

Abstract
Type 2 diabetes mellitus is associated with accelerated cognitive decline. The underlying pathophysiological mechanisms still remain to be elucidated although it is known that insulin signaling modulates neurotransmitter activity, including inhibitory γ-aminobutyric acid (GABA) and excitatory glutamate (Glu) receptors. Therefore, we examined whether levels of GABA and Glu are related to diabetes status and cognitive performance.
Forty-one participants with type 2 diabetes and 39 participants without type 2 diabetes underwent detailed cognitive assessments and 3-Tesla proton MR spectroscopy. The associations of neurotransmitters with type 2 diabetes and cognitive performance were examined using multivariate regression analyses controlling for age, sex, education, BMI, and percentage gray/white matter ratio in spectroscopic voxel.
Analysis revealed higher GABA+ levels in participants with type 2 diabetes, in participants with higher fasting blood glucose levels and in participants with higher HbA1c levels, and higher GABA+ levels in participants with both high HbA1c levels and less cognitive performance.
To conclude, participants with type 2 diabetes have alterations in the GABAergic neurotransmitter system, which are related to lower cognitive functioning, and hint at the involvement of an underlying metabolic mechanism.
Keywords: γ-aminobutyric acid, cognition, glutamate, magnetic resonance spectroscopy, type 2 diabetes mellitus
1. Introduction
Type 2 diabetes mellitus is an endocrine disorder characterized by attenuated insulin signaling and decreased cellular responsiveness to insulin. Since systemic insulin resistance is accompanied by central insulin resistance, the complications of diabetes not only involve peripheral tissues, but also the central nervous system (CNS).[1,2] Indeed, type 2 diabetes is associated with cognitive deficits,[3] accelerated cognitive decline,[4] and an increased risk for developing dementia and Alzheimer disease.[4–7] Insulin signaling plays an important role in synaptic plasticity by modulating neurotransmitter channel activity, including excitatory, and inhibitory receptors such as γ-aminobutyric acid (GABA) and glutamate (Glu) receptors.[8,9] Therefore, defects in brain insulin signaling may give rise to neuronal dysfunction and impaired cognitive performance.[10,11]
Proton MR spectroscopy (1H-MRS) provides the unique opportunity to assess noninvasively the concentrations of neurometabolites including neurotransmitters GABA and Glu in vivo, through the identification and quantification of spectral peaks. GABA is the major inhibitory neurotransmitter, whereas Glu is the major excitatory neurotransmitter. At clinical field strengths (≤3.0T), GABA and Glu are difficult to quantify due to the spectral overlap with the signals of other metabolites, including n-acetyl aspartate (NAA), total creatine (tCr), and glutamine (Gln). However, advanced spectral editing methods have been developed that enable the detection of metabolites with strong spectral overlap. For example, the MEscher-GArwood-point resolved spectroscopy sequence (MEGA-PRESS) edited 1H-MRS method can be used for the quantification of GABA.[12]
As 1H-MRS facilitates the assessment of neurotransmitters GABA and Glu, it is a suitable technique to explore whether an altered neurotransmitter metabolism underlies cognitive problems in type 2 diabetes. A previous 1H-MRS study with respect to neurotransmitters in depressed type 2 diabetes has reported decreased Glu levels[13] compared with healthy controls. Moreover, lower GABA levels were observed in patients with diabetic neuropathy.[14]
To our knowledge, no studies explored the neurotransmitter metabolism of impaired cognitive performance in type 2 diabetes. Therefore, the current study was designed to assess whether neurotransmitters (GABA and Glu) are related to type 2 diabetes status, cognitive performance, and the potential interaction between type 2 diabetes and cognitive performance.
2. Materials and methods
2.1. Study population
Forty-seven participants with type 2 diabetes and 41 participants without type 2 diabetes were recruited from the first 866 participants of the Maastricht Study for additional brain MRI measurements. The Maastricht Study is an ongoing observational, prospective, population-based cohort study that focuses on the etiology, pathophysiology, complications, and comorbidities of type 2 diabetes. Participants are between 40 and 75 years of age and live in the southern part of the Netherlands.[15] Participants are considered to have diabetes according to the WHO 2006 criteria if they use diabetes medication or if they have a fasting blood glucose ≥7.0 mmol/L or a 2-hours blood glucose ≥11.1 mmol/L. Participants without type 2 diabetes are characterized by fasting blood glucose <6.1 mmol/L and a 2-hours blood glucose <7.8 mmol/L. At baseline inclusion, participants underwent an extensive battery of measurements, including cognitive performance tasks, blood pressure measurements, and blood sampling. A detailed overview is provided in Schram et al.[15] After their baseline measurements of the Maastricht Study, participants were invited to participate in this MRI study.
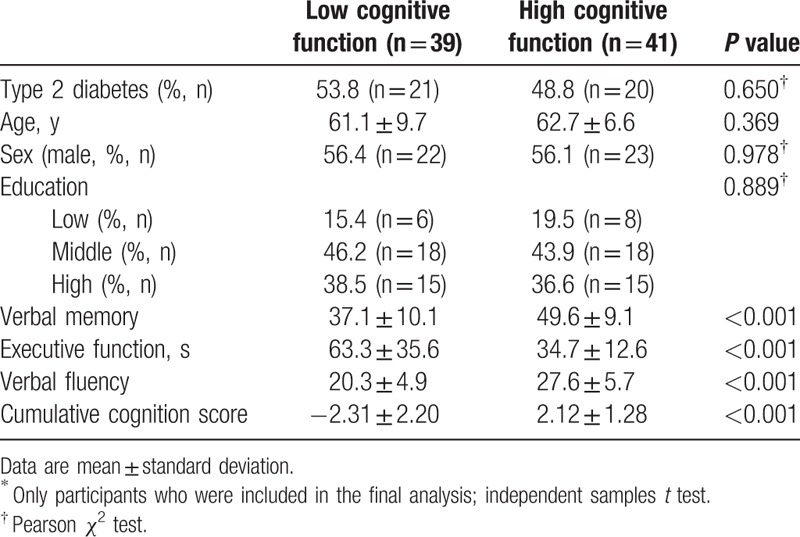
Participants with the highest and lowest cognitive scores were selected from the first 866 participants to increase the probability of finding MRI differences associated with cognitive decrements (Table 1). A detailed selection procedure is provided in van Bussel et al.[16] In brief, the division of participants in a low and high cognition group was based on a cumulative score of three neuropsychological tests covering the domains of verbal memory, attention and flexibility, and executive functioning (Table 1). For matching, the scores for each neuropsychological test were adjusted for age, sex, and education level by linear regression and the cumulative cognition score was calculated by adding the corresponding z-scores (standardized residuals) of the three neuropsychological tests. Exclusion criteria for participants were: a known history of stroke or neurological disease, if the time span between enrollment in the Maastricht Study and MRI was >1.5 years, incomplete cognitive assessments, type 1 diabetes mellitus, mild cognitive impairment, participants with the metabolic syndrome, participants with color blindness, participants with an unknown diabetes status, and for participants without type 2 diabetes, an impaired fasting blood glucose level. The low and high cognition groups were matched on age, sex, and education level, and display a similar distribution of participants with and without type 2 diabetes (Table 1).
Table 1.
Characteristics of the two cognition groups∗.
After taking into account those who declined the invitation and exclusion of participants with MRI contra-indications, a total of 47 and 41 participants with and without type 2 diabetes were included, respectively.
Prior to MRI, all participants underwent a general cognitive function test (Mini-Mental State Examination, MMSE[17]) to assess clinically significant differences in cognitive performance compared with the baseline cognitive tests at enrollment in the Maastricht Study. None of the participants were excluded based on a MMSE score of ≤24. Structural brain and MR spectroscopy scans were obtained from all participants. This study was approved by the Medical Ethics Committee of the Maastricht University Medical Center (MUMC+), the Netherlands, and all participants gave written informed consent. The study is registered at http://www.clinicaltrials.gov with identifier NCT01705210.
2.2. Magnetic resonance imaging
MRI data were acquired on a 3T scanner (Achieva TX, Philips Healthcare, Best, The Netherlands) using a 32-element head coil for parallel imaging. The MRI protocol consisted of structural scans for neuroradiological evaluation (including T1-, T2-, T2∗-weighted, and fluid attenuated inversion recovery (FLAIR) sequences) and 1H-MRS scans. A 3-dimensional T1-weigthed (T1) fast field echo sequence (TR/TE 8.1/3.7 ms, 1.00 mm isotropic voxel size, 170 continuous slices, matrix size of 240 × 240, and 7:56 minutes acquisition time) was acquired and used for the positioning of the spectroscopic voxel and voxel segmentation. 1H-MRS were acquired from a 3 × 3 × 3 cm3 voxel located in the occipital lobe (Fig. 1A and B) due to its favorable signal to noise profile using a single voxel PRESS sequence (TR/TE 2000/38 ms, 128 averages, MOIST water suppression, and 4:52 minutes acquisition time). Additionally, a spectrum (16 averages) was recorded of unsuppressed water. For GABA, a MEGA-PRESS sequence (TR/TE = 2000/68 ms, 320 averages, editing pulses at 1.9 (ON), and 7.46 ppm (OFF) interleaved in 40 blocks, MOIST water suppression, and 10:40 minutes acquisition time) was acquired.[12]
Figure 1.
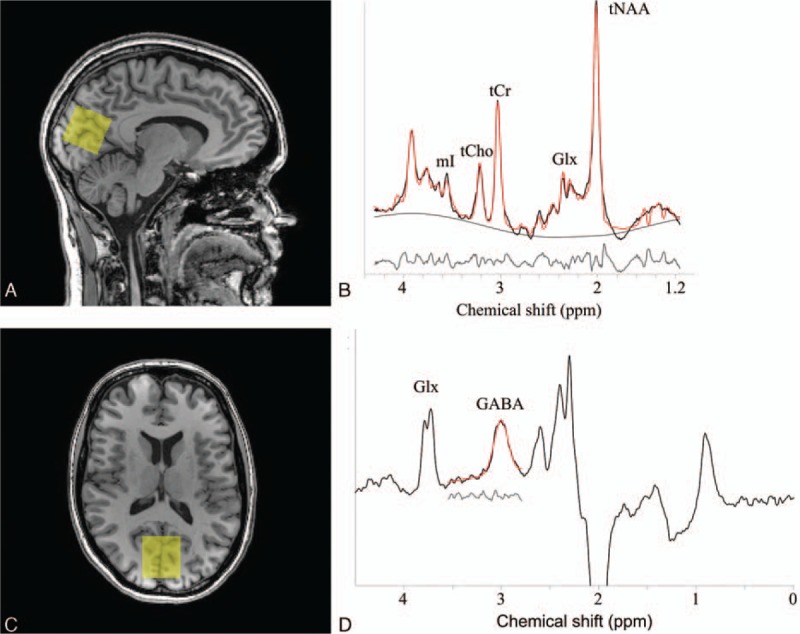
(A) Sagittal and (B) axial view of T1-weighted image of participant without diabetes indicating the 1H-MRS voxel (yellow) in the occipital lobe. (C) Representative PRESS spectrum (black line) and LCModel fit (red line). The smooth black line shows the estimated baselines by LCModel and the gray line shows the residuals between the raw data (black line) and the fit (red line). The peaks in the spectrum represent mI, tCho, tCr, Glx, and tNAA metabolites. (D) Representative MEGA-PRESS spectrum (black line) and the Gannet fit (red line), yielding GABA+ concentration. The gray line shows the residuals between the raw data and the fit.
2.3. Data analysis
The metabolite concentrations within the PRESS voxel were analyzed using the LCModel (linear combination of model spectra) software package (version 6.3-1B), which analyzes the in vivo MR spectra as a linear combination of the spectra of the individual metabolites.[18] LCModel performs water-scaling automatically and uses a simulated basis set. The simulated basis set was provided by Dr Provencher and included the following 16 metabolites: alanine, aspartate, creatine (Cr), GABA, glucose, Gln, Glu, glycerophosphocholine (GPC), phosphocholine (PCh), myo-inositol (mI), lactate, NAA, n-acetylaspartylglutamate (NAAG), scyllo-inositol, taurine, and guanine. Metabolite estimates were excluded from analysis if the Cramér-Rao lower bound, an estimate of the error in metabolite quantification and used as reliability indicator, was greater than 15% range. Glu satisfies the requirements of the Cramér–Rao lower bound <15%. Post hoc analyses were performed for all other metabolites which fulfilled the Cramér-Rao criteria: Glx (the combined signal of Glu and Gln), mI, total choline (tCho; sum of GPC and PCh), total NAA (tNAA; sum of NAA and NAAG), and tCr (sum of Cr and phosphocreatine). Figure 1C shows a typical spectrum and its best-fit model. In this manuscript, metabolic concentrations are reported relative to water[19] and expressed in institutional units (i.u.). To this end, after the in vivo measurement, the signal from unsuppressed tissue water was recorded from the same voxel, which served as an endogenous concentration reference. No corrections for relaxation were performed.
The GABA concentration (quantified in i.u. relative to the unsuppressed water signal from the same volume) was estimated by analyzing the MEGA-PRESS spectrum using the Gannet 2.0 toolkit, a Matlab-based quantification batch analysis tool for analyzing GABA MEGA-PRESS spectra.[12] A detailed overview is provided in Edden et al.[12] As the GABA concentration estimation likely contains contribution from macromolecules and homocarnosine, it will therefore be referred to as GABA+. Figure 1D shows the fitted GABA+ signal. From here on, the GABA+/H2O ratio is referred to as GABA+ level.
The T1 images were used for the 1H-MRS voxel segmentation (Fig. 1A and B) to account for differences in tissue composition which may influence the metabolite concentrations. The 1H-MRS voxel was coregistered to the T1 image and automatically segmented as white matter (WM), gray matter (GM), or cerebrospinal fluid (CSF) using FSL FAST (FMRIB's Automated Segmentation Tool, Oxford University, Oxford, UK).[20] Then, percentages of WM, GM, and CSF in the 1H-MRS voxel were calculated and the metabolite concentrations were corrected for CSF content. Furthermore, the ratio of GM to WM in the voxel was used as a covariate in linear regression analyses.[21]
After careful analyses, data from 41 type 2 diabetes participants and 39 participants without type 2 diabetes remained suitable for final analysis, as data from 8 participants were excluded due to claustrophobia (n = 2), impaired fasting blood glucose levels (n = 2), parkinsonism (n = 1), brain injury due to an accident (n = 1), an incidental finding (i.e., tumor, n = 1), and unreliable data (n = 1). In addition, 1 included participant had a missing GABA measurement.
2.4. Statistical analysis
Descriptive participant characteristics are reported as mean ± standard deviation. Group characteristics were tested using independent samples t tests and Pearson χ2 tests using SPSS (Statistical Package for Social Sciences, version 20, IBM Corp, Armonk, NY), with α = 0.05.
Linear regression analyses, adjusted for age, sex, education level, BMI,[22] and percentage GM/WM ratio were performed to assess the association of the neurotransmitter concentrations (GABA+ and Glu) with type 2 diabetes status and cognition status. Furthermore, the interaction term between type 2 diabetes and cognition status (lower vs higher cognitive performance) was added to the linear regression model to investigate the combined effect of type 2 diabetes and less cognitive performance on metabolite concentrations.
All analyses were repeated replacing the dichotomous type 2 diabetes status by either fasting blood glucose levels or glycated hemoglobin (HbA1c), a measure for long-term blood glucose control. In addition, post hoc analyses were also performed for other metabolite concentrations (Glx, mI, tCho, tNAA, and tCr).
3. Results
3.1. Characteristics
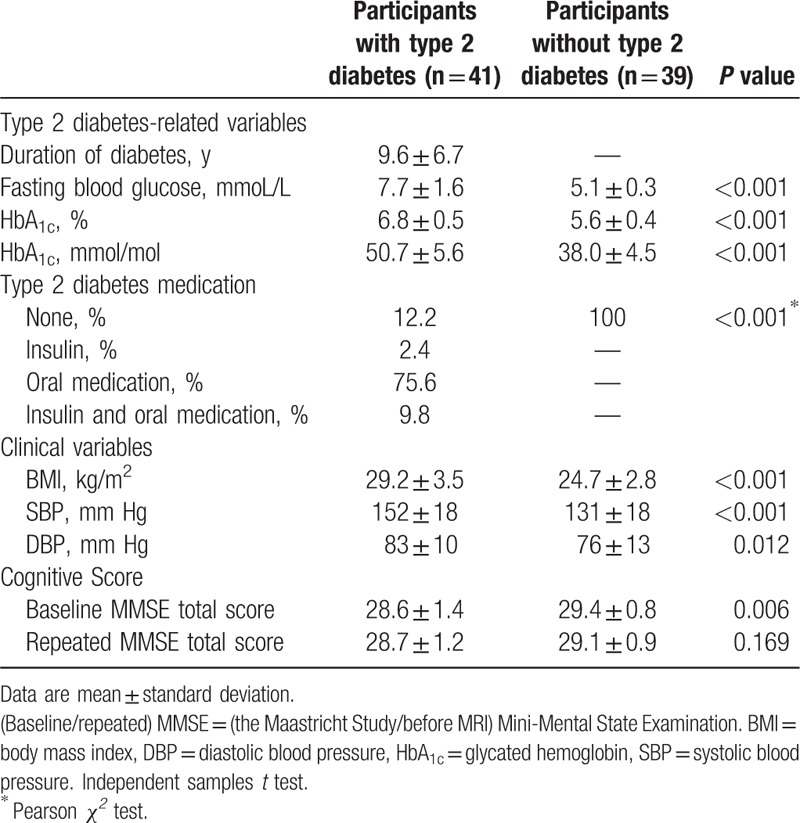
Table 1 shows the baseline characteristics of the low and high cognition groups, as participants were selected based on cognitive performance. The groups were matched on age, sex, education, and type 2 diabetes status, but score different on cognition. Table 2 shows the clinical characteristics of participants based on diabetes status. As expected, participants with type 2 diabetes had higher fasting blood glucose levels, higher HbA1c levels, higher BMI, and both higher systolic and diastolic blood pressure (Table 2). Participants with type 2 diabetes scored significantly worse on baseline MMSE score (P = 0.006). Baseline and repeated MMSE did not differ for all participants (P = 0.280).
Table 2.
Clinical characteristics of participants with and without type 2 diabetes.
3.2. 1H-MRS
Linear regression (Table 3) revealed higher GABA+ levels in participants with type 2 diabetes (P = 0.041), but no differences in Glu concentrations. Linear regression analyses that included the interaction term (diabetes status × cognition status) revealed no significant interaction.
Table 3.
Relationship between the neurotransmitters GABA+ and Glu with type 2 diabetes (either dichotomous status, fasting blood glucose levels, or HbA1c) and cognitive performance and their corresponding interaction term.

Additional linear regression analyses replacing type 2 diabetes status by fasting blood glucose levels or HbA1c showed similar results (Table 3): higher GABA+ levels in participants with higher fasting blood glucose levels and in participants with higher HbA1c levels. Furthermore, linear regression analyses that included the interaction term (HbA1c levels times cognition status) revealed higher GABA+ levels in participants with both higher HbA1c levels and less cognitive performance.
3.3. Post hoc analyses
Post hoc analyses (data not shown) revealed lower tNAA levels in participants who scored less on cognitive performance (β = 0.504, P = 0.030). Other metabolite concentrations did not show a significant analyses with type 2 diabetes or cognitive status (P > 0.267). In addition, higher tCho (β = 0.518, P = 0.013) levels were observed in participants with both higher HbA1c levels and less cognitive performance (interaction analyses).
4. Discussion
The current study examined whether neurotransmitter levels are related to type 2 diabetes, cognitive performance, and the potential interaction between diabetes and cognitive performance. The main findings were that GABA+ is higher in diabetes, as well as in participants with higher fasting blood glucose levels and in participants with higher HbA1c levels, that GABA+ concentrations were higher in participants with both higher HbA1c levels and less cognitive performance. To our knowledge, this is the first study to investigate the role of neurotransmitters in type 2 diabetes and cognitive decrements.
Higher GABA+ levels were found in participants with type 2 diabetes, in participants with higher fasting blood glucose levels, and in participants with both higher HbA1c levels and less cognitive performance. Only one small diabetes study on GABA 1H-MRS is available that reports lower GABA levels in patients with diabetic neuropathy, although translation of this result to the current type 2 diabetes population is not straightforward.[14] In addition, our study population is relatively healthy in terms of good treatment control according to glucose levels and diabetic neuropathy is not yet reported in this group. Nevertheless, increased GABA levels have been found in a type 2 diabetic rat model.[23] GABAergic inhibition is involved in the control of many behaviors, such as anxiety, psychosis, aggression, depression, mood, and cognition.[24] One possible explanation of how GABA affects cognition is by means of its inhibitory function on dopamine release in the mesocortical dopamine pathway, which partly projects to prefrontal cortex. It has previously been hypothesized that this pathway regulates cognition and executive functions.[24] Thus, GABA could downregulate the dopamine activity which might eventually lead to impaired cognitive performance. This issue cannot be resolved with the results of the current study, future studies are needed to confirm this hypothesis.
No significant changes in Glu levels were found for participants with type 2 diabetes, less cognitive performance, nor their interaction, whereas a previous 1H-MRS study did report decreased subcortical Glu levels in type 2 diabetes.[13] In contrary to our participants, these patients were suffering from a major depression, which could explain the difference in Glu concentrations. In addition, an important methodological difference is that the voxel placement and field strength (1.5T) differs compared with our study. Rather than excluding a potential role for the glutamatergic neurotransmitter system in less cognitive performance and type 2 diabetes, it could be the case that our applied technique for detecting Glu is simply not sensitive enough. An alternative 1H-MRS technique, making use of spectral editing, might yield better results, due to an enhanced sensitivity.[25]
The post hoc analyses revealed significant results regarding tNAA and tCho, and although this study was not designed specifically to investigate these metabolites, the results are still interesting. For participants who scored less on cognitive performance, we observed lower tNAA levels. tNAA is a surrogate marker of normal functioning neurons and these results indicate that a decline in neuronal integrity is associated with cognitive deficits. Similar results have been found in patients with mild cognitive impairment, which might suggest that tNAA could be a predictor for cognitive deficits.[26] With respect to cognition and type 2 diabetes, one 1H-MRS study observed no differences in NAA metabolite concentrations and concluded that cognitive decline cannot be explained by this metabolite.[27] Nevertheless, this study was performed at lower field strength (1.5 Tesla), which could make the quantification less sensitive to NAA differences.
Furthermore, our study also observed higher tCho levels in participants with both higher HbA1c levels and less cognitive performance. Choline is involved is membrane turnover, which is a process of loss and replacement of cellular membrane, inflammatory processes, astrocytosis, and in the synthesis of the neurotransmitter acetylcholine and all these processes could affect cognitive performance.[24,28] Interestingly, Sahin et al[29] found higher choline levels in participants with poor glycemic control. Similar higher choline levels have been found in patients with Alzheimer Disease characterized by poor recognition memory performance.[30] Therefore, altered membrane metabolism seems to underlie cognitive decrements both in Alzheimer Disease and type 2 diabetes, which may indicate a shared mechanism.
This study has several strengths: first, to our knowledge, it is the first study to investigate the neurotransmitter system in participants with type 2 diabetes in relationship with cognitive functioning. Second, the participants are extensively characterized. Third, the detection and quantification of the neurometabolites was applied at higher field strength (3T) and using editing, which results in improved spectral resolution, compared with 1.5 T in most other studies in type 2 diabetes.
A number of limitations also need to be addressed. First, the study had a cross-sectional design, so the results should be interpreted cautiously in terms of causality. Nevertheless, the first results are promising and open directions to future longitudinal studies assessing neurotransmitter metabolism in type 2 diabetes and cognitive decrements. Second, our voxel was placed in the occipital lobe, chosen for optimal spectral quality, rather than neuropsychological relevance.[4] However, the fact that we do find significant effects, despite the occipital placement of the voxel, might indicate that metabolic effects are global. Future studies could apply spectroscopic imaging to explore multiple brain regions to assess whether spatial variations could be related to specific cognitive domains. Third, the association between receptor activity and metabolite concentration is not necessarily linear but rather intricate. Fourth, as the interaction analyses only observed an effect with GABA for the continuous HbA1c levels rather than the dichotomous diabetes status, it seems that HbA1c levels are more sensitive to detect an interaction effect.
4.1. Clinical perspectives
Unfortunately, our study was not able to measure cerebral insulin levels and therefore we were not able to link whether insulin causes directly the increase in GABA+ levels. Other studies already have shown the involvement of insulin on increased activity of dopamine neurons, and increased expression of the GABA receptors.[8,9,31] Thus insulin could also indirectly (independent of GABA+) be involved in the process of cognitive decrements in participants with type 2 diabetes. Therefore an important question to be addressed in future studies is whether insulin modulates directly the levels of GABA+ in the brain of participants with type 2 diabetes and causes cognitive decrements. Furthermore, our results open directions for future (longitudinal and pharmacological) studies, which are needed to unravel the underlying mechanism of cognitive decrements in type 2 diabetes. It would be interesting to investigate and validate whether induced changes in the GABAergic system or choline mechanism, for instance with drug therapies (GABAergic drugs or choline agonists), can lead to improvements in cognitive performance or prevention of cognitive deficits in participants with type 2 diabetes. Interestingly, drug treatment with Xanomeline, a muscarinic acetylcholine receptor agonist, has already been shown to provide promising results in improving cognitive performance in Alzheimer disease, which was reflected by the normalization of cerebral choline to normal levels.[32]
In conclusion, this study revealed alterations in GABAergic neurotransmitter system in participants with type 2 diabetes, which were related to lower cognitive functioning.
Acknowledgments
The authors acknowledge Marc Geerlings and Jos Slenter (Radiology and Nuclear Medicine, Maastricht University Medical Center, Maastricht, The Netherlands) for continuous hardware and software support. The authors also acknowledge Alfons Kessels (Clinical Epidemiology and Medical Technology Assessment, Maastricht University Medical Center, Maastricht, The Netherlands) for statistical support.
Footnotes
Abbreviations: 1H-MRS = proton magnetic resonance spectroscopy, CNS = central nervous system, Cr = creatine, CSF = cerebrospinal fluid, GABA = γ-aminobutyric acid, Gln = glutamine, Glu = glutamate, Glx = combined Glu and Gln signals, GM = gray matter, GPC = glycerophosphocholine, MEGA-PRESS = Mescher-Garwood-point resolved spectroscopy sequence, mI = myo-inositol, MMSE = mini-mental state examination, NAA = n-acetyl aspartate, NAAG = n-acetylaspartylglutamate, PCh = phosphocholine, tCho = total choline, tCr = total creatine, tNAA = total n-acetyl aspartate, WM = white matter.
All authors gave final approval of the version to be published and agreed to the be listed as authors.
JFAJ was funded by VENI research grant 916.11.059 from The Netherlands Organization for Scientific Research (NWO) and The Netherlands Organization for Health Research and Development (ZonMw). Additionally, this work was supported by “Stichting de Weijerhorst” foundation. This study applies tools developed under NIH R01 EB016089 and P41 EB015909; RAEE and NAJP also receive salary support from these grants.
The authors have no conflicts of interest to disclose.
References
- 1.McNay EC, Recknagel AK. Brain insulin signaling: a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem 2011; 96:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reagan LP. Diabetes as a chronic metabolic stressor: causes, consequences and clinical complications. Exp Neurol 2012; 233:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Berg E, Reijmer YD, de Bresser J, et al. A 4 year follow-up study of cognitive functioning in patients with type 2 diabetes mellitus. Diabetologia 2010; 53:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Awad N, Gagnon M, Messier C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J Clin Exp Neuropsychol 2004; 26:1044–1080. [DOI] [PubMed] [Google Scholar]
- 5.Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer's disease: an epidemiological perspective. Eur J Pharmacol 2008; 585:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kloppenborg RP, van den Berg E, Kappelle LJ, et al. Diabetes and other vascular risk factors for dementia: which factor matters most? A systematic review. Eur J Pharmacol 2008; 585:97–108. [DOI] [PubMed] [Google Scholar]
- 7.Spauwen PJ, Kohler S, Verhey FR, et al. Effects of type 2 diabetes on 12-year cognitive change: results from the Maastricht Aging Study. Diabetes Care 2013; 36:1554–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleinridders A, Ferris HA, Cai W, et al. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014; 63:2232–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao WQ, Chen H, Quon MJ, et al. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol 2004; 490:71–81. [DOI] [PubMed] [Google Scholar]
- 10.Karczewska-Kupczewska M, Tarasow E, Nikolajuk A, et al. The effect of insulin infusion on the metabolites in cerebral tissues assessed with proton magnetic resonance spectroscopy in young healthy subjects with high and low insulin sensitivity. Diabetes Care 2013; 36:2787–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Felice FG, Lourenco MV, Ferreira ST. How does brain insulin resistance develop in Alzheimer's disease? Alzheimers Dement 2014; 10 (1 suppl):S26–32. [DOI] [PubMed] [Google Scholar]
- 12.Edden RA, Puts NA, Harris AD, et al. Gannet: a batch-processing tool for the quantitative analysis of gamma-aminobutyric acid-edited MR spectroscopy spectra. J Magn Reson Imaging 2014; 40:1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ajilore O, Haroon E, Kumaran S, et al. Measurement of brain metabolites in patients with type 2 diabetes and major depression using proton magnetic resonance spectroscopy. Neuropsychopharmacology 2007; 32:1224–1231. [DOI] [PubMed] [Google Scholar]
- 14.Petrou M, Pop-Busui R, Foerster BR, et al. Altered excitation-inhibition balance in the brain of patients with diabetic neuropathy. Acad Radiol 2012; 19:607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schram MT, Sep SJ, van der Kallen CJ, et al. The Maastricht Study: an extensive phenotyping study on determinants of type 2 diabetes, its complications and its comorbidities. Eur J Epidemiol 2014; 29:439–451. [DOI] [PubMed] [Google Scholar]
- 16.van Bussel FC, Backes WH, Hofman PA, et al. On the interplay of microvasculature, parenchyma, and memory in type 2 diabetes. Diabetes Care 2015; 38:876–882. [DOI] [PubMed] [Google Scholar]
- 17.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198. [DOI] [PubMed] [Google Scholar]
- 18.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 1993; 30:672–679. [DOI] [PubMed] [Google Scholar]
- 19.Barker PB, Soher BJ, Blackband SJ, et al. Quantitation of proton NMR spectra of the human brain using tissue water as an internal concentration reference. NMR Biomed 1993; 6:89–94. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Brady M, Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Trans Med Imaging 2001; 20:45–57. [DOI] [PubMed] [Google Scholar]
- 21.Harris AD, Puts NA, Edden RA. Tissue correction for GABA-edited MRS: considerations of voxel composition, tissue segmentation, and tissue relaxations. J Magn Reson Imaging 2015; 42:1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maudsley AA, Govind V, Arheart KL. Associations of age, gender and body mass with 1H MR-observed brain metabolites and tissue distributions. NMR Biomed 2011; 25:580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sickmann HM, Waagepetersen HS, Schousboe A, et al. Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem Int 2012; 60:267–275. [DOI] [PubMed] [Google Scholar]
- 24.Stahl SM. Stahl's Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 3rd edCambridge: Cambridge University Press; 2008. [Google Scholar]
- 25.Lee HK, Yaman A, Nalcioglu O. Homonuclear J-refocused spectral editing technique for quantification of glutamine and glutamate by 1H NMR spectroscopy. Magn Reson Med 1995; 34:253–259. [DOI] [PubMed] [Google Scholar]
- 26.Targosz-Gajniak MG, Siuda JS, Wicher MM, et al. Magnetic resonance spectroscopy as a predictor of conversion of mild cognitive impairment to dementia. J Neurol Sci 2013; 335:58–63. [DOI] [PubMed] [Google Scholar]
- 27.Tiehuis A, van der Meer F, Mali W, et al. MR spectroscopy of cerebral white matter in type 2 diabetes; no association with clinical variables and cognitive performance. Neuroradiology 2010; 52:155–161. [DOI] [PubMed] [Google Scholar]
- 28.Ross AJ, Sachdev PS. Magnetic resonance spectroscopy in cognitive research. Brain Res Brain Res Rev 2004; 44:83–102. [DOI] [PubMed] [Google Scholar]
- 29.Sahin I, Alkan A, Keskin L, et al. Evaluation of in vivo cerebral metabolism on proton magnetic resonance spectroscopy in patients with impaired glucose tolerance and type 2 diabetes mellitus. J Diabetes Complications 2008; 22:254–260. [DOI] [PubMed] [Google Scholar]
- 30.Pfefferbaum A, Adalsteinsson E, Spielman D, et al. In vivo brain concentrations of N-acetyl compounds, creatine, and choline in Alzheimer disease. Arch Gen Psychiatry 1999; 56:185–192. [DOI] [PubMed] [Google Scholar]
- 31.Wan Q, Xiong ZG, Man HY, et al. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 1997; 388:686–690. [DOI] [PubMed] [Google Scholar]
- 32.Frederick B, Satlin A, Wald LL, et al. Brain proton magnetic resonance spectroscopy in Alzheimer disease: changes after treatment with xanomeline. Am J Geriatr Psychiatry 2002; 10:81–88. [PubMed] [Google Scholar]