antibodies against NFE2L2 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and NRF‐1 was obtained from Cell Signaling Technology (Beverly, MA, USA). 5‐Aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) was purchased from Adipogen AG (Liestal, Switzerland). Unless stated otherwise, all other reagents were purchased from Sigma‐Aldrich Chemicals (St Louis, MO, USA).
N‐acetylcysteine (NAC) and l‐N G‐nitroarginine methyl ester (l‐NAME) treatment of mice
For experiments involving the inhibition of NO synthase and scavenging of ROS mice received l‐NAME (1 g l−1 ) (Wadley & McConell, 2007) or NAC (10 g l−1) (Chen et al. 2010), respectively (Cobley et al. 2015), in their drinking water for 3 days prior to the experiment.
Real‐time PCR
RNA was extracted from cells or frozen muscle samples using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and mRNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). For quantification of mtDNA, total DNA was isolated by standard proteinase K and phenol–chloroform extraction. Quantitative real‐time PCR was performed on a ViiA™ 7 Real‐Time PCR System (Applied Biosystems) using the SYBR green select master mix (Applied Biosystems). Reactions were performed in triplicate and relative quantification was achieved using the ΔΔCt method with 18S ribosomal RNA as an internal control. The primer sequences used are listed in Table 1.
Table 1.
Mouse quantitative PCR primer sequences
| Gene name | Forward primer | Reverse primer |
|---|---|---|
| 18S | GATCCATTGGAGGGCAAGTCT | CCAAGATCCAACTACGAGCTTTTT |
| CAT | TGAGAAGCCTAAGAACGCAATTC | CCCTTCGCAGCCATGTG |
| COX4 | ACTACCCCTTGCCTGATGTG | GCCCACAACTGTCTTCCATT |
| GSR | GCTATGCAACATTCGCAGATG | AGCGGTAAACTTTTTCCCATTG |
| GST | GCTCTTACCACGTGCAGCTT | GGCTGGGAAGAGGAAATGGA |
| mtTFA | CACCCAGATGCAAAACTTTCAG | CTGCTCTTTATACTTGCTCACAG |
| NFE2L2 | CGAGATATACGCAGGAGAGGTAAGA | GCTCGACAATGTTCTCCAGCTT |
| NRF‐1 | AATGTCCGCAGTGATGTCC | GCCTGAGTTTGTGTTTGCTG |
| NRF‐2 | TGAAGTTCGCATTTTGATGGC | CTTTGGTCCTGGCATCTCTAC |
| PGC1α | AAGTGTGGAACTCTCTGGAACTG | GGGTTATCTTGGTTGGCTTTATG |
| Prx1 | GATCCCAAGCGCACCATT | TAATAAAAAGGCCCCTGAAAGAGAT |
| SOD1 | GTGATTGGGATTGCGCAGTA | TGGTTTGAGGGTAGCAGATGAGT |
| SOD2 | TTAACGCGCAGATCATGCA | GGTGGCGTTGAGATTGTTCA |
| Trx1 | CCGCGGGAGACAAGCTT | GGAATGGAAGAAGGGCTTGATC |
Cell culture
C2C12 myoblasts were grown in DME media containing 5.5 mm d‐glucose and 20% fetal bovine serum at 37°C in 5% CO2/21% O2. Short hairpin RNA (shRNA) knockdown of NFE2L2 was achieved by transfecting pooled shRNA clones (TRCN0000012128, TRCN0000012129, TRCN0000012130; Broad Institute RNAi consortium, Cambridge, MA, USA) using Lipofectamine 2000 (Invitrogen) before antibiotic selection (10 day, 1 μg ml−1 puromycin). Transient knockdown of NRF‐1 and PGC1α gene expression was achieved by transfection with the indicated concentration of the corresponding specific small interfering RNAs (siRNAs) (Santa Cruz Biotechnology) using Lipofectamine 2000 and cells were collected 24 h after transfection. Prolonged intermitted treatment of C2C12 cells with AICAR (1 mm), diethylenetriamine/nitric oxide adduct (Deta/NO) (100 μm) or H2O2 (5 μm) involved applying the compound for 5 h day–1 for 5 days. The doses and treatment times were based on concentrations used in previous studies investigating mitochondrial biogenesis in immortalized muscle cells (McConell et al. 2010; Lira et al. 2010 a), as well as preliminary studies that accessed the optimal dose required to elevate NFE2L2 expression without causing any cell death (data not shown).
Immunoblotting
Immunoblotting was preformed essentially as described previously (Merry et al. 2014). Briefly, cells were scrapped from their dish or snap‐frozen mouse tissue samples were homogenized using an electrical handheld homogenizer in 10–20 volumes of ice‐cold RIPA lysis buffer (50 mm Hepes, pH 7.4, 1%, v/v Triton X‐100, 1% v/v sodium deoxycholate, 0.1% v/v SDS, 150 mm NaCl, 10% v/v glycerol, 1.5 mm MgCl2, 1 mm EGTA, 50 mm sodium fluoride, protein inhibitor cocktail (Roche, Basel, Switzerland), 1 mm phenylmethysulphonyl fluoride, 1 mm sodium vanadate), incubated for 20 min on ice and centrifuged at 20,000 g for 60 min at 4°C. The supernatants were resolved by SDS‐PAGE and processed for immunoblotting by standard procedures.
SOD and citrate synthase activity assays
Frozen muscle was ground in a nitrogen‐chilled mortar together with 50 mm phosphate buffer + 1 mm EDTA, and sonicated. Lysates were cleared by centrifugation for 15 min at 12,000 g and 4°C. The supernatant was used for the subsequent measurement of superoxide dismutase as described previously (Weimer et al. 2014). Citrate synthase activity was measured in supernatant by examining the increase of 5,5‐dithiobis‐2‐nitrobenzoate at a wavelength of 412 nm (Srere, 1969).
Glutathione and protein carbonylation
Total and oxidized glutathione levels in whole blood were measured using a BIOXYTECH GSH/GSSG‐412 assay kit (Oxis International, Inc., Tampa, FL, USA) and GSH:GSSG ratios were determined in accordance with the manufacturer's instructions. Protein carbonylation was monitored using an OxyBlot Protein Oxidation Detection Kit (Merck Millipore, Billerica, MA, USA) in accordance with the manufacturer's instructions.
Statistical analysis
All data are presented the as mean ± SEM. Statistical significance was determined using an unpaired two‐tailed Student's t test and one‐ and two‐way ANOVA with Fisher's least significant difference post hoc analysis as indicated. P < 0.05 was considered statistically significant.
Results
Skeletal muscle NFE2L2 expression is increased by exercise
Exercise increases skeletal muscle production of ROS and NO (Powers & Jackson, 2008) and the transcription factor NFE2L2 is sensitive to changes in cellular ROS and NO levels (Kensler et al. 2007). Therefore, we first determined whether exercise regulates the expression of NFE2L2 in skeletal muscle of mice. Thirty minutes following acute treadmill exercise mRNA for skeletal muscle NFE2L2, as well as that of some of its known downstream targets [glutathione S‐transferase (GST) and glutathione reductase (GSR)], was increased by more than 3‐fold (Fig. 1 A). This increase was transient and no different from rested muscle at 2 or 16 h post‐exercise (Fig. 1 B). Furthermore, NFE2L2, GST and GSR mRNA were elevated in skeletal muscle of exercise‐trained mice (Fig. 1 C). Despite the testing of several commercially available antibodies, we were unable to identify one that specifically detected NFE2L2 in tissue samples (Fig. 1 D); however, we confirmed that NFE2L2 KO mice did not express NEF2L2 through the determination of NFE2L2 mRNA levels in skeletal muscle, liver and heart (Fig. 1 E). The blot shown in Fig. 1 D was obtained using NFE2L2 antibody #12721 (Cell Signaling Technologies) but is representative of the antibodies #8882 (Cell Signaling Technologies) and H300 and C20 (Santa Cruz Biotechnology) that were tested in mouse muscle, heart and liver samples. It is important to acknowledge that, although NFE2L2 has a predicted molecular of ∼65–70 kDa, it is recognized to be detected at ∼95–110 kDa (Lau et al. 2013). Therefore, we were unable to confirm that NFE2L2 mRNA expression resulted in increased protein levels in tissue samples. However, because NFE2L2 primarily acts a transcription factor, increases in mRNA for its own and other targets (GST and GSR) confirm that it is regulated by exercise.
Figure 1. NFE2L2 expression in skeletal muscle following exercise .
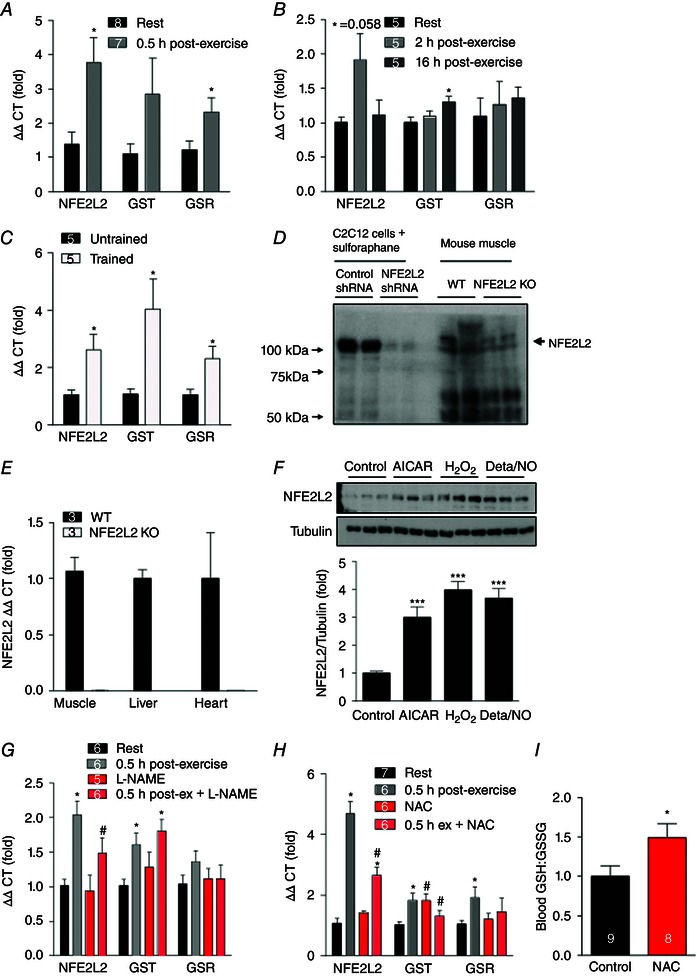
Mouse skeletal muscle (gastrocnemius) NFE2L2 and NFE2L2 target (GST and GSR) mRNA was increased 30 min following an acute (1 h) bout of treadmill exercise (A and B) and following 6 weeks of treadmill exercise training (C). Representative blot showing that NFE2L2 antibodies tested detected a band at the reported size (∼95–110 kDa) of NFE2L2 in cell lysates that was not seen in NFE2L2 shRNA‐treated cells; however, these antibodies were unable to detect a band in tissue lysates at the correct molecular weight that was not seen in lysates from NFE2L2 KO mice (D). NFE2L2 KO mice did not express NFE2L2 mRNA in skeletal muscle, liver or heart (E). Acute (5 h) treatment of C2C12 myoblasts with AICAR (1 mm) and H2O2 (50 μm) or Deta/NO (100 μm) increased NFE2L2 protein expression (F). Three days of treatment of mice with a NO synthase inhibitor (G) or anti‐oxidant (NAC) (H) attenuates acute (1 h) exercise‐induced increases in skeletal muscle (gastrocnemius) NFE2L2, GST and GSR mRNA expression and (I) increases the blood GSH:GSSG ratio. Results are shown as the mean ± SE (n = 5–7 per group for the cell culture experiments; n for mouse experiments is shown individually). Significance was determined using a two‐tailed Student's t test or one‐way ANOVA with Fisher's least significant difference post hoc analysis. * P < 0.05 and *** P < 0.001 vs. rest/control. # P < 0.05 vs. untreated of the same condition. [Colour figure can be viewed at wileyonlinelibrary.com]
Having established that acute exercise and exercise training increases skeletal muscle NFE2L2 mRNA, we next investigated the stimuli that may be inducing NFE2L2 expression during exercise. Treatment of C2C12 myoblasts with compounds that mimic aspects of exercise, including an 5‘ adenosine monophosphate‐activated protein kinase (AMPK) activator (AICAR), hydrogen peroxide (H2O2) and the NO donor Deta/NO increased NFE2L2 protein expression (Fig. 1 F). To determine whether ROS and NO regulate exercise‐induced increases in NFE2L2 mRNA in vivo, prior to acute exercise, we treated mice with the anti‐oxidant NAC or the NO synthase inhibitor l‐NAME. NAC increased the blood reduced glutathione (GSH) to oxidized glutathione (GSSG) ratio, indicating an anti‐oxidant effect and a reduction in systemic oxidative stress (Fig. 1 I). Exercise‐induced increases in skeletal muscle NFE2L2 mRNA were attenuated by l‐NAME and NAC, with NAC also attenuating exercise‐induced increases in NFE2L2 target GST and GSR mRNA (Fig. 1 G and H).
NEF2L2 is required for exercise training‐induced mitochondrial biogenesis markers
ROS and NO have been implicated in the regulation of exercise‐induced mitochondrial biogenesis (Gomez‐Cabrera et al. 2005; Gomez‐Cabrera et al. 2008; Paulsen et al. 2014 a), whereas NFE2L2 is required for mitochondrial biogenesis stimulated by stress in cardiac muscle and liver (Piantadosi et al. 2008; Piantadosi et al. 2011). Therefore, we investigated whether NFE2L2 is required for exercise‐induced mitochondrial biogenesis in skeletal muscle. Exercise training increased mtDNA and citrate synthase activity (markers of mitochondrial volume) in skeletal muscle of WT but not NFE2L2 KO mice (Fig. 2 A and B). Consistent with this, mitochondrial biogenesis‐associated genes mRNA were increased in skeletal muscle of trained WT but not trained NFE2L2 KO mice compared to untrained mice of the same genotype (Fig. 2 C). However, as a result of large individual variability, we were unable to detect any training or genotype differences in skeletal muscle protein levels of PGC1α, mtTFA or cytochrome c oxidase subunit 4 (COX4) (data not shown).
Figure 2. NFE2L2 is required for exercise training induced mitochondrial biogenesis .
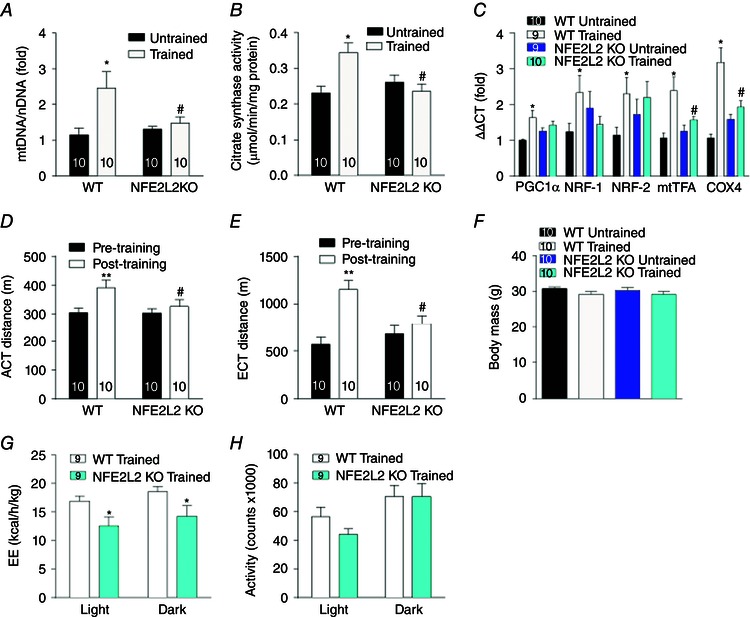
mtDNA (A), citrate synthase activity (B) and mitochondrial biogenesis‐associated genes mRNA (C) were measured in the skeletal muscle (gastrocnemius) of WT and NFE2L2 KO mice following 6 weeks of treadmill exercise training (trained) or normal sedentary behaviour (untrained). Four‐week treadmill trained NRFE2L2 KO mice had reduced exercise performance (D and E) and energy expenditure (G) compared to trained WT mice. WT and NFE2L2 KO mice had a similar body weight and activity levels (F and H). Results are shown as the mean ± SE (n is shown individually). Significance was determined using one‐way ANOVA with Fisher's least significant difference post hoc analysis. * P < 0.05 ** P < 0.001 vs. untrained or pre‐training of the same genotype. # P < 0.05 vs. WT of the same condition. EE, energy expenditure. [Colour figure can be viewed at wileyonlinelibrary.com]
In agreement with the mitochondrial biogenesis data, exercise training improved the aerobic and endurance capacity of WT but not NFE2L2 KO mice (Fig. 2 D and E). Despite there being no effect of training on WT or NFE2L2 KO mice body mass (Fig. 2 F), whole body energy expenditure of trained WT mice was greater than that of trained NFE2L2 mice (Fig. 2 G), an effect that was not observed in untrained mice and was not the result of differences in activity levels (data not shown) (Fig. 2 H).
ROS and NO act via NFE2L2 and NRF‐1 to induce mitochondrial biogenesis
To further investigate the role of NFE2L2 in exercise‐induced mitochondrial biogenesis, we assessed the effect of acute exercise on mitochondrial biogenesis‐associated genes mRNA in WT and NFE2L2 KO mice (Fig. 3 A). PGC1α and COX4 mRNA was similarly increased in the skeletal muscle of WT and NFE2L2 KO mice following acute exercise, whereas NRF‐1 and mtTFA mRNA only increased in skeletal muscle of WT mice (Fig. 3 A).
Figure 3. NFE2L2 is required for acute exercise, NO and H2O2 induced increases in mRNA of mitochondrial biogenesis‐associated genes .
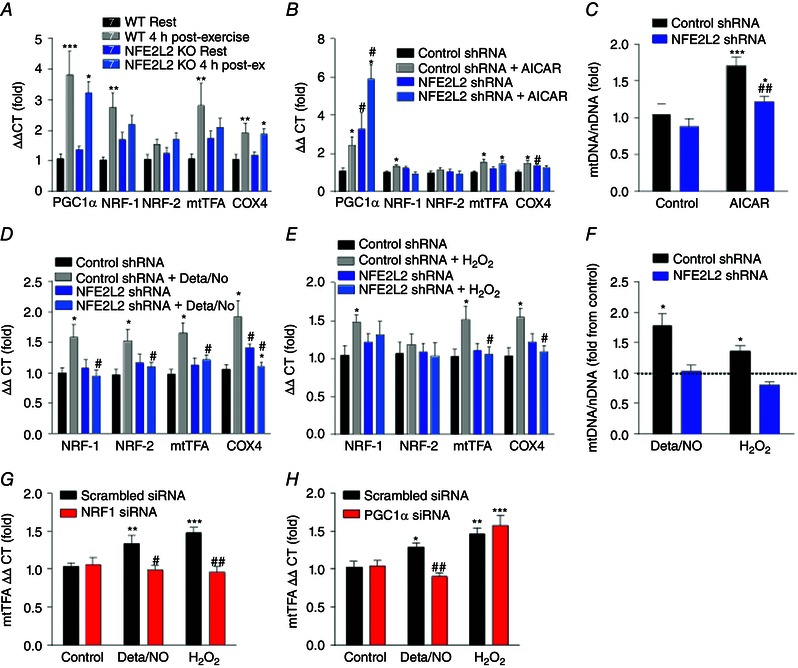
Mitochondrial biogenesis‐associated genes mRNA levels in the skeletal muscle (gastrocnemius) of WT and NFE2L2 KO mice 4 h following acute exercise (A). Mitochondrial biogenesis‐associated genes mRNA expression in control shRNA or NFE2L2 shRNA C2C12 myoblasts following 5 h treatment with AICAR (1 mm) (B), Deta/NO (100 μm) (D) or H2O2 (50 μm) (E). mtDNA was determined in control shRNA or NFE2L2 shRNA C2C12 myoblasts following 5 days (5 h day–1) treatment with AICAR (1 mm) (C), Deta/NO (100 μm) or H2O2 (5 μm) (F). mtTFA mRNA expression was assessed in C2C12 myoblasts stimulated for 5 h with Deta/NO (100 μm) or H2O2 (50 μm) 24 h following control, NRF‐1 (100 nm) (G) or PGC1α (10 nm) (H) siRNA transfection. Results are shown as the mean ± SE (n = 6–8 per group for cell culture experiments; n for mouse experiments is shown individually). Significance was determined using one‐way ANOVA with Fisher's least significant difference post hoc analysis. * P < 0.05, ** P < 0.01 *** P < 0.001 vs. rest or untreated of same genotype or si/shRNA. # P < 0.05 vs. control si/shRNA of same condition. [Colour figure can be viewed at wileyonlinelibrary.com]
We then utilized the C2C12 myoblast muscle cell model to examine the pathway through which NFE2L2 may act to facilitate mitochondrial biogenesis in skeletal muscle. Control and cells deficient in NFE2L2 protein (NFE2L2 shRNA) (Fig. 1 D) were acutely treated with the AMPK activator (AICAR). Although AICAR treatment increased PGC1α and mtTFA mRNA levels in both control and NFE2L2 deficient cells, NFE2L2 shRNA prevented AICAR‐induced increases in NRF‐1 and COX4 mRNA levels (Fig. 3 B). In agreement, NFE2L2 shRNA also attenuated prolonged intermittent AICAR‐induced increases in mtDNA (Fig. 3 C). To determine the role of NFE2L2 in NO‐ or ROS‐mediated mitochondrial biogenesis, control and NFE2L2 shRNA cells were exposed to acute (Fig. 3 D and E) or prolonged intermittent (Fig. 3 F) treatments with the NO donor Deta/NO or H2O2. Acute Deta/NO or H2O2 treatment increased mitochondrial biogenesis genes mRNA in control but not NFE2L2 shRNA cells. Similarly, prolonged intermitted treatment with Deta/NO or H2O2 increased mtDNA in control but not NFE2L2 shRNA cells (Fig. 3 F).
Because the promoter region of NRF‐1 contains a NFE2L2 binding site (Piantadosi & Suliman, 2006) and NFE2L2 deficiency does not affect acute exercise or AICAR‐induced PGC1α expression (Fig. 3 A and B), we used siRNAs to determine whether NRF‐1 and PGC1α are required for NO‐ and ROS‐mediated increases in the downstream mitochondrial biogenesis mediator, mtTFA mRNA. Deta/NO and H2O2 induced increases in mtTFA mRNA were prevented by siRNA knockdown of NRF‐1 (Fig. 3 G) and PGC1α siRNA knockdown prevented Deta/NO but not H2O2 induced increases in mtTFA (Fig. 3 H).
ROS, NO and NFE2L2 regulate the exercise‐induced anti‐oxidant response
Acute exercise‐induced increases in skeletal muscle anti‐oxidant genes superoxide dismutase (SOD)1, SOD2, catalase, Prx1 and Trx1 mRNA were prevented by the NO synthesis inhibitor l‐NAME and the anti‐oxidant NAC (Fig. 4 A and B). Although SOD1 mRNA levels were elevated, and SOD2 tended to be elevated in non‐exercised NFE2L2 skeletal muscle, acute exercise did not increase their levels further, nor did it increase catalase mRNA (Fig. 4 C). By contrast, acute exercise increased the mRNA levels of SOD1, SOD2 and catalase in the skeletal muscle of WT mice (Fig. 4 C). Following exercise training, SOD1, SOD2 and catalase mRNA was increased in the skeletal muscle of WT but not NFE2L2 KO mice (Fig. 4 D), whereas Prx1 mRNA was only increased by training in the skeletal muscle of NFE2L2 KO mice. Exercise training increased skeletal muscle SOD activity of WT but not NFE2L2 KO mice (Fig. 4 E). Despite having lower anti‐oxidant levels, protein oxidation (as assessed by the OxyBlot protein carbonylation assay) was not elevated in the skeletal muscle of NFE2L2 KO mice, nor were protein oxidation levels altered by exercise training (Fig. 4 F).
Figure 4. NFE2L2, ROS and NO regulate the exercise‐induced anti‐oxidant response .
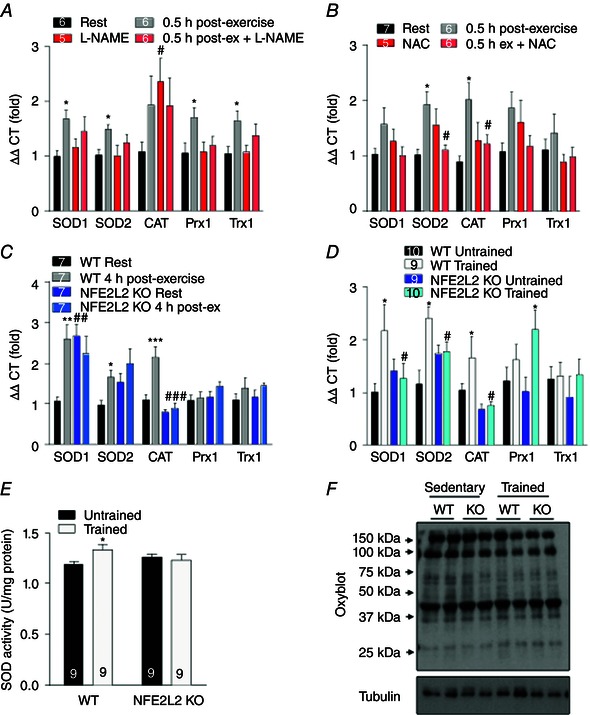
Mice were treated for 3 days with the NO synthase inhibitor (l‐NAME) (A) or the anti‐oxidant (NAC) (B) and skeletal muscle (gastrocnemius) anti‐oxidant gene mRNA expression was assessed 30 min following acute (1 h) treadmill exercise. Anti‐oxidant genes mRNA or SOD activity were determined in the skeletal muscle (gastrocnemius) of WT and NFE2L2 KO mice 4 h following an acute treadmill exercise (C) or following 6 weeks of treadmill exercise training (trained) or normal sedentary behaviour (untrained) (D and E). WT and NFE2L2 KO gastrocnemius muscle showed similar levels of protein carbonylation and this was not affected by exercise training (F). Results are shown as the mean ± SE (n is shown individually). Significance was determined using one‐way ANOVA with Fisher's least significant difference post hoc analysis. * P < 0.05 and *** P < 0.001 vs. rest or untrained. # P < 0.05 vs. untreated or WT of the same condition. CAT, catalase; Prx, peroxiredoxin; Trx, theroxiredoxin. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
In the present study, we show that NO and ROS regulate NFE2L2 mRNA levels following acute exercise stress and that disruption of NFE2L2 expression impairs acute exercise‐ and exercise training‐induced increases in skeletal muscle mitochondrial biogenesis markers and the anti‐oxidant response. Consistent with these findings, exercise‐trained mice that lack NFE2L2 had reduced whole body energy expenditure and exercise performance. The present study provides the first causative evidence of a mechanism through which exercise‐induced increases in NO and ROS may be acting to promote adaptation to exercise stress.
Anti‐oxidant supplementation in humans (Gomez‐Cabrera et al. 2008; Ristow et al. 2009; Paulsen et al. 2014 a) and rodent models (Gomez‐Cabrera et al. 2005; Strobel et al. 2011) has been shown to attenuate exercise‐induced increases in markers of skeletal muscle mitochondrial biogenesis. The present study furthers these observations by providing evidence that anti‐oxidant supplementation during exercise attenuates the normal activation of NFE2L2, and that NFE2L2 is a redox sensitive transcriptional regulator of the mitochondrial biogenesis response to exercise. Exercise activates AMPK and p38 mitogen‐activated protein kinase (MAPK) in skeletal muscle, and these protein kinases are considered to act via PGC1α to trigger a response from the transcription factors NRF‐1, NRF‐2 and mtTFA to induce mitochondrial biogenesis (Bassel‐Duby & Olson, 2006). Although anti‐oxidant supplementation can attenuate acute exercise‐induced increases in AMPK and p38 MAPK phosphorylation and PGC1α expression (Gomez‐Cabrera et al. 2005), this does not always prevent exercise training‐induced increases in mitochondrial biogenesis (Wadley et al. 2013). Furthermore, mice deficient in either AMPKα2 or PGC1α show normal mitochondrial biogenesis responses to exercise (Jorgensen et al. 2007; Leick et al. 2008). This suggests that other redox sensitive pathways are probbaly involved in regulating mitochondrial biogenesis in response to exercise.
Our finding that disruption of NFE2L2 expression impairs exercise, NO and H2O2 induced increases in mitochondrial mass markers (citrate synthase and mtDNA) is supported by previous studies showing that NFE2L2 regulates resveratrol, sepsis and carbon monoxide induced mitochondrial biogenesis (Piantadosi et al. 2008; Piantadosi et al. 2011; Kim et al. 2014). Mechanistically, the promoter region of NRF‐1 contains a NFE2L2 binding site (Piantadosi et al. 2011), suggesting that NFE2L2 activation can induce NRF‐1 transcription, and that NRF‐1 can then interact with mtTFA to drive mitochondrial biogenesis. Acute exercise, NO and H2O2 induced increases in NRF‐1 and mtTFA were dependent on NFE2L2 and, in muscle cells, NO and H2O2 induced increases in mtTFA were prevented by disruption of NRF‐1. Consistent with our muscle cell data, NO synthase inhibition and anti‐oxidant supplementation in rodents reduces mtTFA expression after acute exercise (Wadley & McConell, 2007; Wadley et al. 2013) and, in rat hepatoma cells, oxidative stress increases the binding of NRF‐1 to mtTFA (Piantadosi & Suliman, 2006). This suggests that ROS and NO may act via NFE2L2 and NRF‐1 to promote the mitochondrial biogenesis that is regulated by mtTFA.
In support of the concept that NFE2L2 is part of a pathway that is downstream of PGC1α in the mitochondrial biogenesis signalling cascade, in the absence of NFE2L2 both AICAR and acute exercise stimulated normal increases in PGC1α mRNA. Interestingly, however, siRNA knockdown of PGC1α prevented NO but not H2O2 mediated increases in mtTFA mRNA. Considering also that AICAR stimulated NFE2L2 expression and AICAR‐induced mitochondrial biogenesis was partially prevented by NFE2L2 shRNA knockdown, we speculate that there is probably some level of co‐operation between pathways involving PGC1α and NFE2L2 in the promotion of mitochondrial biogenesis. Previous studies have shown that NO promotes mitochondrial biogenesis through a pathway that involves the formation of cGMP and induction of PGC1α (Nisoli et al. 2003; Lira et al. 2010 b). Although NO interacts with Keap1 cysteine 151 to inhibit degradation of NFE2L2 (McMahon et al. 2010), and thus promote its expression, it is not clear whether this is the mechanism through which exercise‐induced NO promotes NFE2L2 interaction with NRF‐1 or whether intermediary steps are involved, such as the formation of cGMP. Regardless, the notion that PGC1α and NFE2L2 may act in concert and/or have a redundancy basis, as a result of H2O2 appearing to act independently of PGC1α, requires further attention.
Several groups, however, have reported that anti‐oxidant supplementation does not affect the normal skeletal muscle exercise response (Yfanti et al. 2010; Higashida et al. 2011; Wadley et al. 2013). Although variable anti‐oxidant supplementation (type, duration, and dose) and exercise training (mode, duration, intensity) are probably significant contributing factors (Merry & Ristow, 2015), these findings may also support redundancies in the exercise mitochondrial biogenesis pathway. Indeed, we show that both ROS and NO contribute to the activation of NFE2L2 following acute exercise, perhaps suggesting that, although scavenging one may be effective in reducing mitochondrial biogenesis response to signal bout of exercise, such inhibition is overcome and/or alternative pathways are up‐regulating, with subsequent bouts resulting in normal training responses. Furthermore, it must be acknowledged that, in the present study, we utilized whole body NFE2L2 deficient mice, although further in vivo research should consider whether the blunt exercise responses reported in these mice were the result of a muscle‐specific effect or systemic regulation.
NFE2L2 regulates both basal anti‐oxidant expression and the anti‐oxidant response to stress in organisms ranging from yeast and Caenorhabditis elegans to mammals (Kensler et al. 2007; Nguyen et al. 2009). Recently, NFE2L2 has been shown to be required for increases in anti‐oxidant mRNA expression immediately following exhaustive exercise in the skeletal muscle of old mice (Narasimhan et al. 2014) and cardiac muscle of young mice (Muthusamy et al. 2012). We add to these findings by showing that both ROS and NO contribute to NFE2L2 activation during exercise, and that NFE2L2 is not only responsible for normal increases in anti‐oxidant expression and activity following acute exercise, but also as a result of exercise training. We also report that expression of SOD1 and 2 is upregulated in the basal (rested) state and, unlike in WT mice, peroxiredoxin 1 expression was increased with exercise training in NFE2L2 KO mice. Although this did not translate to increases in basal SOD activity in NFE2L2 KO mice, it is probably a compensatory response to the reduced expression of catalase and the genes associated with glutathione metabolism (data not shown; Narasimhan et al. 2014), suggesting that alternative mechanisms also regulate skeletal muscle anti‐oxidant expression. One probable candidate is nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB), which is known to be involved in the transcriptional regulation of anti‐oxidant expression, with vitamin C and allopurinol supplementation suppressing exercise‐induced increases in NF‐κB expression (Gomez‐Cabrera et al. 2005; Gomez‐Cabrera et al. 2008; Ji, 2008).
In a response known as hormesis (Southam & Ehrlich, 1943), acute increases in ROS and NO stress promote adaptations that protect the cell from the potential deleterious effects of subsequent stress, and extend the lifespan of C. elegans and Drosophila melanogaster (Ristow & Zarse, 2010). Ablation of the C. elegans NFE2L2 homologue, skin‐1, abrogates the lifespan extending effect of interventions such as the impairment of insulin/insulin‐like growth factor‐1 signalling (Zarse et al. 2012), glucosamine supplementation (Weimer et al. 2014) and glycolysis inhibition (Schulz et al. 2007). Importantly, these interventions require increases in ROS to induce adaptations, which include increased mitochondrial respiration and anti‐oxidant expression, extending lifespan. Because regular physical exercise also increases life expectancy (Warburton et al. 2006), and enhanced mitochondrial respiration and increased endogenous anti‐oxidant expression can prevent metabolic disease (Koves et al. 2008; Anderson et al. 2009), it is tempting to speculate that NFE2L2 may be involved in the organism‐wide health promoting effects of exercise. Indeed, NFE2L2 activators have been shown to extend the lifespan of C. elegans and D. melanogaster (Suckow & Suckow, 2006; Lee et al. 2010) and protect against metabolic disease in mice (Yu et al. 2011; He et al. 2012). Surprisingly, however, fat‐specific and whole body deletion of NFE2L2 also reduces genetic‐ and diet‐induced obesity (Chartoumpekis et al. 2011; Xue et al. 2013). This may indicate a tissue‐specific effect of NFE2L2 in regulating metabolic responses, and suggest a differential effect associated with chronic deletion vs. acute activation. Regardless, these data and previously published work in humans (Gomez‐Cabrera et al. 2008; Ristow et al. 2009; Paulsen et al. 2014 a; Paulsen et al. 2014 b; Cobley et al. 2015) suggest that ROS produced under normal physiological conditions, and particularly during exercise, are required for optimal adaptation and the resulting protection against future cellular stressor. This advises against the unnecessary use of anti‐oxidants in humans. Although speculative, our data may also suggest that the ingestion of nutrients that activate NFE2L2 may have protective effects on cell function similar to that of exercise; however, this remains to be established.
Taken together, our data suggest that NO and ROS produced during exercise activate skeletal muscle NFE2L2. Furthermore, NFE2L2 expression is required for normal mitochondrial biogenesis and the anti‐oxidant transcriptional response to acute exercise and exercise training. These results are consistent with the hormesis theory, where acute increases in ROS/NO stress initiate cell‐ and possibly system‐wide adaptive responses that improve tolerance to prospective stress.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
TLM contributed to the designed and execution of experiments and the interpretation of the results. MR contributed to the study design and the interpretation of the results. TLM drafted the manuscript. MR edited and revised the manuscript. All authors have approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the research program of the Jena Centre for Systems Biology of Aging (JenAge) funded by the German Ministry for Education and Research (Bundesministerium für Bildung und Forschung/BMBF 0315581), the German Research Association (Deutsche Forschungsgemeinschaft, DFG) Research Training Group 1715 Molecular Signatures of Adaptive Stress Responses, Jena, Germany, the Swiss National Science Foundation (Schweizerischer Nationalfonds, SNF 31003A_156031), the Novartis Foundation (Biomedical research fund; 14C149) and the European Foundation for the Study of Diabetes (EFSD/Lilly Research Fellowship Programme).
Acknowledgements
We are also thankful to Doris Pohlmann and Beate Laube for their expert technical assistance.
References
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd , Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH & Neufer PD (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel‐Duby R & Olson EN (2006). Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem 75, 19–37. [DOI] [PubMed] [Google Scholar]
- Chan P, Di Monte DA, Luo JJ, DeLanney LE, Irwin I & Langston JW (1994). Rapid ATP loss caused by methamphetamine in the mouse striatum: relationship between energy impairment and dopaminergic neurotoxicity. J Neurochem 62, 2484–2487. [DOI] [PubMed] [Google Scholar]
- Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Sykiotis GP & Habeos IG (2011). Nrf2 represses FGF21 during long‐term high‐fat diet‐induced obesity in mice. Diabetes 60, 2465–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Johansson E, Yang Y, Miller ML, Shen D, Orlicky DJ, Shertzer HG, Vasiliou V, Nebert DW & Dalton TP (2010). Oral N‐acetylcysteine rescues lethality of hepatocyte‐specific Gclc‐knockout mice, providing a model for hepatic cirrhosis. J Hepatol 53, 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobley JN, McHardy H, Morton JP, Nikolaidis MG & Close GL (2015). Influence of vitamin C and vitamin E on redox signaling: implications for exercise adaptations. Free Radic Biol Med 84, 65–76. [DOI] [PubMed] [Google Scholar]
- Egan B & Zierath JR (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184. [DOI] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Borras C, Pallardo FV, Sastre J, Ji LL & Vina J (2005). Decreasing xanthine oxidase‐mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J Physiol 567, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J & Vina J (2008). Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training‐induced adaptations in endurance performance. Am J Clin Nutr 87, 142–149. [DOI] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Salvador‐Pascual A, Cabo H, Ferrando B & Vina J (2015). Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic Biol Med 86, 37–46. [DOI] [PubMed] [Google Scholar]
- He HJ, Wang GY, Gao Y, Ling WH, Yu ZW & Jin TR (2012). Curcumin attenuates Nrf2 signaling defect, oxidative stress in muscle and glucose intolerance in high fat diet‐fed mice. World J Diabetes 3, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida K, Kim SH, Higuchi M, Holloszy JO & Han DH (2011). Normal adaptations to exercise despite protection against oxidative stress. Am J Physiol Endocrinol Metab 301, E779–E784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji LL (2008). Modulation of skeletal muscle antioxidant defense by exercise: role of redox signaling. Free Radic Biol Med 44, 142–152. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF & Richter EA (2007). Role of AMPKalpha2 in basal, training‐, and AICAR‐induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab 292, E331–E339. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N & Biswal S (2007). Cell survival responses to environmental stresses via the Keap1‐Nrf2‐ARE pathway. Annu Rev Pharmacol Toxicol 47, 89–116. [DOI] [PubMed] [Google Scholar]
- Kim SK, Joe Y, Zheng M, Kim HJ, Yu JK, Cho GJ, Chang KC, Kim HK, Han J, Ryter SW & Chung HT (2014). Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production. Antioxid Redox Signal 20, 2589–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD & Muoio DM (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7, 45–56. [DOI] [PubMed] [Google Scholar]
- Lau A, Tian W, Whitman SA & Zhang DD (2013). The predicted molecular weight of Nrf2: it is what it is not. Antioxid Redox Signal 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Lee BS, Semnani S, Avanesian A, Um CY, Jeon HJ, Seong KM, Yu K, Min KJ & Jafari M (2010). Curcumin extends life span, improves health span, and modulates the expression of age‐associated aging genes in Drosophila melanogaster . Rejuvenation Res 13, 561–570. [DOI] [PubMed] [Google Scholar]
- Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J & Pilegaard H (2008). PGC‐1alpha is not mandatory for exercise‐ and training‐induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab 294, E463–E474. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C & Spiegelman BM (2005). Metabolic control through the PGC‐1 family of transcription coactivators. Cell Metab 1, 361–370. [DOI] [PubMed] [Google Scholar]
- Lira VA, Benton CR, Yan Z & Bonen A (2010. a). PGC‐1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am J Physiol Endocrinol Metab 299, E145–E161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH & Criswell DS (2010. b). Nitric oxide and AMPK cooperatively regulate PGC‐1 in skeletal muscle cells. J Physiol 588, 3551–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConell GK, Ng GP, Phillips M, Ruan Z, Macaulay SL & Wadley GD (2010). Central role of nitric oxide synthase in AICAR and caffeine‐induced mitochondrial biogenesis in L6 myocytes. J Appl Physiol (1985) 108, 589–595. [DOI] [PubMed] [Google Scholar]
- McMahon M, Lamont DJ, Beattie KA & Hayes JD (2010). Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci USA 107, 18838–18843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merry TL & Ristow M (2015). Do antioxidant supplements interfere with skeletal muscle adaptation to exercise training? J Physiol 594, 5135–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merry TL, Tran M, Stathopoulos M, Wiede F, Fam BC, Dodd GT, Clarke I, Watt MJ, Andrikopoulos S & Tiganis T (2014). High‐fat‐fed obese glutathione peroxidase 1‐deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxid Redox Signal 20, 2114–2129. [DOI] [PubMed] [Google Scholar]
- Muthusamy VR, Kannan S, Sadhaasivam K, Gounder SS, Davidson CJ, Boeheme C, Hoidal JR, Wang L & Rajasekaran NS (2012). Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med 52, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan M, Hong J, Atieno N, Muthusamy VR, Davidson CJ, Abu‐Rmaileh N, Richardson RS, Gomes AV, Hoidal JR & Rajasekaran NS (2014). Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free Radic Biol Med 71C, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Nioi P & Pickett CB (2009). The Nrf2‐ARE signaling pathway and its activation by oxidative stress. J Biol Chem 284, 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S & Carruba MO (2003). Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899. [DOI] [PubMed] [Google Scholar]
- Paulsen G, Cumming KT, Holden G, Hallen J, Ronnestad BR, Sveen O, Skaug A, Paur I, Bastani NE, Ostgaard HN, Buer C, Midttun M, Freuchen F, Wiig H, Ulseth ET, Garthe I, Blomhoff R, Benestad HB & Raastad T (2014. a). Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double‐blind randomized controlled trial. J Physiol 592, 1887–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen G, Hamarsland H, Cumming KT, Johansen RE, Hulmi JJ, Borsheim E, Wiig H, Garthe I & Raastad T (2014. b). Vitamin C and E supplementation alters protein signalling after a strength training session, but not muscle growth during 10 weeks of training. J Physiol 592, 5391–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Carraway MS, Babiker A & Suliman HB (2008). Heme oxygenase‐1 regulates cardiac mitochondrial biogenesis via Nrf2‐mediated transcriptional control of nuclear respiratory factor‐1. Circ Res 103, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA & Suliman HB (2006). Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem 281, 324–333. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty‐Wolf KE & Suliman HB (2011). Heme oxygenase‐1 couples activation of mitochondrial biogenesis to anti‐inflammatory cytokine expression. J Biol Chem 286, 16374–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK & Jackson MJ (2008). Exercise‐induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M & Zarse K (2010). How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 45, 410–418. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR & Blüher M (2009). Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Nat Acad Sci USA 106, 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M & Ristow M (2007). Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Southam CM & Ehrlich J (1943). Effects of extract of western red‐cedar heartwood on certain wood‐decaying fungi in culture. Phytopathology 33, 517–524. [Google Scholar]
- Srere PA (1969). Citrate synthase. Methods Enzymol 13, 3–11. [Google Scholar]
- Strobel NA, Peake JM, Matsumoto A, Marsh SA, Coombes JS & Wadley GD (2011). Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Med Sci Sports Exerc 43, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Suckow BK & Suckow MA (2006). Lifespan extension by the antioxidant curcumin in Drosophila melanogaster . Int J Biomed Sci 2, 402–405. [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kumagai T, Goto A & Sugiura T (1998). Increase in intracellular hydrogen peroxide and upregulation of a nuclear respiratory gene evoked by impairment of mitochondrial electron transfer in human cells. Biochem Biophys Res Commun 249, 542–545. [DOI] [PubMed] [Google Scholar]
- Wadley GD & McConell GK (2007). Effect of nitric oxide synthase inhibition on mitochondrial biogenesis in rat skeletal muscle. J Appl Physiol (1985) 102, 314–320. [DOI] [PubMed] [Google Scholar]
- Wadley GD, Nicolas MA, Hiam DS & McConell GK (2013). Xanthine oxidase inhibition attenuates skeletal muscle signaling following acute exercise but does not impair mitochondrial adaptations to endurance training. Am J Physiol Endocrinol Metab 304, E853–E862. [DOI] [PubMed] [Google Scholar]
- Warburton DE, Nicol CW & Bredin SS (2006). Health benefits of physical activity: the evidence. Can Med Ass J (CMAJ) 174, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer S, Priebs J, Kuhlow D, Groth M, Priebe S, Mansfeld J, Merry TL, Dubuis S, Laube B, Pfeiffer AF, Schulz TJ, Guthke R, Platzer M, Zamboni N, Zarse K & Ristow M (2014). D‐glucosamine supplementation extends lifespan of nematodes and of ageing mice. Nat Commun 5, 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue P, Hou Y, Chen Y, Yang B, Fu J, Zheng H, Yarborough K, Woods CG, Liu D, Yamamoto M, Zhang Q, Andersen ME & Pi J (2013). Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes 62, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yfanti C, Akerstrom T, Nielsen S, Nielsen AR, Mounier R, Mortensen OH, Lykkesfeldt J, Rose AJ, Fischer CP & Pedersen BK (2010). Antioxidant supplementation does not alter endurance training adaptation. Med Sci Sports Exerc 42, 1388–1395. [DOI] [PubMed] [Google Scholar]
- Yu Z, Shao W, Chiang Y, Foltz W, Zhang Z, Ling W, Fantus IG & Jin T (2011). Oltipraz upregulates the nuclear factor (erythroid‐derived 2)‐like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high‐fat diet in C57BL/6 J mice. Diabetologia 54, 922–934. [DOI] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Guthke R, Platzer M, Kahn CR & Ristow M (2012). Impaired insulin/IGF1‐signaling extends life span by promoting mitochondrial L‐proline catabolism to induce a transient ROS signal. Cell Metab 15, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]