

Abstract
ADS‐5102 is a long‐acting, extended‐release capsule formulation of amantadine HCl administered once daily at bedtime. This study investigated the safety, efficacy, and tolerability of ADS‐5102 in Parkinson's disease (PD) patients with levodopa‐induced dyskinesia. This was a randomized, double‐blind, placebo‐controlled, parallel‐group study of 83 PD patients with troublesome dyskinesia assigned to placebo or one of three doses of ADS‐5102 (260 mg, 340 mg, 420 mg) administered daily at bedtime for 8 weeks. The primary efficacy analysis compared change from baseline to week 8 in Unified Dyskinesia Rating Scale (UDysRS) total score for 340 mg ADS‐5102 versus placebo. Secondary outcome measures included change in UDysRS for 260 mg, 420 mg, Fatigue Severity Scale (FSS), Movement Disorder Society Unified Parkinson's Disease Rating Scale (MDS‐UPDRS), patient diary, Clinician's Global Impression of Change, and Parkinson's Disease Questionnaire (PDQ‐39). ADS‐5102 340 mg significantly reduced dyskinesia versus placebo (27% reduction in UDysRS, P = 0.005). In addition, ADS‐5102 significantly increased ON time without troublesome dyskinesia, as assessed by PD patient diaries, at 260 mg (P = 0.004), 340 mg (P = 0.008) and 420 mg (P = 0.018). Adverse events (AEs) were reported for 82%, 80%, 95%, and 90% of patients in the placebo, 260‐mg, 340‐mg, and 420‐mg groups, respectively. Constipation, hallucinations, dizziness, and dry mouth were the most frequent AEs. Study withdrawal rates were 9%, 15%, 14%, and 40% for the placebo, 260‐mg, 340‐mg, and 420‐mg groups, respectively. All study withdrawals in the active treatment groups were attributable to AEs. ADS‐5102 was generally well tolerated and resulted in significant and dose‐dependent improvements in dyskinesia in PD patients. © 2015 Adamas Pharmaceuticals, Inc. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: clinical trial, randomized controlled trial, Parkinson's disease, levodopa‐induced dyskinesia, amantadine
Levodopa is considered the gold standard treatment for Parkinson's disease (PD); however, long‐term treatment with levodopa is complicated by motor fluctuations and dyskinesia.1 Approximately 40% of patients develop motor complications and dyskinesia after 4 to 6 years of levodopa treatment,2 yet there are no approved medical treatments for dyskinesia in PD.1
Amantadine is an antiviral agent that was discovered to be a potential treatment for PD motor symptoms in 1968.3 More recently, amantadine was reported to improve levodopa‐induced dyskinesia (LID).4, 5, 6, 7 The clinical use of amantadine is limited because of tolerability issues, although doses up to a maximum of 400 mg/d, in divided doses, are approved for use in PD.
ADS‐5102 is an investigational, extended‐release capsule formulation of amantadine HCl in development for the treatment of LID associated with PD. ADS‐5102 is administered once daily at bedtime, which may increase convenience and patient adherence. The maximum concentration is achieved 12 to 14 hours after dosing. This results in sustained concentrations throughout the morning and mid‐day, when patients are active and LID can be bothersome. There is a gradual decline to low concentrations during the evening and overnight, to reduce the potential for sleep‐related side effects. The present study was designed to evaluate the safety and efficacy of three dose levels of ADS‐5102 oral capsules dosed once daily at bedtime for the treatment of LID in PD patients.
Methods
This trial (EASED Study, Adamas Pharmaceuticals, ADS‐PAR‐AM201, NCT 01397422) was conducted between July 2011 and April 2013 at 31 sites in the United States, and in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. Before initiating the study, all participating sites received approval from an institutional review board, and all study participants provided written informed consent. See Acknowledgments for full list of sites that enrolled study participants.
Patients
Key inclusion criteria included age between 30 and 85 years, inclusive, diagnosis of PD based on the United Kingdom Parkinson's Disease Society Brain Bank Clinical Diagnostic Criteria,8 score of at least 2 on part IV, item 4.2 (functional impact of dyskinesia) of the Movement Disorder Society Unified Parkinson's Disease Rating Scale (MDS‐UPDRS)9 at screening and at day 1 (baseline), and at least two half‐hour periods between 9:00 am and 4:00 pm documented as ON time with troublesome dyskinesia on a 24‐h PD patient diary10 on each of 2 consecutive days just before day 1. All antiparkinsonian medications, including levodopa preparations, were unchanged for at least 30 days before screening and during study participation. Levodopa preparations had to be administered at least three times daily.
Key exclusion criteria included history of dyskinesia that was exclusively diphasic, off state, myoclonic, dystonic, or akathetic without peak dose dyskinesia, neurosurgical intervention related to PD, atypical parkinsonism, levodopa or dopamine agonist‐induced psychosis, cognitive impairment as evidenced by a Mini‐Mental Status Examination score of less than 24 during screening, estimated glomerular filtration rate (GFR) less than 50 mL/min/1.73 m2, use of amantadine within 30 d before screening, documented inability to tolerate or lack of dyskinesia response to prior amantadine treatment, current treatment with apomorphine or dopamine receptor blocking agents, clinically significant electrocardiogram abnormalities, use of rimantadine or history of hypersensitivity or allergic reaction to amantadine, rimantidine, or memantine.
Design
This was a multicenter, randomized, double‐blind, placebo‐controlled, four‐arm parallel group study. Eligible patients were randomized on day 1 in a 1:1:1:1 ratio to receive placebo, 260 mg ADS‐5102, 340 mg ADS‐5102, or 420 mg ADS‐5102. Randomization was stratified by the baseline Fatigue Severity Scale Score (FSS > 4 or ≤ 4). The randomization list was generated and validated by inVentiv Health Clinical (Cary, NC). Randomization was accomplished through an interactive web‐based response system, which also allowed for unblinding of treatment assignment if necessary for patient safety.
Study medication was administered as three capsules, identical in appearance, once daily at bedtime for 8 weeks. Patients randomized to placebo or 260 mg ADS‐5102 received that nightly dose throughout the 8‐week dosing period. Patients randomized to 340 mg ADS‐5102 or 420 mg ADS‐5102 all received 260 mg ADS‐5102 during week 1, and 340 mg ADS‐5102 during week 2, and the 420 group then went on to receive 420 mg ADS‐5102 beginning week 3. Assigned doses were to be maintained without dose adjustment for the remainder of the treatment period. Patients were evaluated at baseline and after 1, 2, 4, 6, and 8 weeks of treatment. A final safety follow‐up visit occurred approximately 14 d after treatment completion.
Prequalified raters completed training and were certified to administer efficacy scales. Rater training used the MDS teaching modules for the scales. Patients needed to demonstrate ability to complete PD home diaries, and concordance in observed ON time with dyskinesia between study staff and patient was required before randomization. If possible, the same rater conducted efficacy assessments at least 30 minutes after the patient's regularly scheduled levodopa dose, when the patient was ON (PD medications providing good effect on motor symptoms) and experiencing typical dyskinesia. During study conduct, the protocol was amended to allow inclusion of patients up to 85 years of age, and the time window for the PD patient diary dyskinesia inclusion criterion was widened from 9:00 am to 2:00 pm to 10:00 am to 4:00 pm.
Study Assessments
Outcome Measures
The primary outcome measure was the change from baseline to week 8 in the Unified Dyskinesia Rating Scale (UDysRS) total score (four parts, total possible score of 104). The primary efficacy analysis compared the 340‐mg dose level with placebo. Secondary outcome measures included the change from baseline to week 8 in the following (in order of priority): Fatigue Severity Scale (FSS) score, MDS‐UPDRS combined score (I, II, III), UDysRS total objective score (III, IV), ON time without troublesome dyskinesia (ON time without dyskinesia plus ON time with non‐troublesome dyskinesia), ON time with troublesome dyskinesia, total ON time with dyskinesia (non‐troublesome plus troublesome), MDS‐UPDRS, part IV, items 4.1 (time spent with dyskinesia) and 4.2 (functional impact of dyskinesia), MDS‐UPDRS, individual part scores (I, II, III, IV), Clinician's Global Impression of Change (CGI‐C), and health‐related quality of life as measured by a Parkinson's Disease Questionnaire (PDQ‐39). The UDysRS, MDS‐UPDRS, FSS, and CGI‐C were evaluated at the baseline (except CGI‐C), week 2, week 4, and week 8 visits; PDQ‐39 was completed at the baseline, week 4, and week 8 visits. During screening, potential patients and any designated caregivers or study partners were trained on the proper completion of PD home diaries. After training, a set of two consecutive 24‐hour diaries (48 hours total) were completed before day 1 (day of randomization), serving as the baseline diary score, and repeated before the week 2, week 4, week 6 and week 8 visits.
Safety Measures
Safety assessments included adverse events (AEs), reasons for discontinuation, physical examinations, vital signs, and clinical laboratory testing. Study safety data were periodically reviewed by an independent data monitoring committee.
Pharmacokinetics
Samples for amantadine plasma concentration analyses were obtained on day 1, week 1, week 2, week 4, week 6 and week 8 (if the week 6 sample was missed), between 9:00 am and 4:00 pm to correspond with amantadine plateau concentrations.
Statistical Analyses
All statistical methods were based on the International Conference on Harmonization E9 Guidance for Industry “Statistical Principles for Clinical Trials.”11
The efficacy analysis population (modified intent to treat, mITT) consisted of patients who met all of the following: had PD, satisfied the MDS‐UPDRS and diary study entry criteria for LID, were randomized, received at least one dose of study medication, and provided at least one post–baseline efficacy assessment. The sample size was based on providing 80% power, using a two‐sided, two‐sample test at the 5% level of significance.
An analysis of covariance (ANCOVA) model with treatment as a factor and baseline UDysRS value as a covariate was used for the primary efficacy analysis of the 340‐mg dose level versus placebo. Except for CGI‐C, analysis of secondary end points also used an ANCOVA model.
Key secondary analyses of the UDysRS (260‐mg and 420‐mg dose levels vs. placebo) and the FSS (all dose levels vs. placebo) were conducted using a hierarchical procedure12 to control the overall type 1 error. Within the same ANCOVA model used for the primary endpoint analysis, a test of dose–response across the four groups (placebo, 260 mg ADS‐5102, 340 mg ADS‐5102, 420 mg ADS‐5102) was carried out. The dose–response test was implemented using the scores 0, 260, 340, and 420 and additionally using equally spaced scores for the treatment groups.
Prespecified directions for the handling of individual missing data values related to each efficacy outcome measure, including imputation (where allowable), and the appropriate utilization of a Last Observation Carried Forward approach, were included in the Statistical Analysis Plan.
Results
Demographics and PD characteristics at baseline are shown in Table 1. The treatment groups appeared to be reasonably well balanced at study entry for these parameters. A total of 83 patients were randomized in the study. The mITT population included 80 patients; one patient was excluded from each of the active treatment groups according to the predefined mITT criteria. Figure 1 depicts patient disposition. The most common reasons for screen failure were insufficient dyskinesia, inadequate renal function, and use of a prohibited medication. The patient study completion rate was 81% (91% placebo, 77% ADS‐5102). The most common reason for study drug discontinuation was adverse events. Five of the eight patients in the 420‐mg group who discontinued study drug because of adverse events withdrew during initial dose titration (before week 3) and did not receive the 420‐mg assigned dose. Treatment assignment was not revealed for any patient during study conduct. The daily levodopa dosage at week 8 was unchanged from baseline for all treatment groups. At baseline and week 8, respectively, 98.8% and 100% of UDysRS records, and 96.4% and 100% of MDS‐UPDRS records, were complete and evaluable, and 100% and 97.0% of PD home diaries were evaluable, with four or fewer 30‐min intervals of missing or illegible data.
Table 1.
Patient baseline characteristics
| Placebo (n = 22) | 260 mg ADS‐5102 (n = 20) | 340 mg ADS‐5102 (n = 21) | 420 mg ADS‐5102 (n = 20) | All Patients (n = 83) | ||
|---|---|---|---|---|---|---|
| Age (y), Mean (SD) | 65.5 (10.2) | 67.5 (8.6) | 64.7 (10.0) | 66.4 (9.4) | 66.0 (9.5) | |
| Sex, n | Male/female | 14/8 | 8/12 | 13/8 | 10/10 | 45/38 |
| Ethnicity, n (%) | Not Hispanic | 21 (95.5) | 18 (90.0) | 21 (100.0) | 18 (90.0) | 78 (94.0) |
| Hispanic | 1 (4.5) | 2 (10.0) | 0 | 2 (10.0) | 5 (6.0) | |
| Race, n (%) | Amer. Indian or Alaska Native | 0 | 2 (10.0) | 0 | 0 | 2 (2.4) |
| Asian | 1 (4.5) | 0 | 0 | 3 (15.0) | 4 (4.8) | |
| Black or African American | 1 (4.5) | 0 | 1 (4.8) | 0 | 2 (2.4) | |
| White | 20 (90.9) | 18 (90.0) | 20 (95.2) | 17 (85.0) | 75 (90.4) | |
| Time since PD diagnosis (y), mean (SD) | 10.7 (7.1) | 8.9 (3.4) | 9.3 (4.9) | 9.0 (3.5) | 9.5 (5.0) | |
| PD medication | Levodopa | 22 (100.0) | 20 (100.0) | 21 (100.0) | 20 (100.0) | 83 (100.0) |
| Dopamine agonist | 14 (63.6) | 14 (70.0) | 10 (47.6) | 16 (80.0) | 54 (65.1) | |
| MAO‐B inhibitor | 14 (63.6) | 11 (55.0) | 12 (57.1) | 12 (60.0) | 49 (59.0) | |
| COMT inhibitor | 12 (54.5) | 7 (35.0) | 9 (42.9) | 9 (45.0) | 37 (44.6) | |
| Anticholinergic | 1 (4.5) | 0 | 0 | 1 (5.0) | 2 (2.4) | |
| Duration of levodopa treatment (y), mean (SD) | 9 (7.0) | 6.9 (3.7) | 8.2 (5.3) | 8.3 (3.2) | 8.1 (5.1) | |
| Levodopa daily dose (mg), mean (SD) | 801.1 (431.9) | 714.5 (449.3) | 694.0 (278.4) | 862.5 (585.9) | 768.6 (444.4) | |
| FSS, mean (SD) | 4.9 (1.2) | 4.4 (1.5) | 4.8 (1.4) | 4.8 (1.1) | 4.7 (1.3) | |
| MMSE, mean (SD) | 28.6 (1.8) | 28.6 (2.0) | 28.8 (1.5) | 28.2 (2.0) | 28.5 (1.8) | |
| Hoehn and Yahr, mean (SD) | 2.5 (0.7) | 2.5 (0.9) | 2.5 (0.6) | 2.4 (0.8) | 2.5 (0.7) | |
| UDysRS, total score, mean (SD) | 39.2 (17.8) | 39.8 (13.5) | 43.8 (12.1) | 41.9 (12.0) | 41.1 (14.0) | |
| MDS‐UPDRS (part IV) mean (SD) | 11.7 (3.1) | 10.7 (2.6) | 11.7 (2.8) | 10.8 (3.0) | 11.2 (2.9) | |
| PD home diary, mean (SD) | ON time without troublesome dyskinesia, h | 6.9 (3.8) | 6.7 (3.9) | 7.9 (3.5) | 9.2 (3.1) | 7.7 (3.7) |
| ON time with troublesome dyskinesia, h | 6.1 (3.2) | 6.3 (3.3) | 4.3 (2.9) | 5.0 (2.5) | 5.4 (3.1) | |
| ON time with dyskinesia, total, h | 10.2 (3.0) | 10.1 (3.6) | 7.9 (3.2) | 10.2 (3.4) | 9.6 (3.4) | |
| OFF time, h | 3.2 (2.7) | 2.7 (2.6) | 4.1 (2.7) | 2.2 (1.6) | 3.1 (2.5) | |
| Sleep time, h | 7.8 (1.3) | 8.3 (2.1) | 7.7 (1.6) | 7.6 (1.6) | 7.8 (1.7) | |
PD, Parkinson's disease; SD, standard deviation; FSS, Fatigue Severity Scale; MMSE, Mini‐Mental Status Examination; UDysRS, Unified Dyskinesia Rating Scale; MDS‐UPDRS, Movement Disorder Society—Unified Parkinson's Disease Rating Scale.
Figure 1.
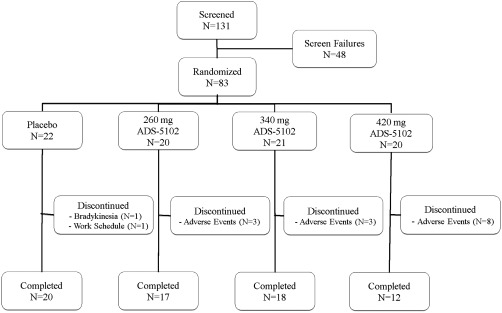
Patient Disposition
Efficacy
The primary efficacy analysis compared 340 mg ADS‐5102 with placebo in mean change in UDysRS total score from baseline to week 8. A significant decrease (improvement in LID) was observed for 340 mg ADS‐5102 (least‐square [LS] mean treatment difference = –11.3 [95% CI: −19.1, −3.5], P = 0.005). For the 340‐mg dose, this represented a 27% reduction compared with placebo (43% and 16% reductions from baseline for 340 mg and placebo, respectively). The 420‐mg ADS‐5102 dose also met statistical significance (LS mean treatment difference = –10.0 [95% confidence interval (CI): −17.8, −2.2], P = 0.013), a 25% reduction compared with placebo, whereas 260 mg ADS‐5102 did not (LS mean treatment difference = –5.6 [95% CI: −13.4, 2.2], P = 0.159). A dose–response was also confirmed for the mean change from baseline to week 8 in UDysRS total score (P < 0.006 from both scoring methods). Results are shown in Figure 2.
Figure 2.
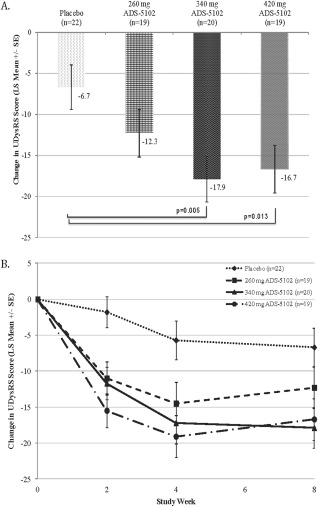
Change in Unified Dyskinesia Rating Scale (UDysRS) total score (LS Mean +/‐SE), Baseline to Week 8 (Panel A; a decrease indicates improvement) and over time (Panel B).
Additional efficacy results are presented in Table 2. The 24‐h PD patient diaries showed that ON time without troublesome dyskinesia increased by 3.3 h for 260 mg ADS‐5102, 3.0 h for 340 mg ADS‐5102, and 2.7 h for 420 mg ADS‐5102 (LS mean treatment differences vs. placebo, all statistically significant). In addition, for 260 mg and 340 mg, a mean decrease occurred in OFF time (−1.3 and −0.9 h, respectively, relative to placebo), although this did not reach statistical significance.
Table 2.
Secondary efficacy outcomes, changes from baseline to week 8
| LS Mean Treatment Difference vs. Placebo (95% CI) | |||
|---|---|---|---|
| Outcome Measure | 260 mg ADS‐5102 (n = 19) | 340 mg ADS‐5102 (n = 20) | 420 mg ADS‐5102 (n = 19) |
| ON time without troublesome dyskinesia, h | 3.3 (1.1, 5.5) p=0.004 | 3.0 (0.8, 5.2) p=0.008 | 2.7 (0.5, 5.0) p=0.018 |
| ON time with troublesome dyskinesia, h | −1.3 (−3.1, 0.6) p=0.169 | −1.8 (−3.6, 0.0) p=0.055 | −2.8 (−4.6, −0.9) p=0.003 |
| ON time with dyskinesia, total, h | −1.1 (−3.7, 1.5) p=0.408 | −2.1 (−4.8, 0.5) p=0.117 | −3.1 (−5.8, −0.5) p=0.021 |
| OFF time, h | −1.3 (−2.7, 0.1) p=0.074 | −0.9 (−2.3, 0.5) p=0.199 | 0.1 (−1.4, 1.5) p=0.934 |
| Sleep time, h | −0.8 (−1.8,0.2) p=0.099 | −0.4 (−1.4,0.5) p=0.367 | −0.3 (−1.2,0.7) p=0.573 |
| UDysRS, total objective score (parts III, IV) | −2.5 (−6.0, 0.9) p=0.147 | −5.2 (−8.7, −1.7) p=0.004 | −6.4 (−9.8, −2.9) p<0.001 |
| MDS‐UPDRS (part IV) | −0.7 (−2.9, 1.5) p=0.520 | −2.4 (−4.6, −0.3) p=0.026 | −3.4 (−5.6, −1.2) p=0.003 |
| MDS‐UPDRS (part IV, item 4.1) ‐Time spent with dyskinesia | −0.2 (−0.8, 0.5) p=0.630 | −0.6 (−1.2, 0.1) p=0.100 | −0.6 (−1.3, 0.0) p=0.057 |
| MDS‐UPDRS, (part IV, item 4.2) ‐Functional impact of dyskinesia | −0.8 (−1.4, −0.2) p=0.014 | −1.0 (−1.6, −0.4) p=0.002 | −1.3 (−2.0, −0.7) p<0.001 |
| MDS‐UPDRS (parts I, II, III, combined) | 1.2 (−7.7, 10.1) p=0.786 | −2.2 (−11.2, 6.9) p=0.636 | 1.7 (−7.2, 10.6) p=0.705 |
| Fatigue Severity Score | 0.2 (−0.6, 1.0) p=0.630 | −0.3 (−1.1, 0.5) p=0.431 | 0.3 (−0.5, 1.0) p=0.522 |
| PDQ‐39 (summary index score) | −0.3 (−8.7, 8.0) p=0.942 | −3.4 (−11.5, 4.7) p=0.406 | 2.2 (−5.9, 10.3) p=0.590 |
| Outcome Measure | Placebo (n = 22) | 260 mg ADS‐5102 (n = 19) | 340 mg ADS‐5102 (n = 20) | 420 mg ADS‐5102 (n = 19) | |
|---|---|---|---|---|---|
| CGI‐C, n (%) | Marked improvement | 1 (4.5) | 2 (10.5) | 7 (35.0) | 4 (21.1) |
| Moderate improvement | 6 (27.3) | 8 (42.1) | 8 (40.0) | 6 (31.6) | |
| Minimal improvement | 4 (18.2) | 5 (26.3) | 1 (5.0) | 5 (26.3) | |
| No change | 10 (45.5) | 3 (15.8) | 4 (20.0) | 2 (10.5) | |
| Minimal worsening | 1 (4.5) | 1 (5.3) | 0 | 0 | |
| Moderate worsening | 0 | 0 | 0 | 2 (10.5) | |
| Marked worsening | 0 | 0 | 0 | 0 | |
| P‐valuea | N/A | 0.1042 | 0.0036 | 0.2158 | |
The P‐value was determined using the Cochran‐Mantel‐Haenszel mean score test (using equally spaced scores)
Abbreviations: LS, least square; CI, confidence interval; UDysRS, Unified Dyskinesia Rating Scale; MDS‐UPDRS, Movement Disorder Society Unified Parkinson's Disease Rating Scale; PDQ‐39, Parkinson's Disease Questionnaire; CGI‐C, Clinician Global Impression of Change.
Little change was seen in the MDS‐UPDRS total score (parts I, II, III, combined) vs placebo at all dose levels, suggesting that no worsening of PD occurred during ADS‐5102 dosing. The CGI‐C results indicated that 75% of patients in the 340‐mg dose group had a moderate to marked improvement in their clinical status (related to overall PD, including but not limited to LID) at week 8, versus 32% of placebo patients (see Table 2).
Safety Results
Overall, 87% of the 83 patients experienced AEs: 82% of placebo patients, and 80%, 95%, and 90% of patients in the 260‐mg, 340‐mg, and 420‐mg ADS‐5102 treatment groups, respectively. The most frequent AEs are shown in Table 3. A similar proportion of patients in all three ADS‐5102 treatment groups reported hallucinations, most often visual, an adverse event that was not reported by placebo patients. Eight of the 13 patients who experienced hallucinations ultimately discontinued treatment. Five patients had a total of seven serious adverse events (SAEs), including one patient in the 260‐mg group with lobar pneumonia and mental status changes, and four patients in the 420‐mg group with skin hypersensitivity and psychotic disorder, urinary tract infection, lower extremity cellulitis, and subdural hematoma.
Table 3.
Adverse events (>10% or >2 patients in any ADS‐5102 group)
| Adverse Event n (%) | Placebo (n = 22) | 260 mg ADS‐5102 (n = 20) | 340 mg ADS‐5102 (n = 21) | 420 mg ADS‐5102 (n = 20) |
|---|---|---|---|---|
| Constipation | 2 (9.1) | 7 (35.0) | 5 (23.8) | 3 (15.0) |
| Dizziness | 1 (4.5) | 3 (15.0) | 6 (28.6) | 3 (15.0) |
| Hallucination | 0 | 4 (20.0) | 5 (23.8) | 4 (20.0) |
| Dry mouth | 0 | 3 (15.0) | 4 (19.0) | 2 (10.0) |
| Fall | 3 (13.6) | 1 (5.0) | 3 (14.3) | 3 (15.0) |
| Confusion | 1 (4.5) | 1 (5.0) | 3 (14.3) | 2 (10.0) |
| Headache | 1 (4.5) | 1 (5.0) | 3 (14.3) | 1 (5.0) |
| Nausea | 1 (4.5) | 1 (5.0) | 3 (14.3) | 1 (5.0) |
| Asthenia | 1 (4.5) | 0 | 3 (14.3) | 1 (5.0) |
Fourteen patients discontinued treatment because of AEs (including some of the previously mentioned SAEs). These included three patients in the 260‐mg group (2 patients with hallucination, and 1 patient with psychotic disorder, mental status changes, and suicidal ideation), three patients in the 340‐mg group (1 patient with hallucination and muscle spasms, 1 patient with hallucination, urinary hesitation, and renal impairment, 1 patient with hallucination and peripheral edema), and eight patients in the 420‐mg group (3 patients with hallucination, 1 patient with balance disorder, confusional state, and dry mouth, 1 patient with urinary tract infection, 1 patient with skin hypersensitivity, 1 patient with subdural hematoma, and 1 patient with constipation). No differences were seen across treatment groups in laboratory results and vital signs.
Mean (standard deviation) steady‐state amantadine plasma concentrations (ng/mL) were 1,383 (354), 1,431 (707), and 1,677 (512) in the 260‐mg, 340‐mg, and 420‐mg groups, respectively. In the 260‐mg group, mean concentrations were similar across all time points. During the titration period for the 340‐mg and 420‐mg groups, a dose proportional increase in the mean concentrations was generally seen. Concentrations were below limit of quantification in all placebo patients at all time points measured.
Discussion
The study demonstrated that ADS‐5102 at 340 mg administered once daily at bedtime was safe and effective in improving dyskinesia in PD patients with LID as measured by the UDysRS, PD patient diaries, and MDS‐UPDRS, part IV. The highest dose of 420 mg was efficacious, but more patients withdrew from this group (primarily during initial dose titration) because of AEs. The 340‐mg dose level provided the best balance between efficacy and safety. Improvement was evident by 2 weeks of dosing, was further improved at 4 weeks, and was maintained throughout the study.
The most frequent AEs in ADS‐5102–treated patients were constipation, dizziness, hallucinations, and dry mouth. One patient developed livedo reticularis (340 mg), and no patient who received ADS‐5102 reported an impulse control disorder. Central nervous system AEs occurred in all treatment groups, including placebo, and primarily during the first 3 weeks of dosing (Kaplan‐Meier survival analysis, data not shown). Most AEs that resulted in ADS‐5102 discontinuation were central nervous system related, and frequently involved hallucinations, which are part of amantadine's known safety profile.
Although hallucinations did not occur in the placebo group, they commonly occur in PD, and additional factors such as age, cognitive impairment, other co‐morbidities, or concomitant medications also may influence the risk of this event in this population.13, 14
The change in the UDysRS total score was the primary outcome measure for this study. The UDysRS has not been widely used in clinical trials. Goetz et al.15 recently evaluated the UDysRS and several other instruments for their effectiveness in detecting treatment effects in PD patients with LID and demonstrated that the UDysRS, having both subjective and objective ratings, was superior in detecting treatment effects. The authors reported a reduction in the UDysRS total score of −6.6 points with immediate‐release amantadine (mean dose: 284 mg/d) compared with placebo. In this study, which did not include a direct comparison to immediate‐release amantadine, ADS‐5102 at 340 mg and 420 mg significantly reduced the UDysRS total score by −11.3 points and −10.0 points compared with placebo, respectively.
In this study, the functional impact of dyskinesia (assessed by MDS‐UPDRS part IV, duration of ON time with troublesome dyskinesia, and duration of ON time with non‐troublesome dyskinesia) was also improved. The CGI‐C results also showed significant improvement at the 340‐mg dose level.
No worsening of PD symptoms as assessed by the MDS‐UPDRS was seen. A numerical reduction in OFF time, a secondary endpoint, suggested possible improvement in underlying PD symptoms. Previous studies have reported improvement in motor fluctuations4 with amantadine, but the improvement in OFF time seen in our study did not reach statistical significance.
One of the limitations of immediate‐release amantadine is that adverse effects, in particular, sleep disturbances, increase in frequency at higher doses (300 mg/d or higher),16, 17, 18 leading to avoidance of amantadine dosing beyond the hours of 2:00 to 4:00 pm.19 In this study, once‐daily dosing at bedtime of ADS‐5102 was well tolerated. Duration of sleep was not impacted, and there was no difference in reported insomnia, nightmares, or abnormal dreams relative to placebo.
In summary, ADS‐5102 was generally well tolerated and resulted in significant and dose‐dependent improvements in LID in PD patients. Although the study was small, it was adequately powered, and results were significant and consistent as measured by the UDysRS, MDS‐UPDRS part IV, PD patient diaries, and the CGI‐C. The types of adverse events were consistent with PD and the known amantadine safety profile. Any conclusions regarding the relative efficacy and safety of ADS‐5102 and immediate‐release amantadine would require a direct comparison of these agents in a randomized trial. ADS‐5102 at 340 mg administered once daily at bedtime provided the best balance between efficacy and safety in improving LID in patients with PD.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
R.P.: 1A, 1B, 1C, 3B, 3A
C.M.T.: 1A, 1B, 1C, 3B
R.A.H.: 1A, 1B, 1C, 3B
K.S.: 1A, 1B, 1C, 3B
S.I.: 1C, 3B
D.T.: 1C, 3B
L.S.: 1C, 3B
A.E.R.: 1B, 1C, 3B
N.L.M.: 1A, 2A, 2C, 3B
G.T.W.: 1A, 2C, 3B
M.J.S.: 1A, 1C, 2A, 2C, 3B
Financial Disclosures
Rajesh Pahwa, MD, is or has been a consultant for Acadia, Adamas, Impax, St Jude Medical, Teva Neuroscience, Medtronic, and US World Meds. He has received honoraria from Medtronic, Teva Neuroscience, UCB, and US World Meds. He has received research grants from Acadia, Adamas, Avid, NIH/NINDS, NPF, and PSG/University of Rochester. He has also served on the data monitoring committee for Ceregene. He has received personal compensation as the Co‐Editor‐in‐Chief of the International Journal of Neuroscience. Caroline M. Tanner, MD, has received honoraria or payments for consulting at AbbVie and Pfizer Pharmaceuticals. Robert A. Hauser, MD, is supported in part by a center grant from the National Parkinson's Disease Foundation. Dr. Hauser received payment from Adamas for participating as a Steering Committee Member and has received honoraria or payments for consulting from advisory boards, or speaking services from AbbVie, Allergan, AstraZeneca, Biotie Therapeutics, Ceregene, Chelsea Therapeutics, Cleveland Clinic, Eli Lilly, GE Healthcare, Heptares, Gerson Lehrman Group, Impax Laboratories, Neurocrine, Indus, Ispen Biopharmaceuticals, Lundbeck, L&M Healthcare, Merck/MSD, Noven Pharmaceuticals, Pfizer, Straken Pharmaceuticals, Targacept, Teva Pharmaceuticals Industries, Ltd., Teva Neuroscience, Upsher‐Smith Laboratories, UCB, UCB Pharma SA, University of Houston, US World Meds, Xenoport and Zambon Company Sp. Kapil Sethi, MD, is a consultant for Teva Neuroscience, Synosia, Veloxis, and Nupathe. He has received grant and research support from Abbot, Acadia, Impax, Phytopharm, NIH, and NPF. He has received honoraria from Teva Neuroscience. He has ownership interest in Elan and receives salary from Merz Pharma as a part‐time Senior Medical Expert. Stuart Isaacson, MD, has received honoraria for CME, consultant, research grants, or promotional speaker on behalf of AbbVie, Acadia, Addex, Allergan, Allon, AstraZeneca, Biotie, Britannia, Chelsea Therapeutics, Civitas, Eisai, GE Healthcare, GSK, Impax, Ispen, Kyowa, Lilly, Lundbeck, Merck Schering‐Plough, Medtronic, Merz, Michael J. Fox Foundation, Novartis, Neurocrine, NIH, Novartis, Orion, Parkinson Study Group, Pfizer, Phytopharm, Purdue, Roche, Santhera, Serono, Shire, Teva, UCB, US World Meds, Vanda and Xenoport. Daniel Truong, MD, has received research grants from Ispen, Merz, Auspex, Diichi Sankyo Pharma, AbbVie, National Institute of Neurological Disorders and Stroke, Kyowa, and Neurocrine. Lynn Struck, MD, has received research grants from Merz and UCB. Lynn Struck MD, has also received honoraria from Teva Neurosciences as a speaker and from Allergan for physician training. April Ruby, Natalie McClure, PhD, and Greg Went, PhD, employees of Adamas, and Mary Jean Stempien, MD, a consultant to Adamas, received compensation and stock options. Mary Jean Stempien, MD, has also received consultancy payments from Cymabay Therapeutics, Inc. and Adheron Therapeutics.
Acknowledgments
We acknowledge and thank the study participants, the EASED Study Investigators, including the Steering Committee, and their staff, the Independent Data Monitoring Committee, Charles Davis PhD, CSD Biostatistics, who provided statistical and analysis support, and Christopher Goetz, MD, Rush University, who trained raters on the proper administration and scoring of the UDysRS.
EASED Principal Investigators/Sites that enrolled study participants included Pinky Agarwal, MD, Booth Gardner Parkinson's Care Center, Evergreen Hospital Medical Center, Kirkland, Washington; Matthew S. Boyce, MD, Neurological Associates, Inc., Richmond, Virginia; Lawrence W. Elmer, MD, PhD, University of Toledo, Toledo, Ohio; Alan Freeman, MD, Emory University, Atlanta, Georgia; Ramon A. Gil, MD, Parkinson's Disease Treatment Center of SW Florida, Port Charlotte, Florida; Robert A. Hauser, MD, University of South Florida Medical Center, Tampa, Florida; Bonnie P. Hersh, Harvard Vanguard Medical Associates, Boston, Massachusetts; Keith L. Hull, Jr., MD, Raleigh Neurology Associates, Raleigh, North Carolina; Robert Hutchman, MD, Neurosearch, Inc., Reseda, California; Stuart Isaacson, MD, Parksinson's Disease and Movement Disorders Center, Boca Raton, Florida; Srinath Kadimi, MD, FRCS, Associated Neurologists of Southern CT, Fairfield, Connecticut; Kevin J. Klos, MD, The Movement Disorders Clinic of Oklahoma, Tulsa, Oklahoma; Peter A. LeWitt, MD, Henry Ford West Bloomfield Hospital, West Bloomfield, Michigan; Grace S. Lin Liang, MD, The Parkinson's Institute, Sunnyvale, California; Jerome P. Lisk, MD, Neurosearch, Pasadena, California; Paul A. Nausieda, MD, Wisconsin Institute for Neurologic and Sleep Disorders, Milwaukee, Wisconsin; Omid Omidvar, MD, Collaborative Neuroscience Network, Inc., Long Beach, California; Rajesh Pahwa, MD, University of Kansas Medical Center, Kansas City, Kansas; Eric J. Pappert, MD, Neurology Associates, San Antonio, Texas; Michael Rezak, MD, PhD, Central DuPage Hospital, Winfield, Illinois; William L. Severt, MD, Beth Israel Medical Center, New York, New York; Robert J. Shorr, MD, Neurosearch II, Ventura, California; Lynn Struck, MD, Iowa Health Physicians, Des Moines, Iowa; Daniel Truong, MD, Parkinson's & Movement Disorder Institute, Fountain Valley, California; Leonard A. Verhagen Metman, MD, PhD, Rush University Medical Center, Chicago, Illinois; Ronald E. Wilson, MD, Brentwood Neurology PLC, Brentwood, Tennessee.
The Independent Data Monitoring Committee included Richard Dewey, MD, University of Texas Southwestern Medical Center, Dallas, Texas; Stewart Factor, DO, Emory School of Medicine, Atlanta, Georgia; Andrew Siderowf, MD, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania.
The copyright line for this article was changed on 12 February 2015 after original online publication.
Funding agencies: This study was supported by Adamas Pharmaceuticals, Inc.
Relevant conflicts of interest/financial disclosures: Rajesh Pahwa, Caroline Tanner, Robert Hauser and Kapil Sethi were Steering Committee members and received honoraria for this service. Rajesh Pahwa, Robert Hauser, Stuart Isaacson, Daniel Truong and Lynn Struck were Study Investigators and received research support for study conduct. April Ruby, Natalie McClure and Greg Went, employees of Adamas, and Mary Jean Stempien, a consultant to Adamas, received compensation and stock options.
Full financial disclosures and author roles may be found in the online version of this article.
References
- 1. Suh DC, Pahwa R, Mallya U. Treatment patterns and associated costs with Parkinson's disease levodopa induced dyskinesia. J Neurol Sci 2012;319:24–31. [DOI] [PubMed] [Google Scholar]
- 2. Ahlskog JE, Muenter MD. Frequency of levodopa‐related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001;16:448–458. [DOI] [PubMed] [Google Scholar]
- 3. Schwab RS, England AC, Jr ., Poskanzer DC, Young RR. Amantadine in the treatment of Parkinson's disease. JAMA 1969;208:1168–1170. [PubMed] [Google Scholar]
- 4. Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson's disease. Neurology 1998;50:1323–1326. [DOI] [PubMed] [Google Scholar]
- 5. Verhagen Metman L, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa‐induced dyskinesias: a 1‐year follow‐up study. Arch Neurol 1999;56:1383–1386. [DOI] [PubMed] [Google Scholar]
- 6. Goetz CG, Stebbins GT, Tilley BC. Calibration of unified Parkinson's disease rating scale scores to Movement Disorder Society‐unified Parkinson's disease rating scale scores. Mov Disord 2012;27:1239–1242. [DOI] [PubMed] [Google Scholar]
- 7. Wolf E, Seppi K, Katzenschlager R, et al. Long‐term antidyskinetic efficacy of amantadine in Parkinson's disease. Mov Disord 2010;25:1357–1363. [DOI] [PubMed] [Google Scholar]
- 8. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society‐sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 10. Hauser RA, Deckers F, Lehert P. Parkinson's disease home diary: further validation and implications for clinical trials. Mov Disord 2004;19:1409–1413. [DOI] [PubMed] [Google Scholar]
- 11.ICH Harmonised Tripartite Guideline E9: Statistical Principles for Clinical Trials. 1998:Current Step 4 version.
- 12. Westfall PH, Krishen A. Optimally weighted, fixed sequence and gatekeeper multiple testing procedures. J Statistical Planning Inference 2001;99:25–40. [Google Scholar]
- 13. Forsaa EB, Larsen JP, Wentzel‐Larsen T, Goetz CG, Stebbins GT, Aarsland D, Alves G. A 12‐year population‐based study of psychosis in Parkinson disease. Arch Neurol 2010;67:996–1001. [DOI] [PubMed] [Google Scholar]
- 14. Lee AH, Weintraub D. Psychosis in Parkinson's disease without dementia: common and comorbid with other non‐motor symptoms. Mov Disord 2012;27:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goetz CG, Stebbins GT, Chung KA, et al. Which dyskinesia scale best detects treatment response? Mov Disord 2013;28:341–346. [DOI] [PubMed] [Google Scholar]
- 16. Silver DE, Sahs AL. Double blind study using amantadine hydrochloride in the therapy of Parkinson's disease. Trans Am Neurol Assoc 1971;96:307–308. [PubMed] [Google Scholar]
- 17. Hayden FG, Gwaltney JM Jr., Van de Castle RL, Adams KF, Giordani B. Comparative toxicity of amantadine hydrochloride and rimantadine hydrochloride in healthy adults. Antimicrob Agents Chemother 1981;19:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackson GG, Stanley ED, Muldoon RL. Chemoprophylaxis of viral respiratory disease. Bull Pan Am Health Org 1967:595–603. [Google Scholar]
- 19.Merz Pharmaceuticals GmbH. PK‐Merz®film‐coated tablet. 2003:11.