

Abstract
N-methyl D-aspartate receptor subtype 2B (GluN2B), encoded by GRIN2B, is one of the components of the N-methyl D-aspartate receptor protein. Aberrations in GRIN2B have been reported to be responsible for various types of neurodevelopmental disorders. We report a Japanese boy with an ~2 Mb interstitial deletion in 12p13 involving the entire GRIN2B gene, who presented with intellectual disability, motor developmental delay and marked macrocephaly.
The N-methyl D-aspartate receptor (NMDAR) is one of the ionotropic glutamate receptors involved in intermediate excitatory neurotransmission in the mammalian central nervous system.1,2 NMDARs are associated with higher cerebral function or cellular apoptosis after ischemic brain disease. They consist of three subunits, GluN1, GluN2 and GluN3, and the GluN2 subunit is further classified into GluN2A, GluN2B, GluN2C and GluN2D. GRIN2B is located on 12p13.1 and encodes GluN2B. Variants in GRIN2B are associated with various psychiatric disorders, including schizophrenia,3 Parkinson’s disease4 and bipolar disorder.5 GRIN2B mutations are also associated with an autosomal dominant form of intellectual disability (MRD6, OMIM#613970) and early infantile epileptic encephalopathy (EIEE27, OMIM#616139),6 but these are rare diseases involving partial or entire GRIN2B deletions.
Here we report the case of a boy with mild intellectual disability and macrocephaly without epilepsy who has an ~2 Mb interstitial deletion in 12p13, including the entire GRIN2B. The patient is a 6-year-old Japanese boy who is the first child of non-consanguineous parents. His gestational age was 40 weeks and 0 days. His birth weight was 4,112 g (+2.91 s.d.), his length was 52 cm (+1.48 s.d.), and his head circumference was 36 cm (+2.2 s.d.). His parents and younger sister are healthy. He was referred to our hospital at an age of 7 months because of a motor developmental delay and intellectual disability. At that time, he was unable to hold his head up. He could not sit alone at the age of 1 year. At 2 years and 0 months, his height was 90.9 cm (+1.8 s.d.), his weight was 13.28 kg (+1.4 s.d.; Figure 1a), and his head circumference was 54.0 cm (+4.8 s.d.; Figure 1b). He showed macrocephaly, but his face was not disease-specific, and he had no epileptic convulsions. Cranial magnetic resonance imaging revealed no significant abnormality. His total developmental quotient, as assessed by the Kyoto Scale of Psychological Development 2001, was 60 (at 3 years and 6 months), 65 (at 5 years of age), and 67 (at 6 years and 1 month).7 He was also diagnosed with autism spectrum disorder (ASD). Standard G-band karyotyping revealed a normal male karyotype (46, XY).
Figure 1.

Standard growth curve of the patient. He had a normal height and bodyweight (a) but a large head circumference (b). y.o. years-old.
To identify the cause of his developmental delay, we performed chromosomal and genetic analysis after receiving written informed consent from his parents. All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine and were performed in accordance with the ethical standards of the Declaration of Helsinki. We first performed fluorescence in situ hybridization and direct sequencing for NSD1 (the nuclear receptor binding SET domain protein 1 gene on 5q35) because the patient’s developmental delay and macrocephaly suggested that he had Sotos syndrome (SOTOS1; OMIM#117550). However, no aberrations were identified.
Next, we performed direct sequencing of NFIX (nuclear factor I/X gene on 19p13.3), whose mutation can cause Sotos syndrome-2 (SOTOS2; OMIM#614753).8 We identified a heterozygous missense mutation: NM_002501.2: c.830G>A, p.Arg277His. c.830G>A was not registered in the healthy control database in dbSNP (http://www.ncbi.nlm.nih.gov/snp), the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/) or ExAC (http://exac.broadinstitute.org/). Moreover, p.Arg277His was found to be a pathogenic amino-acid change through use of the functional prediction websites PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org/). However, the patient inherited this from one of his healthy parents; thus, we determined that c.830G>A was non-pathogenic.
We then used the TruSight One Sequencing Panel on a MiSeq platform (Illumina, San Diego, CA) following the manufacturer’s instructions to analyze 4,813 genes. However, no mutations were associated with the patient’s neurological disorder. Array-based comparative genomic hybridization (array CGH, 180 k CGH+SNP, Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions identified an ~2 Mb heterozygous deletion in chromosome 12p (Figure 2a): arr[hg19]12p13.2p13.1 (12,482,956-14,436,199) x1. This region included 21 genes: MANSC1, LOH12CR2, LOH12CR1, DUSP16, CREBL2, GPR19, CDKN1B, APOLD1, MIR613, DDX47, RPL13AP20, GPRC5A, MIR614, GPRC5D, HEBP1, HTR7P1, KIAA1467, GSG1, EMP1, C12orf36 and GRIN2B (Figure 2b). The deletion was not detected in either parent and was therefore considered to be de novo. Although one healthy control sample (nsv826262) in the database of genomic variants (http://dgv.tcag.ca/dgv/app/home)9 carried a small deletion including GRIN2B exon 4, we concluded that the GRIN2B mutations have a central role in the phenotype of this patient.
Figure 2.
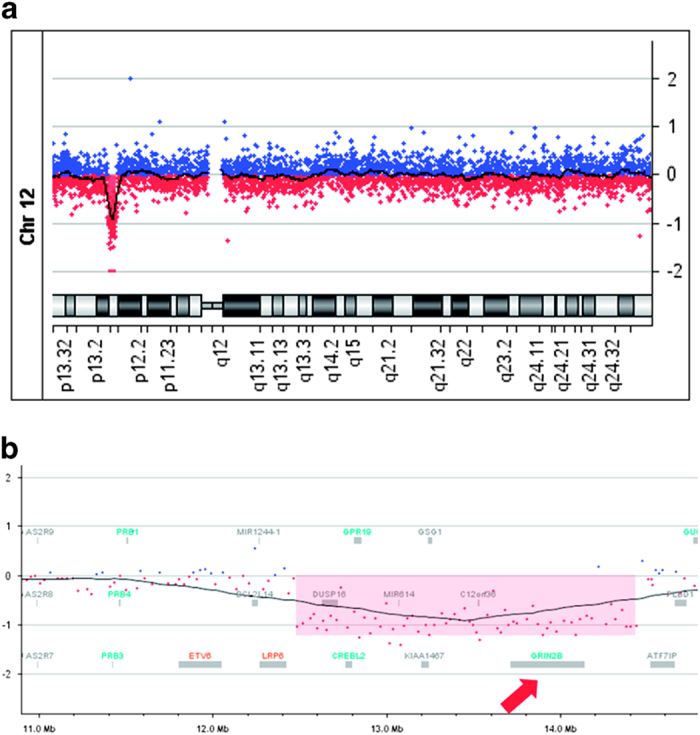
An approximately 2 Mb deletion on the short arm of chromosome 12 was revealed by array CGH analysis (a). This region contains 21 genes, including GRIN2B (red arrow; b).
NMDARs are commonly assembled as two GluN1 subunits and two GluN2 subunits.10 During the fetal and early postnatal period, GluN1/2B exists predominantly in the mammalian brain, and GluN1/2A is later dominant in the adult brain.11 GRIN2B is essential for neural pattern formation, synaptic plasticity, short-term memory and language acquisition.12 Moreover, disruptions or mutations of GRIN2B have been reported to be responsible for mild to severe intellectual disability6 and autism.13 Endele et al.6 have previously reported two patients with chromosomal translocations of GRIN2B and 4 out of 468 patients with intellectual disability that had GRIN2B mutations. All patients showed behavioral anomalies including ASD, although none had epilepsy, facial dysmorphism or macrocephaly; patients with GRIN2A mutations frequently have epileptic seizures.6 Disruption of GRIN2A is responsible for focal epilepsy and speech disorders, with or without intellectual disability (OMIM#245570).14 GRIN2C and GRIN2D have not been shown to be associated with neurodevelopmental disorders.
Dimassi et al.12 were the first to report patients with a 12p13.1 microdeletion including all or part of GRIN2B. The one female and two male patients affected presented with clinical phenotypes of moderate intellectual disability, delayed language development and mild facial defects, including an oval-shaped face, arched eyebrows and a high forehead.12 Their growth was almost normal, including their head circumferences. Our patient showed macrocephaly but lacked the dolichocephaly and prominent head seen in Sotos syndrome. Because intellectual disability, language developmental delay and macrocephaly are common features of ASD patients, the 12p13 interstitial deletion should be considered as one responsible for non-specific ASD and intellectual disability.
In conclusion, array CGH is useful for disease diagnosis in non-specific neurodevelopmental patients. Our results should provide insight into the relationship between glutamate receptor expression and neurodevelopmental disease.
Acknowledgments
The authors acknowledge the cooperation of the patient and his family.
Footnotes
KI has received grants from Daiichi Sankyo, Japan Blood Product Organization, Miyarisan Pharmaceutical, AbbVie LLC, CSL Behring, JCR Pharmaceuticals, Teijin Pharma, Novo Nordisk Pharma, AIR WATER MEDICAL, Astellas Pharma, Takeda Pharmaceutical Company and Taisho Toyama Pharmaceutical, as well as lecture fees from Meiji Seika Pharma, Novartis Pharma K.K., Zenyaku Kogyo, Chugai Pharmaceutical, Daiichi Sankyo, Springer Japan, Asahi Kasei Pharma, Boehringer Ingelheim, Medical Review, NIKKEI RADIO BROADCASTING CORPORATION, Japan Blood Product Organization and CSL Behring, manuscript fees from Chugai Pharmaceutical and consulting fees from Zenyaku Kogyo, Chugai Pharmaceutical, Astellas Pharma, Takeda Pharmaceutical Company and Ono Pharmaceutical.
References
- Cull-Candy S , Brickley S , Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 2001; 11: 327–335. [DOI] [PubMed] [Google Scholar]
- Yamamoto H , Hagino Y , Kasai S , Ikeda K. Specific Roles of NMDA Receptor Subunits in Mental Disorders. Curr Mol Med 2015; 15: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki T , Sakurai K , Dou H , Toru M , Yamakawa-Kobayashi K , Arinami T. Mutation analysis of the NMDAR2B (GRIN2B) gene in schizophrenia. Mol Psychiatry 2001; 6: 211–216. [DOI] [PubMed] [Google Scholar]
- Wu SL , Wang WF , Shyu HY , Ho YJ , Shieh JC , Fu YP et al. Association analysis of GRIN1 and GRIN2B polymorphisms and Parkinson's disease in a hospital-based case-control study. Neurosci Lett 2010; 478: 61–65. [DOI] [PubMed] [Google Scholar]
- Beneyto M , Kristiansen LV , Oni-Orisan A , McCullumsmith RE , Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007; 32: 1888–1902. [DOI] [PubMed] [Google Scholar]
- Endele S , Rosenberger G , Geider K , Popp B , Tamer C , Stefanova I et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 2010; 42: 1021–1026. [DOI] [PubMed] [Google Scholar]
- Aoki S , Hashimoto K , Ikeda N , Takekoh M , Fujiwara T , Morisaki N et al. Comparison of the Kyoto Scale of Psychological Development 2001 with the parent-rated Kinder Infant Development Scale (KIDS). Brain Dev 2015; 38: 481–490. [DOI] [PubMed] [Google Scholar]
- Yoneda Y , Saitsu H , Touyama M , Makita Y , Miyamoto A , Hamada K et al. Missense mutations in the DNA-binding/dimerization domain of NFIX cause Sotos-like features. J Hum Genet 2012; 57: 207–211. [DOI] [PubMed] [Google Scholar]
- Park H , Kim JI , Ju YS , Gokcumen O , Mills RE , Kim S et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet 2010; 42: 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow NG , Siegler Retchless B , Johnson JW. Molecular bases of NMDA receptor subtype-dependent properties. J Physiol 2015; 593: 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature 1992; 357: 686–689. [DOI] [PubMed] [Google Scholar]
- Dimassi S , Andrieux J , Labalme A , Lesca G , Cordier MP , Boute O et al. Interstitial 12p13.1 deletion involving GRIN2B in three patients with intellectual disability. Am J Med Genet A 2013; 161A: 2564–2569. [DOI] [PubMed] [Google Scholar]
- Tarabeux J , Kebir O , Gauthier J , Hamdan FF , Xiong L , Piton A et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry 2011; 1: e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reutlinger C , Helbig I , Gawelczyk B , Subero JIM , Tonnies H , Muhle H et al. Deletions in 16p13 including GRIN2A in patients with intellectual disability, various dysmorphic features, and seizure disorders of the rolandic region. Epilepsia 2010; 51: 1870–1873. [DOI] [PubMed] [Google Scholar]
Data Citations
- Morisada Naoya.HGV Database. 2016. 10.6084/m9.figshare.hgv.845. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Morisada Naoya.HGV Database. 2016. 10.6084/m9.figshare.hgv.845. [DOI]