

Abstract
Objective
To investigate the expression and adenosine‐generating activity of the ecto‐5′‐nucleotidase CD73 on synovial fluid mononuclear cells (SFMCs) and peripheral blood mononuclear cells (PBMCs) from children with juvenile idiopathic arthritis (JIA).
Methods
Given the role of CD73 protein in the production of antiinflammatory adenosine and its intersection with inflammatory biologic pathways, the expression of CD73 on SF and PB lymphocytes from patients with JIA and PB lymphocytes from healthy control subjects was determined by flow cytometry. The AMPase activity of CD73 on PBMCs and SFMCs was measured by high‐performance liquid chromatography. The effects of cell activation on CD73 expression were examined by in vitro culture of PBMCs.
Results
CD8+ and CD19+ SFMCs from patients with JIA expressed decreased levels of CD73 when compared to paired PBMCs from JIA patients and PBMCs from healthy controls. When the percentages of CD73+ synovial lymphocytes were compared between the 2 clinical forms of oligoarthritis, children with extended oligoarthritis showed lower CD73 expression compared to those with the milder form of the disease. CD8+ SFMCs had a lower ability to produce adenosine from etheno‐AMP compared to CD8+ PBMCs. T cell activation through the T cell receptor (TLR) of CD8+CD73+ cells and B cell activation through TLR‐9 resulted in reduced expression of CD73. This down‐regulation occurred on dividing cells.
Conclusion
These findings show that low CD73 expression on T and B cells in the inflamed site is related to cell proliferation and is correlated with the clinical severity of oligoarticular JIA. The decreased CD73 expression on SFMCs, in turn, results in reduced adenosine production, which leads to a decreased potential for antiinflammatory activity.
The autoimmune disorder juvenile idiopathic arthritis (JIA) is an exclusion diagnosis for chronic childhood arthritis of unknown etiology, characterized by swelling of the joints and thickening of the synovial lining (1). Oligoarticular‐onset JIA has a wide spectrum of outcomes and is considered relatively benign, with fewer than 5 joints affected during the first 6 months of disease. If the disease continues on a milder course, it is known as persistent oligoarthritis. When more than 4 joints become affected after 6 months, the disease, defined as extended oligoarthritis, is more severe and complex to control, usually requiring disease‐modifying antirheumatic drugs. Children who have 5 or more joints involved in the first 6 months are defined as having polyarticular JIA (1).
One of the aberrant immune phenomena seen in the inflamed joint in JIA is accumulation and retention of T and B lymphocytes, as well as monocytes and granulocytes (2). The potent proinflammatory nucleotide ATP is released into the extracellular environment during inflammation after cell damage (3) and following ligation of the T cell receptor (TCR) (4). ATP activates the inflammasome, leading to secretion of proinflammatory interleukin‐1β (IL‐1β), the expression of which is elevated in the synovial fluid (SF) of patients with JIA (5). ATP mediates its proinflammatory effects via the purinergic P2 receptors, widely expressed on immune cells. Extracellular ATP concentrations are maintained at physiologic levels by the action of the ectoenzymes CD39 and CD73, which sequentially dephosphorylate ATP to adenosine. CD39 metabolizes ATP to ADP and AMP, while CD73 hydrolyzes AMP to adenosine. The nucleoside adenosine is a cytoprotective modulator that inhibits leukocyte activation (6) and modulates release of proinflammatory cytokines (7) by binding to P1 receptors, with the high‐affinity, cAMP‐increasing A2A receptor (A2AR) subtype being the receptor most strongly associated with immunosuppressive activity. The ectoenzymes CD39 and CD73 can affect the inflammatory process in the joint by balancing the ligand availability of the P2 or P1 receptors, which generally exert opposing effects.
We have previously observed that the proportion of CD39+ T cells is elevated in the joints of JIA patients, as compared to that in the blood of JIA patients (8), suggesting that the availability of cells with ATP‐hydrolyzing capacity, which conveys the potential to limit inflammation, is increased in patients with JIA. We therefore questioned whether the expression of CD73 is also increased in patients with JIA.
To date, no studies have systemically defined CD73 expression and function on cell infiltrates in human arthritis, and none have examined the relationship of this protein to the clinical severity of the disease. JIA provides a powerful model in which to investigate how this purinergic pathway impacts human inflammation. The objective of this study was to define the expression and AMPase activity of CD73 on synovial infiltrates and investigate the mechanisms that affect its expression at the inflamed site.
PATIENTS AND METHODS
Study population
Seventy‐two patients with JIA who fulfilled the International League of Associations for Rheumatology updated classification criteria for JIA (1) were evaluated in this study. Samples were obtained at the time of clinical blood testing and therapeutic joint aspirations. In accordance with the local research ethics committee (Great Ormond Street Hospital/Institute of Child Health Research Ethics Committee; reference nos. 95RU04 and 04RU07), samples were obtained with full informed parental consent. Details on the clinical characteristics of the patients are available at http://discovery.ucl.ac.uk/1456634/. For 10 patients, blood was collected just prior to starting treatment with methotrexate (MTX) and again at 6 months after initiation of MTX treatment. The response to MTX treatment was measured using the preliminary definition of improvement in JIA (9). Peripheral blood (PB) samples from 6 healthy children (median age 6 years) and 39 healthy adults were used as controls.
Cell separation
The PB and SF samples were drawn into heparinized tubes. Thereafter, PBMCs and SFMCs were isolated by density‐gradient centrifugation, as previously described (8).
Antibodies and flow cytometry
The following antibodies were used: V450‐conjugated CD3 (clone UCHT1), fluorescein isothiocyanate (FITC)–conjugated CD19 (HIB19), phycoerythrin (PE)–conjugated CD73 (AD2), FITC‐conjugated CD66B (G10F5) (all from BD Biosciences), allophycocyanin (APC)–conjugated CD8 (SK1), PE–Cy7–conjugated CD39 (eBioA1), Alexa 488–conjugated interferon‐γ (IFNγ) (42.B3), APC‐conjugated FoxP3 (PCH101), eFluor 450–conjugated Ki‐67 (20Raj1), PerCP–Cy5.5–conjugated CD14 (61D3), FITC‐conjugated CD14 (61D3) (all from eBioscience), Qdot 605–conjugated CD4 (S3.5) (Invitrogen), and APC‐ and perforin (dG9)/FITC–conjugated CD26 (BA5b) (Biolegend). Dead cells were excluded using Live/Dead fixable blue DEAD Cell Stain (Life Technologies). Flow cytometry data were acquired on an LSRII (BD PharMingen) and analyzed with FlowJo version 7.6.4 (Tree Star).
Cell sorting
Isolation of monocytes and lymphocytes was performed on a BD FACSAria III or Beckman Coulter MoFlo XDP. The cells were stained for CD73 and CD8 and sorted into CD8+CD73+ and CD8+CD73− cell populations. Monocytes (CD14+) were sorted and stained for CD39 expression. CD8+ T cells were separated with a human CD8+ T cell enrichment kit (EasySep; StemCell Technologies). Cell purity was assessed by flow cytometry and typically found to be >90%.
Surface and intracellular staining
PBMCs and SFMCs (each 2 × 105) were stained with the appropriate antibodies at 4°C for 30 minutes. Intracellular staining for CD73, FoxP3, and Ki‐67 was carried out by fixing and permeabilizing the cells according to the manufacturer's instructions (eBioscience). Detection of IFNγ production by T cells following stimulation with 0.05 μg/ml phorbol 12‐myristate 13‐acetate (PMA), 0.5 μg/ml ionomycin, and 5 μg/ml brefeldin A (all from Sigma) was performed as previously described (10). Intracellular perforin expression was detected using the same protocols as used for IFNγ, but without prior stimulation of the cells. Carboxyfluorescein succinimidyl ester (CFSE) dilution was used to assess cell proliferation by flow cytometry, as described previously (11).
Staining of the cells from whole blood or SF was done by incubating 100 μl of freshly isolated PB or SF with each antibody for 30 minutes at 4°C. Red blood cells were lysed with 2 ml of lysis buffer (BD Biosciences) and then vortexed, centrifuged, and incubated at room temperature for a further 20 minutes.
High‐performance liquid chromatography (HPLC) assay
To measure production of adenosine from fluorescent AMP and ATP substrate analogs, 5 × 105 unsorted or CD8+ bead–sorted PBMCs or SFMCs were incubated in RPMI 1640 medium at 37°C in 5% CO2for 30, 45, and 60 minutes, together with 25 μM etheno‐AMP (E‐AMP) or E‐ATP (Biolog) and with or without 10 μM CD73 inhibitor α,β‐methylene ADP. The reaction was stopped by the addition of 15 mM solution of HCl. Culture supernatants were frozen until analyzed by reverse‐phase HPLC with fluorescence detection (excitation wavelength of 290 nm and emission wavelength of 415 nm), as described previously (8). Etheno analogs of ATP and breakdown products (ADP, AMP, adenosine, and adenine) were used as standards to clarify the identity of the peaks in each sample. In the coincubations of CD8+CD73+ T cells with monocytes, the cell subsets were incubated at a 1:1 ratio.
Cell culture and stimulation
Whole PBMCs or sorted CD8+CD73+ and CD8+CD73− cell populations were cultured on plates precoated with 1 μg/ml of anti‐CD3 monoclonal antibodies (mAb) (clone UCHT1; R&D Systems), either alone or with 5 μg/ml plate‐bound anti‐CD28 mAb (clone CD28.2; BD Biosciences). The recombinant cytokines used in the experiments were tumor necrosis factor α (TNFα; BD PharMingen), IL‐1β (Escherichia coli–derived; R&D Systems), and IL‐6 (BD PharMingen), either alone or in combination, at a final concentration of 10 ng/ml. For B cell stimulation assays, PBMCs were cultured with soluble type B CpG‐containing oligonucleotide 2006 (Invivogen) at 1 μM or were cocultured with control Chinese hamster ovary (CHO) cells or CHO cells transfected with CD40L (a kind gift from Prof. C. Mauri, University College London).
Statistical analysis
Results are expressed as the median with interquartile range. Data were analyzed using paired t‐tests for matched paired samples, analysis of variance, or the Kruskal‐Wallis test, as indicated. Data were analyzed using Prism software version 5 (GraphPad).
RESULTS
Reduced CD73 expression on synovial T and B lymphocytes from JIA patients
CD73 expression on PB T cells from healthy controls (healthy adults and children) was compared to that on T cells from the PB and SF of JIA patients (Figure 1). Expression of CD73 was significantly reduced on CD8+ SFMCs from JIA patients compared to CD8+ PBMCs from healthy controls (Figures 1A and B). However, CD73 expression on CD8+ T cells from the blood was not significantly different between healthy children, healthy adults, and patients with JIA. Furthermore, no significant difference was seen between the PB and SF samples from JIA patients with regard to the levels of CD73 on the minority of CD4+ T cells showing detectable expression (P = 0.09) (results not shown).
Figure 1.
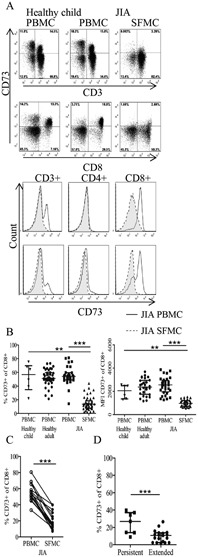
Reduced CD73 expression on CD8+ T cells from patients with juvenile idiopathic arthritis (JIA). A, Top, CD73 expression on CD3+ and CD8+ T cells in peripheral blood mononuclear cells (PBMCs) from healthy children and paired PBMCs and synovial fluid mononuclear cells (SFMCs) from JIA patients, as assessed by flow cytometry gated on live cells. Bottom, Histograms showing CD73 expression on CD3+, CD4+, and CD8+ T cells in paired PBMCs and SFMCs from JIA patients. B, Percentage of CD73+ cells and relative amount (mean fluorescence intensity [MFI]) of CD73 protein expression within the CD3+CD8+ subset on PBMCs from healthy children (n = 6) and healthy adults (n = 39) and PBMCs (n = 31) and SFMCs (n = 46) from JIA patients. Statistical testing was performed using the Kruskal‐Wallis test with Dunn's correction for multiple comparisons and analysis of variance. C, Frequency of CD8+CD73+ cells in paired PBMCs and SFMCs from JIA patients (n = 20). D, Frequency of CD8+ SFMCs expressing CD73 in patients with extended oligoarthritis (n = 19) compared to patients with the less severe persistent oligoarthritis (n = 7). Groups were compared by unpaired t‐test. Bars show the median with interquartile range. ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001.
The decrease in CD73 expression on SF CD8+ T cells was evident in terms of both the frequency of cells positive for CD73 and the protein expression per cell (assessed as the mean fluorescence intensity) (Figure 1B). When paired samples of JIA PBMCs and SFMCs were analyzed, the reduced expression of CD73 on CD8+ T cells from the SF compared to the PB was highly significant (P < 0.0001) (Figure 1C). No significant difference between CD73 surface and total (surface plus intracellular) protein expression on SF CD8+ T cells was observed, showing that protein retention inside the cell does not explain the reduced CD73 expression on JIA synovial lymphocytes (representative results from fluorescence‐activated cell sorter analysis and summary data are available at http://discovery.ucl.ac.uk/1456634/).
We next analyzed whether the decrease in CD73 expression on SF CD8+ T cells was related to disease severity. The JIA patient data (as shown in Figure 1B) were obtained from patients with either extended or persistent oligoarticular‐onset JIA. Comparison of CD73 protein expression between these 2 clinically distinct categories revealed a significantly higher proportion (P = 0.0006) of CD8+ T cells expressing CD73 in the inflamed joints of patients whose disease had remained mild (<5 joints affected) compared to those whose disease had become more severe (extended) (Figure 1D).
We also investigated the expression of CD26, a marker for adenosine deaminase (ADA) (12) that is responsible for adenosine metabolism. CD26 expression was also reduced on SFMCs from JIA patients compared to paired PBMCs from JIA patients and PBMCs from healthy controls (results available at http://discovery.ucl.ac.uk/1456634/).
The percentage of B cells expressing CD73 and their CD73 protein levels were also decreased in the inflamed SF compared to the blood of JIA patients or healthy controls (Figures 2A and B). The reduced percentage of B cells expressing CD73 in the SF was particularly evident for CD19+ B cells from paired JIA patient PB and SF samples (P < 0.0001) (Figure 2C). CD73 expression on B cells was also linked to the clinical subtype of JIA, with higher levels of CD73 on CD19+ synovial cells from patients with the mild, persistent form of oligoarticular JIA compared to patients with the more severe, extended form of oligoarticular JIA (Figure 2D). In addition, low frequencies of CD19+ B cells were found in SF samples compared to paired PB samples from JIA patients (results available at http://discovery.ucl.ac.uk/1456634/).
Figure 2.
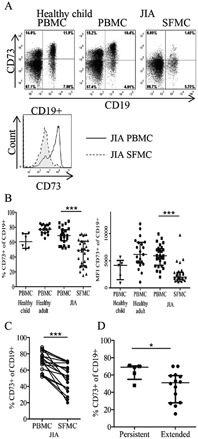
Reduced CD73 expression on CD19+ B cells from patients with JIA. A, Top, CD73 expression on CD19+ B cells in PBMCs from healthy children and paired PBMCs and SFMCs from JIA patients, as assessed by flow cytometry gated on live cells. Bottom, Histogram showing CD73 expression on CD19+ B cells in paired PBMCs and SFMCs from JIA patients. B, Percentage of CD19+CD73+ cells and relative amount (MFI) of CD73 protein expression on CD19+ B cells in PBMCs from healthy children (n = 6) and healthy adults (n = 17) and PBMCs (n = 28) and SFMCs (n = 30) from JIA patients. Statistical testing was performed using analysis of variance and the Kruskal‐Wallis test with Dunn's correction for multiple comparisons. C, Frequency of CD19+CD73+ cells in paired PBMCs and SFMCs from JIA patients (n = 18). D, Frequency of CD19+CD73+ cells in SFMCs from patients with extended oligoarthritis (n = 14) compared to those with the less severe persistent oligoarthritis (n = 5). Groups were compared by unpaired t‐test. Bars show the median with interquartile range. ∗ = P < 0.05; ∗∗∗ = P < 0.0001. See Figure 1 for definitions.
The difference in expression of CD73 on SF CD8+ T cells and SF CD19+ B cells between patients with persistent oligoarticular JIA and those with extended oligoarticular JIA was not simply due to the longer disease duration of those with the more severe disease. Thus, no correlation between CD73 expression by T and B cells and disease duration was found (n = 44 patients with persistent oligoarticular JIA and n = 34 patients with extended oligoarticular JIA; P = 0.14 and P = 0.61, respectively and Spearman's r = 0.22 and r = −0.09, respectively).
Further evidence of the lack of a relationship between disease duration and CD73 expression came from the finding that children with polyarticular arthritis also had a lower percentage of synovial T and B cells expressing CD73 than did children with persistent oligoarticular arthritis, despite the fact that the duration of disease was similar between the 2 groups (mean 2.9 years and 2.4 years, respectively) (results not shown). An inverse relationship, however, was found between CD73 expression and the cumulative swollen and tender joint count (used as a surrogate of disease severity) (results not shown).
Since granulocytes are lost during the preparation of PBMCs or SFMCs, CD73 protein expression was tested in freshly isolated whole SF samples and unseparated healthy PB samples. Neither granulocytes (CD66B+) nor monocytes (CD14+) were found to express CD73 in these samples, although both CD3+ and CD19+ lymphocytes isolated from the inflamed SF showed a reduction in CD73 expression when compared to that in the PB (results available at http://discovery.ucl.ac.uk/1456634/). These results suggest that expression of CD73 was not affected by density centrifugation or cryopreservation.
Reduced adenosine production from AMP in the joints of patients with JIA
The enzymatic function of CD73 is to dephosphorylate AMP, leading to generation of adenosine. The observed reduction in CD73 expression on synovial lymphocytes from the inflamed joints would be predicted to result in reduced AMPase activity and, thus, lower adenosine generation. To test this hypothesis, HPLC was used to examine the hydrolysis of exogenously added AMP to adenosine by unseparated cells and CD8+ sorted cells from healthy control PB samples and JIA patient PB or SF samples.
SFMCs from patients with JIA exhibited reduced AMPase activity compared to either healthy PBMCs or JIA PBMCs, as revealed by their decreased ability to produce E‐adenosine (Figure 3A). This difference was observed in unsorted SFMCs and in CD8+ sorted SFMCs, both of which generated lower levels of E‐adenosine compared to their blood counterparts (Figure 3B), whereas the AMPase activity of JIA PBMCs did not differ from that of PBMCs from healthy controls. The rate at which E‐AMP was broken down by healthy control PBMCs was greater than that by JIA SFMCs, as shown in Figure 3C.
Figure 3.
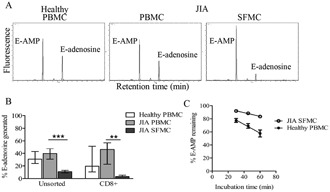
Reduced ability of lymphocytes from the joints of JIA patients to hydrolyze exogenous 5′‐AMP. A, Breakdown of etheno‐AMP (E‐AMP) to E‐adenosine by healthy control PBMCs and JIA PBMCs and SFMCs. Representative chromatograms are shown, with results expressed in arbitrary units. B, Percentage of E‐adenosine generated by unsorted healthy control PBMCs (n = 17) and JIA PBMCs (n = 5) and SFMCs (n = 10), and by CD8+ sorted healthy control PBMCs (n = 7) and JIA PBMCs (n = 4) and SFMCs (n = 7). Statistical testing was performed using analysis of variance. C, Reduced rate of breakdown of E‐AMP by JIA SFMCs (n = 5) compared to healthy control PBMCs (n = 5) at 30, 45, and 60 minutes of incubation. Values in B and C are the median and interquartile range. ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001. See Figure 1 for other definitions.
Reduction in CD73 expression by T and B cell stimulation, but not by joint inflammatory cytokines
The microenvironment of JIA SF is a proinflammatory milieu characterized by elevated levels of cytokines, including IL‐1, IL‐6, and TNFα (5), and oligoclonally expanded T cells (13). To investigate whether CD73 expression on cells in the joints of patients with JIA is altered by the inflammatory environment, PBMCs from healthy controls were cultured in vitro with proinflammatory cytokines and ligands that stimulate T and B cells. As shown in Figure 4A, stimulation with IL‐1, IL‐6, or TNFα failed to alter the expression of CD73 after 5 days in culture, whether tested individually (Figure 4A) or together (results not shown). Conversely, TCR stimulation with anti‐CD3 and CD28 mAb led to reduced expression of CD73 on CD8+ T cells (Figure 4A). Furthermore, CD73 expression was decreased when PBMCs were cultured with anti‐CD3 mAb alone, showing that costimulation was not required to achieve this down‐regulation (results available at http://discovery.ucl.ac.uk/1456634/).
Figure 4.
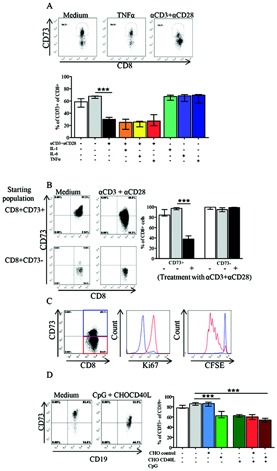
Down‐regulation of CD73 on stimulated T and B cells. A, Top, CD73 expression on PB CD8+ T cells after culture in medium or T cell receptor stimulation with tumor necrosis factor α (TNFα) or anti‐CD3/anti‐CD28 antibodies for 5 days. Representative dot plots are shown. Bottom, CD73 expression on PB CD8+ T cells (n = 3) after culture in medium or stimulation with cytokines (interleukin‐1 [IL‐1], IL‐6, and TNFα) or anti‐CD3/anti‐CD28 antibodies, alone or in combination. B, Left, Sorted CD8+CD73+ and CD8+CD73− cells after 5 days of culture in medium or stimulation with anti‐CD3/anti‐CD28 antibodies, as assessed by flow cytometry. Right, Summary of flow cytometry data (n = 4). C, PB CD8+ T cells cultured with anti‐CD3/anti‐CD28 antibodies and stained for CD73 (left) and for the nuclear protein Ki‐67 (middle) to identify actively cycling cells, and CD8+CD73+ purified cells labeled with carboxyfluorescein succinimidyl ester (CFSE) to examine cell proliferation (right). The red gate and red lines show cells that had down‐regulated CD73 expression, while the blue gate and blue lines show cells that remained CD73 positive. D, Expression of CD73 on CD19+ cells in PBMCs cultured for 3 days in medium or with the Toll‐like receptor 9 ligand CpG in the presence or absence of Chinese hamster ovary (CHO) control cells or CD40 ligand–transfected CHO cells. Representative dot plots (left) and summary data (n = 8) are shown. Bars show the median with interquartile range. In all bar graphs, the open bar represents expression prior to culture. ∗∗∗ = P < 0.0001. See Figure 1 for other definitions.
To test whether the low CD73 expression was due to down‐regulation of CD73 and not to differential cell death of CD73+ T cells, CD8+ T cells were sorted into CD8+CD73+ and CD8+CD73− pure populations. After 5 days of culture in the absence or presence of anti‐CD3 mAb and anti‐CD28 mAb, loss of CD73 expression was observed on cells that were CD73+ at the start of culture. Expression of CD73 on sorted CD73− cells was unaffected by culture or stimulation (Figure 4B).
We next tested whether CD73 down‐regulation was specifically linked to cell proliferation. CD8+CD73+ purified cells were labeled with CFSE and then cultured in the absence or presence of anti‐CD3 and anti‐CD28 mAb and stained for CD73 and the nuclear protein Ki‐67, to identify actively cycling cells. As shown in Figure 4C, at the end of the 5‐day culture, those CD8+ T cells that retained their CD73 expression (45.1% of cells [blue gate in Figure 4C, left]) were Ki‐67 negative and had not proliferated (blue line in Figure 4C, middle and right). In contrast, cells that had become CD73− (54.9% of cells [red gate in Figure 4C, left]) were Ki‐67 positive and had diluted CFSE, thus indicating cell division (red line in Figure 4C, middle and right). These results indicate that upon cell proliferation, CD8+ T cells down‐regulate the expression of CD73.
B cell stimulation with the TLR‐9 ligand CpG or coculture with CD40L‐transfected CHO cells also induced down‐regulation of CD73 on B cells. No synergy was observed in the presence of both signals tested together (Figure 4D). With regard to the effects on T cells, coculture of CD40L‐transfected CHO cells with inflammatory cytokines (IL‐1, IL‐6, and TNFα), either singly or together, did not alter the expression of CD73 on B cells (results not shown).
Effector phenotype of CD8+CD73− lymphocytes
Upon antigen recognition, CD8+ T lymphocytes acquire effector functions that are mediated via the release of cytokines (including IFNγ) and perforin‐dependent pathways. In order to test the effector functions of CD8+ synovial T cells that lacked CD73 expression, IFNγ and perforin were selected as canonical effector molecules. The expression of IFNγ and perforin by CD8+CD73+ T cells and CD8+CD73− T cells was compared. CD8+ T cells from the PB of both healthy controls and JIA patients, which produce IFNγ, were found predominantly within the CD73− population (Figure 5A). Among CD8+ T cells from the site of inflammation, there was a dysregulation in the distribution of IFNγ+ cells, in that both the CD8+CD73+ and CD8+CD73− T cell populations contained cells that were producing IFNγ, although CD8+CD73− T cells still had the highest frequency of IFNγ‐expressing cells.
Figure 5.
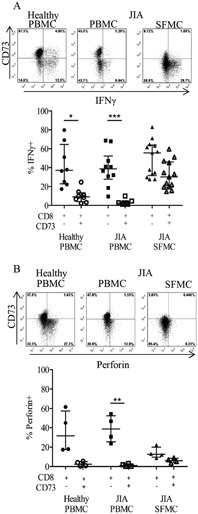
Lack of coexpression of CD73 with the inflammatory and cytotoxic markers interferon‐γ (IFNγ) and perforin on CD8+ T cells. A, Top, Low levels of coexpression of CD73 and IFNγ on CD8+ T cells in healthy control PBMCs or PBMCs and SFMCs from JIA patients after stimulation of the cells with phorbol 12‐myristate 13‐acetate and ionomycin. Representative plots from fluorescence‐activated cell sorter (FACS) analysis gated on CD3+CD8+ cells are shown. Bottom, Percentage of IFNγ+ cells within the CD8+CD73− or CD8+CD73+ population in healthy PBMCs (n = 8) and PBMCs (n = 9) and SFMCs (n = 12) from JIA patients. B, Top, Lack of coexpression of the cytotoxic protein perforin and CD73. Representative FACS plots gated on CD3+CD8+ cells are shown. Bottom, Percentage of perforin‐positive cells among healthy PBMCs (n = 4) and PBMCs (n = 7) and SFMCs (n = 6) from JIA patients. Bars show the median with interquartile range. ∗ = P < 0.05; ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001. See Figure 1 for other definitions.
Production of the cytolytic protein perforin was reduced in SFMCs compared to PBMCs (Figure 5B), as has been observed previously in adult patients with rheumatoid arthritis (14). In both healthy control and JIA patient blood, there was very low coexpression of CD73 and perforin, whereas in SFMCs from patients with JIA, neither CD73+ nor CD73− cells produced meaningful levels of perforin (Figure 5B).
Selective coexpression of ectoenzymes CD73 and CD39 on B cells
Since ATP is metabolized to adenosine via dephosphorylation by CD39 and CD73, both proteins need to be present for the full enzymatic cascade from ATP to adenosine to occur. We have previously shown that CD39‐expressing T cells are highly enriched in the joints of patients with JIA; however, few T cells express both CD39 and CD73 in either healthy control or JIA patient samples (8). Contrary to the findings in murine Treg cells (15), human Treg cells from healthy donor PB and JIA patient PB or SF did not coexpress CD39 and CD73 (results available at http://discovery.ucl.ac.uk/1456634/). B cells coexpressed CD39 and CD73 (Figure 6A) and were therefore able to produce E‐adenosine after incubation with exogenous E‐ATP (Figure 6B), whereas neither CD8+CD73+ nor CD8+ CD73− sorted T cells were able to break down E‐ATP to E‐AMP (Figure 6C).
Figure 6.
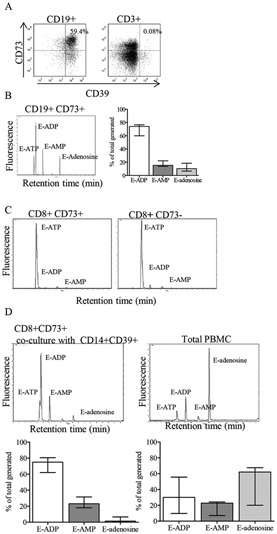
Unique ability of CD19+ B cells to generate adenosine from ATP. A, Coexpression of CD73 and CD39 on CD19+ B cells, but not on CD3+ T cells, in healthy control PBMCs. Representative dot plots are shown. B, Left, Generation of etheno‐adenosine (E‐adenosine) from E‐ATP by sorted CD19+CD73+ cells from healthy control PBMCs, as assessed by high‐performance liquid chromatography (HPLC). Right, Summary HPLC data (n = 4). C, Inability of CD8+CD73+ and CD8+CD73− cells from healthy control PBMCs to generate E‐adenosine from E‐ATP, as assessed by HPLC. D, Top, Failure to generate E‐adenosine from E‐ATP in cocultures of CD8+CD73+ and CD14+CD39+ cells, compared to generation of E‐adenosine by unsorted PBMCs, as assessed by HPLC. Bottom, Summary HPLC data (n = 5). Fluorescence was measured in arbitrary units. Bars show the median with interquartile range. See Figure 1 for other definitions.
We therefore tested whether CD39 and CD73 proteins are required to be coexpressed on the same cell for adenosine generation, or whether nucleotide dephosphorylation can occur with CD73+ and CD39+ cells acting cooperatively. CD8+CD73+ T cells, which were negative for CD39 expression, and CD14+CD39+ (CD73−) monocytes were sorted and cocultured in the presence of E‐ATP, and the resulting culture supernatants were analyzed by HPLC. No E‐adenosine was generated by the combination of CD39+ and CD73+ cells (Figure 6D), whereas unsorted PBMCs were found to be competent in the generation of adenosine, thus demonstrating the importance of CD39 and CD73 coexpression on the same cell for generation of adenosine from ATP.
Effect of MTX therapy on CD73 expression
Since MTX is the main treatment for JIA and can increase adenosine release in a manner dependent on CD73 activity (16), we investigated the effect of MTX on CD73 expression in JIA patients. Expression of CD73 was measured on patient PB CD8+ T and B lymphocytes (n = 10 patients) before treatment and after 6 months of treatment with MTX. Five of these patients were nonresponders or had achieved only a 30% level of response according to the American College of Rheumatology (ACR) Pediatric 30 (Pedi 30) criteria for improvement (9), while the other 5 patients had reached an improvement response of ACR Pedi 70 or higher.
No significant difference in CD73 expression on CD8+ T cells (P = 0.17) or CD19+ B cells (P = 0.5) was observed after treatment with MTX, nor was there a significant difference in CD73 expression between responders and nonresponders either before or after MTX treatment. In addition, analysis of the CD73 expression on SFMCs from patients who had never received treatment with MTX or steroids revealed the same trend of a reduction in frequency of CD73+ cells, which was more severe in patients with extended oligoarticular JIA compared to those with persistent oligoarticular JIA, indicating that the reduction in CD73 was not due to medication usage. All of these results are available at http://discovery.ucl.ac.uk/1456634/.
DISCUSSION
This study was undertaken to characterize the expression and enzymatic function of CD73 on inflammatory cells from patients with JIA. We had previously observed a decrease in expression of CD73 on SFMCs from JIA patients (8), but our earlier study only explored the expression of CD73 on B cells. Herein we analyzed CD73 expression on all SF cells. In addition, we assessed the relationship of CD73 expression to the subtypes of JIA, and investigated signals that may alter CD73 expression during inflammation.
A key finding of this study was the decreased expression of CD73 on SF T and B cells from children with JIA. This reduction resulted in an impaired ability of unsorted or CD8+ sorted SFMCs to metabolize E‐AMP to E‐adenosine. In contrast to the findings in murine studies, for example in a collagen‐induced arthritis model in which elevated CD73 expression was detected on granulocytes and monocytes (17), our study in human autoimmune arthritis demonstrated no CD73 expression on either healthy PB or JIA SF granulocytes and monocytes.
Prevention of the metabolism of AMP to adenosine, which was attributed to the lack of CD73, would result in maintenance of high AMP concentrations in the inflamed synovium. As the A1R has been determined to be the sole receptor through which AMP can signal (18), and presence of the A1R causes inhibition of adenylate cyclase (unlike the A2AR, which stimulates adenylate cyclase), AMP and adenosine would exert opposing effects. Since the CD73 gene promoter contains a cAMP response element (19), it has been suggested that adenosine itself could transcriptionally regulate surface expression of CD73 on some cell types (20).
Because of the observed reduction in CD26+ synovial T cells, the expression of the associated ADA protein is also expected to be decreased on these cells, resulting in a lower potential to metabolize adenosine. CD8+ T cells have previously been found to have an important role in the inflamed joint, as they are major producers of CCL5 (21), a chemokine that recruits and activates T cells and induces production of IFNγ. B cells also contribute to the pathogenesis of autoimmune diseases, via the production of autoantibodies, in addition to acting as antigen‐presenting cells. The analysis of CD73 expression on T and B cells was undertaken in the present study because this ectoenzyme has been found to mediate a regulatory, antisuppressive function via dephosphorylation of AMP to adenosine (22). CD73 is expressed on murine Treg cells (15, 22), together with the ATPase CD39, enabling adenosine to be generated from ATP as a mechanism of immunoregulation (15). In contrast, human CD4+FoxP3+ cells expressed only low levels of CD73, and no coordinated expression with CD39 was present, suggesting that human Treg cells are unable to synthesize adenosine from ATP through these ectoenzymes and are therefore unlikely to contribute to disease remission via the adenosinergic pathway.
Previous studies found a predominance of CD27+ class‐switched memory B cells in the JIA joint (23), and this cell subset in the PB was found to express elevated levels of CD73 (24). Despite this knowledge and the fact that CD73 is considered a B cell maturity marker (25), in our study we detected lower levels of CD73 in synovial B cells.
CD8+ T cells, which were found to express high levels of CD73 in the PB of both healthy controls and JIA patients, can generate E‐adenosine from E‐AMP, suggesting they may have a suppressive and antiinflammatory function. CD73 protein expression on both CD8+ T cells and CD19+ B cells was reduced on SFMCs from JIA patients, and this reduction was more severe in patients with the extended oligoarticular disease subtype compared to that in patients with the persistent oligoarticular subtype. Furthermore, a higher proportion of proinflammatory cells producing perforin and IFNγ was found among those CD8+ cells that were either CD73 low or CD73 negative. The lack of coordinated expression of IFNγ or perforin with CD73 would suggest that CD8+ T cells with a suppressive role may be distinct from effector functions mediated by cytokines and cytolytic pathways. Our data suggest that the ability to produce adenosine, which inhibits proinflammatory mechanisms by repressing T cell functions, could contribute to the mechanism by which milder cases of JIA are prevented from turning to a more severe and destructive form.
In vitro culture of PBMCs with inflammatory cytokines known to be elevated in the SF of patients with JIA (5) did not reduce CD73 expression. In contrast, signaling through the TCR, either alone or together with the costimulatory anti‐CD28 mAb, recapitulated the reduced levels of CD73 observed on SFMCs. It was confirmed that CD73 down‐regulation can be generated in vitro under conditions that induce cell division, which also resulted in increased CD39 expression (results not shown), consistent with the known elevation of CD39 on SFMCs (8).
T cells were found to be unable to metabolize ATP to adenosine, whereas B cells, which express both CD39 and CD73, were capable of adenosine generation from ATP. Because of this, we investigated whether the presence of CD39 and CD73 on the same cell type is required for adenosine production. Our data suggest that the 2 ectoenzymes are required to be coexpressed, since sorted CD14+ monocytes (CD39+) and CD8+ T cells (CD73+) incubated together were unable to produce E‐adenosine from E‐ATP. Given the observed decrease in production of CD19+CD73+ and CD8+CD73+ cells in the SFMCs from JIA patients, combined with the low relative numbers of B cells in the joints compared to the blood, these data together indicate a marked overall reduction in the proportion of lymphocytes that are able to synthesize adenosine in the joint. These results also suggest that in the inflamed JIA joint, the presence of increased numbers of CD39+ cells may lead to a build‐up of AMP, which cannot be further broken down to adenosine. These findings support the hypothesis that adenosine production is reduced in the inflamed joint. Since this nucleoside has antiinflammatory activity, immunoregulation in the joint is reduced. One technical limitation of this study is the inability to accurately measure the levels of adenosine present in the joint, as it has a half‐life in blood of only 1 minute (26), due to its rapid metabolism and turnover.
In conclusion, we have demonstrated that lymphocytes in the JIA joint have a lower potential to produce adenosine, compared to lymphocytes in the PB, which may contribute to the persistence of inflammation in arthritis. Our results could have clinical implications. MTX, a mainstay in the treatment of JIA, has been reported to increase extracellular release of adenosine (27) by a mechanism dependent on CD73 activity (16). In our study, when CD73 expression was measured in the blood of JIA patients before and after treatment with this agent, no difference was found, suggesting that MTX itself is not affecting CD73 expression levels. In addition, the trend toward lower CD73 expression by synovial lymphocytes in patients with extended oligoarthritis compared to those with milder disease was maintained among those who had not received treatment for JIA, suggesting that medication was not a key driver of the observed CD73 expression in the analysis of the 2 groups. Patients lacking CD73 in the joint, therefore, may not benefit from the therapeutic potential of MTX. Further studies are needed to examine the implications of these findings for JIA treatment.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Ms Botta Gordon‐Smith had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Botta Gordon‐Smith, Eaton, Moncrieffe, Wedderburn.
Acquisition of data. Botta Gordon‐Smith, Ursu, Eaton, Wedderburn.
Analysis and interpretation of data. Botta Gordon‐Smith, Ursu, Eaton, Wedderburn.
REFERENCES
- 1. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al.International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001.J Rheumatol 2004;31:390–2. [PubMed] [Google Scholar]
- 2. Murray KJ, Luyrink L, Grom AA, Passo MH, Emery H, Witte D, et al.Immunohistological characteristics of T cell infiltrates in different forms of childhood onset chronic arthritis.J Rheumatol 1996;23:2116–24. [PubMed] [Google Scholar]
- 3. Bodin P, Burnstock G.Increased release of ATP from endothelial cells during acute inflammation.Inflamm Res 1998;47:351–4. [DOI] [PubMed] [Google Scholar]
- 4. Schenk U, Westendorf AM, Radaelli E, Casati A, Ferro M, Fumagalli M, et al.Purinergic control of T cell activation by ATP released through pannexin‐1 hemichannels.Sci Signal 2008;1:6. [DOI] [PubMed] [Google Scholar]
- 5. De Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn L, Kuis W, Prakken BJ.Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross‐sectional study.Ann Rheum Dis 2007;66:589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang S, Apasov S, Koshiba M, Sitkovsky M.Role of A2a extracellular adenosine receptor‐mediated signaling in adenosine‐mediated inhibition of T‐cell activation and expansion.Blood 1997;90:1600–10. [PubMed] [Google Scholar]
- 7. Romio M, Reinbeck B, Bongardt S, Ḧls S, Burghoff S, Schrader J.Extracellular purine metabolism and signaling of CD73‐derived adenosine in murine Treg and Teff cells.Am J Physiol Cell Physiol 2011;301:C530–9. [DOI] [PubMed] [Google Scholar]
- 8. Moncrieffe H, Nistala K, Kamhieh Y, Evans J, Eddaoudi, Eaton S, et al.High expression of the ectonucleotidase CD39 on T cells from the inflamed site identifies two distinct populations, one regulatory and one memory T cell population.J Immunol 2010;185:134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A.Preliminary definition of improvement in juvenile arthritis.Arthritis Rheum 1997;40:1202–9. [DOI] [PubMed] [Google Scholar]
- 10. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al.Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment.Proc Natl Acad Sci U S A 2010;107:14751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pesenacker AM, Bending D, Ursu S, Wu Q, Nistala K, Wedderburn L.CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines.Blood 2013;121:2647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dong RP, Kameoka J, Hegen M, Tanaka T, Xu Y, Schlossman SF, et al.Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response.J Immunol 1996;156:1349–55. [PubMed] [Google Scholar]
- 13. Thompson SD, Murray KJ, Grom AA, Passo MH, Choi E, Glass DN.Comparative sequence analysis of the human T cell receptor beta chain in juvenile rheumatoid arthritis and juvenile spondylarthropathies: evidence for antigenic selection of T cells in the synovium.Arthritis Rheum 1998;41:482–97. [DOI] [PubMed] [Google Scholar]
- 14. Cho BA, Sim JH, Park JA, Kim HW, Yoo WH, Lee SH, et al.Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis.J Clin Immunol 2012;32:709–20. [DOI] [PubMed] [Google Scholar]
- 15. Deaglio S, Dwyer K, Gao W, Friedman D, Usheva A, Erat A, et al.Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression.J Exp Med 2007;204:1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montesinos M, Takedachi M, Thompson L, Wilder TF, Fernandez P, Cronstein B.The antiinflammatory mechanism of methotrexate depends on extracellular conversion of adenine nucleotides to adenosine by ecto‐5′‐nucleotidase: findings in a study of ecto‐5′‐nucleotidase gene–deficient mice.Arthritis Rheum 2007;56:1440–5. [DOI] [PubMed] [Google Scholar]
- 17. Flogel U, Burghoff S, van Lent PL, Temme S, Galbarz L, Ding Z, et al.Selective activation of adenosine A2A receptors on immune cells by a CD73‐dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis.Sci Transl Med 2012;4:108. [DOI] [PubMed] [Google Scholar]
- 18. Rittiner JE, Korboukh I, Hull‐Ryde EA, Jin J, Janzen WP, Frye SV, et al.AMP is an adenosine A1 receptor agonist.J Biol Chem 2012;287:5301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hansen KR, Resta R, Webb CF, Thompson L.Isolation and characterization of the promoter of the human 5′‐nucleotidase (CD73)‐encoding gene.Gene 1995;167:307–12. [DOI] [PubMed] [Google Scholar]
- 20. Narravula S, Lennon PF, Mueller BU, Colgan SP.Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function.J Immunol 2000;165:5262–8. [DOI] [PubMed] [Google Scholar]
- 21. Pharoah DS, Varsani H, Tatham RW, Newton KR, de Jager W, Prakken B, et al.Expression of the inflammatory chemokines CCL5, CCL3 and CXCL10 in juvenile idiopathic arthritis, and demonstration of CCL5 production by an atypical subset of CD8+ T cells.Arthritis Res Ther 2006;8:R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR.T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′‐adenosine monophosphate to adenosine.J Immunol 2006;177:6780–6. [DOI] [PubMed] [Google Scholar]
- 23. Corcione A, Ferlito F, Gattorno M, Gregorio A, Pistorio A, Gastaldi R, et al.Phenotypic and functional characterization of switch memory B cells from patients with oligoarticular juvenile idiopathic arthritis.Arthritis Res Ther 2009;11:R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schena F, Volpi S, Faliti CE, Penco F, Santi S, Proietti M, et al.Dependence of immunoglobulin class switch recombination in B cells on vesicular release of ATP and CD73 ectonucleotidase activity.Cell Rep 2013;3:1824–31. [DOI] [PubMed] [Google Scholar]
- 25. Thompson L, Ruedi JM, O'Connor RD, Bastian JF.Ecto‐5′‐nucleotidase expression during human B cell development: an explanation for the heterogeneity in B lymphocyte ecto‐5′‐nucleotidase activity in patients with hypogammaglobulinemia.J Immunol 1986;137:2496–500. [PubMed] [Google Scholar]
- 26. Dawicki DD, Agarwal KC, Parks RE.Adenosine metabolism in human whole blood: effects of nucleoside transport inhibitors and phosphate concentration.Biochem Pharmacol 1988;37:621–6. [DOI] [PubMed] [Google Scholar]
- 27. Chan ES, Cronstein B.Methotrexate: how does it really work? Nat Rev Rheumatol 2010;6:175–8. [DOI] [PubMed] [Google Scholar]