

Abstract
Background & Aims
Acute liver failure (ALF) is a condition with high mortality and morbidity. Fibrosis in chronic liver disease was extensively researched, whereas fibrosis and underlying mechanism in acute liver failure remains unclear.
Methods
Hepatitis B virus related ALF patients were recruited to investigate if there was ongoing fibrosis by liver histology and liver stiffness measurement(LSM) analysis as well as fibrosis markers assay. Sera HMGB1 were kinetically detected in progression and remission stage of ALF. Hepatic stellate cell(HSC) activation by HMGB1 was explored by testing mRNA and protein level of α‐SMA and collagen 1a1 by using qPCR and western blot. Autophagy induction by HMGB1 was explored by LC3‐II conversion, autophagy flux and fluorescence.
Results
Firstly, ongoing fibrosis in progression stage of ALF was confirmed by histological analysis, LS measurement as well as fibrosis markers detection. HSC activation and autophagy induction in explanted liver tissue also revealed. Next, kinetic monitoring sera HMGB1 revealed elevated HMGB1 in progression stage of ALF vs HBsAg carrier, and drop back to base level in remission stage. Thirdly, rHMGB1 dose dependently activated HSCs, as indicated by increased mRNA and proteins level in α‐SMA and collagen 1a1. Moreover, autophagy was induced in HSC treated with rHMGB1, as illustrated by increased LC3 lipidation, elevated autophagy flux and GFP‐LC3 puncta.
Conclusions
Acute liver failure is accompanied by ongoing fibrosis, HSC activation and autophagy induction. Increased HMGB1 activates HSC via autophagy induction. Those findings integrate HMGB1, HSCs activation, autophagy into a common framework that underlies the fibrosis in ALF.
Keywords: acute liver failure, autophagy, fibrosis, hepatic stellate cell, hepatitis B virus, high mobility group box 1
Abbreviations
- ALF
acute liver failure
- BA
bafilomycin A1
- CHB
chronic hepatitis B
- CIV
collagen IV
- HA
hyaluronic acid
- HBV
hepatitis B virus
- HMGB1
high mobility group box 1
- HSC
hepatic stellate cells
- LN
laminin
- LSM
liver stiffness measurement
- MA
macroautophagy
- PCIII
procollagen III
- α‐SMA
alpha smooth muscle actin
Key Points.
Liver tissue from patients with acute liver failure showed collagen deposition, hepatic stellate cell (HSC) activation and autophagy induction.
In progression stage of acute liver failure, an elevated HMGB1 level were observed when compared with HBsAg carrier, whereas in remission stage, HMGB1 level returned to baseline that is comparable to HBsAg carrier.
In vitro study shown that HMGB1 activates HSC, as measured by increased mRNA and proteins level in α‐SMA and collagen 1a1 in primary rat HSC and HSC‐T6 cell line treated with rHMGB1.
For the first time, we found that HMGB1 induce autophagy in HSC as shown increased LC3 lipidation, autophagosome formation and elevated autophagy flux.
Acute liver failure (ALF) is a rare condition in which rapid deterioration of liver function results in altered mention and coagulopathy in individuals without known pre‐existing liver disease, which often affects young persons and carries a high morbidity and mortality 1, 2. Aside from liver transplantation, there is currently a paucity of effective therapies.
Fibrosis, defined as the accumulation of excessive amounts of extracellular matrix (ECM), is a highly conserved and co‐ordinated protective wound healing response towards acute and chronic tissue injury 3. Activated hepatic stellate cells is primary extracellular ECM‐producing cells in liver 4. In chronic liver injury, fibrosis is widely acknowledged as a damaging process with potential progression to cirrhosis and further sequelae that include liver cancer and hepatic failure. However, data on fibrosis in ALF is still scarce. Recent data support the conception that short‐term occurrence of fibrosis in ALF may be physiological and possible beneficial response by the liver, which serves as a scaffolding that support the parenchyma and maintains hepatic integrity 5, 6. Indeed, blockade of fibrosis by depleting activated HSCs in APAP induced mouse ALF model demonstrated a significantly severe liver damage and declined survival rate 7. In addition, mutation in collagen‐I results in failure of recovery from CCl4‐induced liver fibrosis and diminished hepatocyte regeneration 8.
Hepatic stellate cells are located in the space of Disse between the sinusoidal endothelial cells and hepatic epithelial cells, and account for 5–8% of the cells in the liver 6. In a healthy liver, stellate cells are quiescent and contain numerous vitamin A lipid droplets, constituting the largest reservoir of vitamin A in the body. When the liver is injured due to viral infection or hepatic toxins, hepatic stellate cells receive signals secreted by damaged hepatocytes and immune cells, causing them to transdifferentiate into activated myofibroblast‐like cells, characterized by alpha‐smooth muscle actin (α‐SMA) expression and ECM production 6. Due to the dramatic clinical course of ALF, little research has been done to investigate how HSCs activation is regulated in ALF. HMGB1, a nuclear protein is present in almost all eukaryotic cells,can be passively released from injured/died cell 9, 10, 11. A characteristic feature of ALF is excessive hepatocyte apoptosis and necrosis 12. Previously, studies show that HMGB1 plays a detrimental role in pathogenesis of hepatic inflammation 13, 14, 15. However, recent data shown that hepatocyte‐specific HMGB1 deletion worsens the acute liver injury in ischaemia/reperfusion model 16, suggesting a beneficial effect on acute liver injury. HMGB1 has been involved in pulmonary fibrosis 17. To date, very few studies addressed the role HMGB1 played in HSCs activation and liver fibrosis in ALF.
Autophagy is an evolutionarily conserved process by which cytoplasmic components including macromolecules and organelles are degraded with their own lysosome 18. Autophagy has been connected to human pathophysiology, such as cancer, neurodegeneration, immune response, development and ageing, cell differentiation, tissue remodelling, damage repair and tissue fibrosis 19, 20. Study on liver fibrosis mouse model induced by CCl4 or TAA demonstrates autophagy activation in HSCs 21. In addition, a number of genes modulate autophagy also participate in HSCs activation. The cathepsins B and cathepsins D, lysosomal molecular with well‐defined role in autophagic protein degradation, has been linked to fibrogensis 22. Moreover, HMGB1 can directly interact with autophagy protein Beclin1 and induces autophagy in Panc2.03 and HCT116 cancer cells 23. However, as far as we know, it remains unknown whether autophagy regulate HSC activation and liver fibrosis in the setting of ALF.
Based on this, the aim of this study was to observe if there is ongoing fibrosis during ALF and investigate the underlying mechanism of fibrosis and HSC activation.
Materials and methods
Patients
From April 2011 to November 2013, patients were prospectively studied after admission for acute liver failure (ALF) to the First Affiliated Teaching Hospital, Xian Jiaotong University, Shaanxi, China. A written informed consent was obtained from either patients or their next‐to‐kin, depending on the patient's altered mental status, as part of entry into The Major National Science and Technology Projects for Infectious Diseases (11th and 12th 5 Year, China)(No. 2008ZX10002‐007, No. 2012ZX10002007). Although there were a variety of etiologies of ALF, to better elucidate pathogenesis of ALF, all the participants in this study were homogenously HBV related ALF. Patients with hepatitis B virus (HBV) related ALF were defined as those with (i) evidence of coagulation abnormality, usually an INR≥1.5; (ii) and any degree of mental alteration (encephalopathy); (iii) without pre‐existing cirrhosis and with an illness of <26 weeks duration 2; (iv)evidence of HBV ongoing infection. A total of 67 patients diagnosed as HBV related ALF according to criteria mentioned above. We calculated Model for End‐Stage Liver Disease (MELD) scores according to the equation: 3.78 × ln[serum bilirubin (mg/dl)] + 11.2 × ln(INR) + 9.57 × ln[serum creatinine (mg/dL)] + 6.43. Progression stage of ALF defined as the period from admission to the time point with peak MELD score, whereas remission stage defined as the period from peak MELD score to the lowest in‐hospital MELD score. During the same period, age‐ and gender‐matched HBsAg carrier, CHB patients and compensated cirrhosis patients were recruited as controls for HBV infection, inflammation and cirrhosis respectively. The serum samples of ALF patients in the progression stage and the remission stage, the HBsAg carrier, CHB patients and the cirrhosis patients were collected, as described previously 24, and stored at −80°C in microfuges tubes until assay. Patients with the following conditions were excluded from this study: had a past history of alcoholic liver disease, presence of other causes of liver disease, incomplete data on liver evaluation, refusal providing blood samples, following up for less than 26 weeks. This study protocol was approved by the Human Subject Research Committee from the First Affiliated Hospital of Xi'an Jiaotong University.
Histological sampling
Patients with evidence of pre‐existing cirrhosis were excluded as described previously 25. Tissue samples from three patients received a transplant were retrieved and used for liver histology and immunohistochemistry.
ELISA
The concentrations of HMGB1 in serum were detected by using ELISA kits from Westang Corporation (China) according to the manufacturer's recommendations. Absorbance was measured at 450 nm using Epoch Microplate Spectrophotometer (BioTek instruments, Vermont, USA). The results were calculated using a calibration curve prepared from standards.
Isolation, culturing and treatment of rat primary HSCs
Hepatic Stellate Cells were isolated from rat by sequential digestion of the liver with pronase and collagenase, followed by gradient centrifugation, as previously described 26. Cell viability was determined using Trypan blue exclusion staining. HSC populations were between 90–99% pure and 95% viable. After isolation, rat HSCs were cultured in Dulbecco's Modified Eagles Medium (DMEM) (GIBCO/BRL; Grand Island, NY, USA) supplemented with 10% fetal bovin serum and 2 mM L‐glutamine at 37°C in a 95% air‐5% CO2‐humidified atmosphere. Some cells were treated with 1 μg/ml HMGB1(Sigma, St. Louis, MO, USA), 100 nM bafilomycin A1 (BA) (Sigma, St. Louis, MO, USA) or HMGB1 A box (Sigma, St. Louis, MO, USA).
Culture of HSC‐T6
Hepatic Stellate Cells ‐T6 cells were cultured in the same medium as rat primary HSCs. For Matrigel culture, HSC‐T6 was trypsinized and 5 × 105 cells were seeded on plastic dishes coated with Matrigel (50 μl/cm2 of growth surface) according to the manufacturer's instructions (BD Biosciences, Bedford, MA).
Quantitative real‐time reverse transcript polymerase chain reaction (q‐rt PCR)
Total RNA was isolated by using RNeasy Micro Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instruction. Real‐time quantification was performed by using a FAST real time System (Applied Biosystem, Foster City, CA, USA). Data were normalized to GAPDH or actin. Fold change was determined using the 2‐ΔΔCt method and all reactions were performed in triplicate.
Immunoblot
Protein extracts were prepared by lysing cells in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1.0% Nonidet P‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 1 mM EDTA and 50 mM TrisHCl, pH 8.0) supplemented with Complete EDTA‐free protease inhibitor cocktail tablets (Roche Applied Science, Indianapolis, IN, USA) and phosphatase inhibitor cocktails (Sigma‐Aldrich, St Louis, MO, USA). Cleared lysates were separated on 14% Tris‐glycine gels or 8% to 13% Bis‐Tris gels (Invitrogen) followed by transfer onto nitrocellulose. After blocking in 5% skim milk, blots were probed using the following primary antibodies: rabbit anti‐LC3 (cell signaling, Boston, MA, USA), SQSTM1/p62 (Novus, Littleton, CO, USA), rabbit anti‐type I collagen (Rockland Inc, Gilbertsville, PA, USA), rabbit anti‐α‐SMA (Abcam, cambridge, MA, USA), anti‐GAPDH (Santa Cruz, CA, USA). Bands were detected using horseradish peroxidase–labelled secondary antibodies. Blot were developed using ECL detection system.
Measurement of autophagic flux
Hepatic Stellate Cells ‐T6 cells were plated at 1.5 × 105 cells/well in 12‐well plates the day, and 50 ng of GFP‐LC3 along with Lipofectamine 2000 (Invitrogen, California) was co‐transfected according to the manufacturer's instruction. For the detection of the autophagic flux, HSC‐T6 treated with or without HMGB1 for 24 hours, GFP‐LC3 puncta were visualized using a Leica DM6000B microscope and quantification was carried out using Image J.
Statistical analysis
Results were expressed as mean±standard deviation. Statistical analysis were performed by spss software 13.0(SPSS Inc., Chicago,IL, USA). Student‐t test or one‐way anova followed by post hoc Tukey's analysis or Kruskal–Wallis test depending on the data distribution, were used for comparing the difference in variables between ALF and controls. Reported P values were two‐sided, and P < 0.05 were considered statistically significant.
Results
ALF outcomes
A total of 67 patients diagnosed as HBV related ALF according to criteria mentioned above, of the 67 ALF, 16 died within 4 weeks after diagnosis and 3 underwent liver transplantation, all remaining 45 patients showed recovery with antiviral, immunomodulants and/or supportive medical therapy. Of them, a total of 17 patients enrolled in our observation according to the inclusion and exclusion criteria(referring to Materials and methods). During the same period, age‐ and gender‐ matched HBsAg carrier, Chronic hepatitis B (CHB) patients and cirrhosis patients were recruited as controls for HBV infection, inflammation and cirrhosis respectively. The clinical parameters of HBsAg carrier, CHB patients, cirrhosis patients, patients with ALF in progression stage and ALF in remission stage are listed in Supplementary Table 1.
Table 1.
Comparison of fibrotic markers and liver stiffness in patients with ALF, CHB and cirrhosis
| ALFa | CHB | P 1 | Cirrhosis | P 2 | |
|---|---|---|---|---|---|
| HA (μg/L) | >800 | 179.20 (25.0–591.3) | 0.000 | 243.55 (27.4–800) | 0.000 |
| PCIII (μg/L) | 386.91 (127.5 to >640) | 170.50 (64.7–467.3) | 0.020 | 115.05 (40–280.1) | 0.003 |
| CIV (μg/L) | 194.60 (103.8–312.6) | 84.40 (32.1–127.5) | 0.000 | 86.45 (49.7–217.6) | 0.000 |
| LN (μg/L) | 87.70 (40–170.05) | 73.80 (40–177.1) | 0.344 | 97.60 (40–124.3) | 0.977 |
| LSM (kpa) | 30.80 (14.3–75) | 11.80 (4.1–27.7) | 0.001 | 14.30 (5.3–55.1) | 0.026 |
Progression stage of ALF.
P 1 : P value for comparison between patients with ALF and CHB.
P 2 : P value for comparison between patients with ALF and cirrhosis.
ALF Acute liver failure, CHB chronic hepatitis B, HA hyaluronic acid, PCIII procollagen III, CIV collagen IV, LN Laminin, LSM liver stiffness mesurment.
ALF is accompanied by liver fibrosis
Due to the dramatic clinical course of ALF and the difficulties in obtaining liver samples, the process of fibrosis in ALF remains elucidated. Here, we initially studied whether there is ongoing fibrosis or not during the process of ALF. Firstly, hyaluronic acid (HA), procollagen III (PCIII), collagen IV (CIV) and laminin (LN), which are positively correlated with the histological scores for portal‐periportal activity, were assayed. As shown in Table 1, HA, PCIII and CIV were substantially elevated in patients with ALF when compared with CHB patients and cirrhosis patients(HA level, 800 vs 179.20 μg/L, P = 0.000; PCIII level, 386.91 vs 115.05 μg/L, P = 0.003; CIV level, 194.60 vs 84.40 μg/L, P = 0.000). These findings thus indicate collagen production and tissue fibrogenic in ALF. However, the serum concentration of LN, one of the components of the extracellular matrix, were not different between patients with ALF and those with cirrhosis (87.70 vs 97.60, P = 0.977). Next, liver stiffness were measured by transient elastographay. Consistent with elevated fibrosis markers in ALF, liver stiffness were significantly increased in ALF patients(30.80 vs 14.3 kpa, P = 0.026). Liver stiffness results from a combination of hepatocyte oedema, bilirubin elevation and intrahepatic collagen deposition, liver fibrosis were best detected by biopsy. Then, liver samples of acute liver failure patients were observed under the light microscope. As shown in Fig. 1A–D and Fig. S1A–B, histopathological analysis by H&E, Masson and silver staining revealed that collagen deposition specifically surrounded injured liver tissue. Thus, from the three aspect evidences, fibrosis markers, liver stiffness measurement and liver histology, support the notion that ALF is accompanied by active liver fibrosis.
Figure 1.
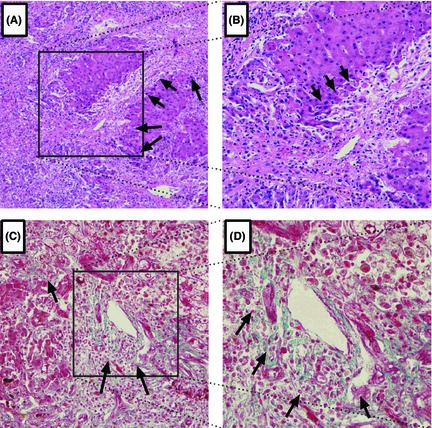
Representative Light Photomicrographs of liver Tissue Showing Collagen Deposition. Original Magnification 100(A, C) or 200(B, D). Histopathological examination of the liver explants by H&E (A, B), and Masson(C, D) staining revealed collagen deposition, as highlighted by arrowhead.
HSCs activation during ALF
Hepatic Stellate Cells is the key cellular source of extracellular matrix synthesis in the liver. Following liver tissue damage, HSCs activated from quiescent towards myofibroblast‐like cells, which are characterized by an increased proliferation and enhanced synthesis of extracellular matrix components. Consistent with increased collagen deposition (Fig. 1A–D and Fig. S1A–B), immunohistochemistry staining for α‐SMA, a marker for HSCs activation, revealed abundant HSC activation (Fig. 2A, B).
Figure 2.
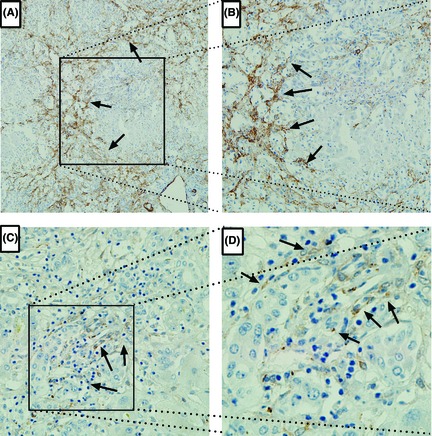
Immunohistochemical α‐smooth muscle actin (α‐SMA)(A, B) and LC3‐II (C, D)staining were conducted in liver tissue from patients with acute liver failure to show hepatic stellate cell (HSC) activation and autophagy induction, as highlighted by arrowhead. Original Magnification 100(A, C) or 200(B, D).
HMGB1 increased in ALF progression stage
Despite the extensive studies on the mechanism of fibrosis in chronic liver injury, mechanism of fibrosis underlying ALF remains elucidated. HMGB1 was originally discovered as a nuclear binding protein, has been involved in pulmonary fibrosis 17, 27. In an effort to elucidate the involvement of HMGB1 in ALF and fibrosis, the concentrations of serum HMGB1 of 17 ALF patients in progression stage and remission stage, sex‐ and gender‐ matched patients with cirrhosis and HBsAg carrier (Table S1) were assayed. As shown in Fig. S2A–B, we observed an increased tendency of serum HMGB1 level in patient with cirrhosis than HBsAg carrier(1.84 ± 0.66 ng/mL vs 1.58 ± 0.52 ng/ml, P = 0.187), although the statistical difference is not significant. Next, an elevated HMGB1 level were observed in progression stage of ALF when compared with HBsAg carrier (2.23 ± 0.74 ng/ml vs 1.58 ± 0.52 ng/ml, P = 0.004), indicating the involvement of HMGB1 in the pathophysiology of ALF. The close temporal correlation of elevated HMGB1 level and liver fibrosis in ALF, suggests that the two events may be linked. Interestingly, in remission stage, HMGB1 level returned to baseline that is comparable to HBsAg carrier, indicating HMGB1 functions in progression stage, but not in remission stage.
HMGB1 activates HSCs
Based on our data that ongoing liver fibrosis and HSCs activation in ALF (Fig. 1, Fig. S1 and Fig. 2A, B) and the close temporal correlation of HMGB1 level with the severity of ALF(Fig. S2A–B), we examined if HSCs could be activated by HMGB1. TGF‐beta1 is known to be a potent inducer of cell proliferation, cell differentiation and fibrogenesis. First, we compared the potent of inducing HSCs activation between HMGB1 and TGF‐beta1, in primary rat HSCs and HSC‐T6 cell line. As shown in Fig. 3A–B, both HMGB1 and TGF‐beta1 alone induced a moderate up‐regulation of α‐SMA and collagen‐I protein expression in primary rat HSCs (Fig. 3A, C left) as well as rat hepatic stellate cell line HSC‐T6 cells(Figrue 3B, 3C right). Costimulation with rHMGB1 and TGF‐beta1 further enhanced α‐SMA and collagen‐I levels (Fig. 3A–B), especially in primary rat HSCs, showing an additive effect. Consistently with increased protein level, after 6 h incubation with HMGB1, the mRNA levels of HSCs activation markers like α‐SMA (Acta2) and collagen 1a1 (Co1a1) are increased 6.83‐fold and 4.38‐fold, respectively, showing that HSCs undergo an activation process(Fig. 3D). Thus, treatment with rHMGB1 increased α‐SMA and collagen 1α1 both at transcript level and protein level, not only in HSC‐T6 cell line but also in primary rat HSCs.
Figure 3.
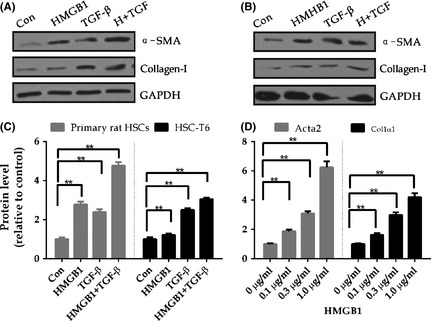
High Mobility Group Box 1 activates HSCs. (A) Primary rat HSCs on day 4 or (B)HSC‐T6 were treated with 1 μg/ml HMGB1, TGF‐beta alone or both as indicated for 24 hours. Western blotting analysis of α‐SMA and Collagen‐1 with specific antibody as indicated, with GAPDH as a loading control. Images presented are representative of three independent experiments. Protein α‐SMA ratios (normalized to GAPDH) were used to quantify fold change relative to control and are shown(C). (D)Quantitative real‐time reverse PCR analysis transcript level of α‐SMA(Acta2) and procollagen 1a1, normalize to housekeeping gene Gapdh in rat hepatic stellate cells treated with indicated concentration of HMGB1 for 6 hours. Data represented the mean value of three experiments, error bars indicate SME, *, P < 0.05,**, P < 0.01.
HMGB1 induces autophagy in HSCs
Recently, autophagy has been implicated in HSCs activation and fibrosis 18. However, it remains unclear whether autophagy play a role in the setting of ALF. Firstly, immunohistochemistry staining for LC3‐II, a well‐defined marker for autophagy, revealed abundant autophagy induction (Fig. 2C–D). Next, to elucidate the effect of HMGB1 on autophagy in HSCs, primary rat HSCs were treated with HMGB1 for indicated period of time and dosage, autophagic activity were assessed. The results shown treatment with HMGB1 induced an increase in LC3 lipidation in time‐ (Fig. S3A–B) and dose‐ (Fig. 4A–B) dependent manner, suggesting HMGB1 is potent of induce autophagy in HSCs. However, LC3‐II itself also a substrate of autophagy, can be degraded by lysosomes. The increase in LC3‐II simply indicates the accumulation of autophagosomes, which may come from enhanced autophagic flux or inhibited autophagic degradation. Effects on autophagy are best assessed by clamping LC3‐II degradation with the lysosomal proton pump inhibitor bafilomycin A1 (BA), a well‐established method for monitoring autophagosome synthesis. If the amount of LC3‐II further accumulates in the presence of BA, this would indicate enhancement of the autophagic flux. If the LC3‐II level were to remain unchanged, it is likely that autophagosome accumulation occurred due to inhibition of autophagic degradation.
Figure 4.
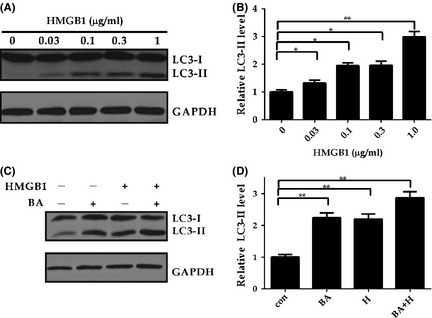
High Mobility Group Box 1 induces autophagy. (A‐B) HMGB1 induces autophagy in a dose dependent manner. Primary rat HSCs on day 4 were treated with different concentration of HMGB1 (0.03–1 μg/ml) for 24 h (A) as indicated, followed by evaluation of autophagy by using immunoblotting with specific antibodies. Protein ratios of LC3‐II(normalized to GAPDH) were used to quantify fold change relative to control and are shown(B), *, P < 0.05,**, P < 0.01. (C–D) HMGB1 induces autophagy flux. Primary rat HSCs on day 4 were incubated with 1 μg/ml HMGB1 for 20 h, followed by 100 nM bafilomycin‐A1 (BA) for 4 hours. Extracts were subjected to autophagy activity and HSCs activation analysis by using immunoblotting with specific antibodies. Protein ratios of LC3‐II(normalized to GAPDH) were used to quantify fold change relative to control and are shown(B), *, P < 0.05,**, P < 0.01.
To investigate whether autophagic flux is induced by HMGB1, primary rat HSCs were incubated with HMGB1 in the presence or absence of BA. This analysis displayed a significantly increased LC3‐II accumulation following HMGB1 treatment in the presence of BA when compared with HMGB1 treatment alone (Fig. 4C–D), indicating increased autophagic flux, but not decreased LC3‐II degradation, accounting for LC3‐II accumulation. To observe the dynamic characteristic of autophagy during HSC activation, GFP‐LC3 plasmid was transfected into HSC‐T6 and then treated with HMGB1. Similarity with increased LC3 lipidation, GFP‐LC3 puncta, reflecting autophagic structures, were strikingly increased in HSC treated with HMGB1 than untreated control (Fig. S3A–B). Thus, we conclude that HMGB1 activates autophagy in HSCs.
Discussion
With the dramatic clinical syndrome, study of liver fibrosis in acute liver failure have been limited. The availability of multiple specimens of explanted livers tissue and serum from patients developed HBV‐associated ALF gave us a unique opportunity to study liver fibrosis in ALF. First, we confirmed the ongoing fibrosis and HSC activation in progression stage of ALF. Secondly, serum HMGB1 assayed showed strikingly increased level in progression stage of ALF. The close temporal correlation of liver fibrosis and increased HMGB1 level, suggests that the two events may be linked. It is well‐established that HSCs activation plays a central role in liver fibrosis. Our in vitro data demonstrated that HMGB1 activate HSCs. Moreover, we found, for the first time, that HMGB1 induce autophagy during HSC activation, indicating that HMGB1 promote HSC activation via autophagy induction. Our results integrate HMGB1, HSCs activation, autophagy into a common framework that underlies the molecular mechanism of fibrosis in ALF.
Fibrosis is an intrinsic response to injury, maintaining organ integrity when extensive necrosis or apoptosis occurs. Such an understanding suggests that fibrosis is not a vicious process per se. For myocardial infarction, collagen fibres are morphologically evident at the end of first week, which maintain heart structure and preventing heart from rupture 28. In kidney, following acute ischaemic/reperfusion injury, interstitial collagen deposition was evident 29. In patients with H5N1 influenza, it was demonstrated the presence of interstitial fibrosis 30. However, little is known about fibrosis in ALF. In the present study, we found collagen deposition, elevated fibrosis markers as well as increased LSM. More important, histopathology detection demonstrated a remarkable collagen deposition. Those findings clearly demonstrated ongoing fibrogenesis in progression stage of ALF.
Recently, raised the notion that fibrosis in response to ALF is not an injurious process. Short‐term production of collagen and the occurrence of fibrosis in response to ALF may be physiological and possible beneficial response to the liver 5, and HSCs is dispensable for liver regeneration and remoulding. 6 Indeed, blockade of fibrosis by depleting activated HSCs in APAP induced mouse ALF model demonstrated a significantly severe liver damage and declined survival rate. 7 In animal model, mutation in collagen‐I results in failure of recovery from CCl4‐induced liver fibrosis and diminished hepatocyte regeneration 8. Because of the rarity and severity of this disease, the in vivo fibrosis in ALF remain largely unknown.
Differ from chronic fibrosis, which can obliterate the architecture and function of the underlying organ or tissue 3, fibrosis in ALF may be reflecting a reaction response to maintain liver architecture and avoiding collapse, and supply collagen for hepatocyte proliferation. Activated hepatic stellate cells are primary extracellular matrix–producing (ECM‐producing) cells in liver. Hepatic stellate cells are located in the space of Disse between the sinusoidal endothelial cells and hepatic epithelial cells, and account for 5–8% of the cells in the liver. In a healthy liver, stellate cells are quiescent and contain numerous vitamin A lipid droplets, constituting the largest reservoir of vitamin A in the body. When the liver is injured due to viral infection or hepatic toxins, hepatic stellate cells receive signals secreted by damaged hepatocytes and immune cells, causing them to transdifferentiate into activated myofibroblast‐like cells 6. Our in vivo study showed HSCs activation in the liver tissue and HMGB1 elevation in serum in patients with ALF. Moreover, our in vitro study clearly showed that HSC can be activated by HMGB1, as indicated by striking increase in α‐SMA and collagen 1a1 at transcript and protein level in rat primary HSCs as well as HSC‐T6 cell line incubated with rHMGB1. Those finding indicate that the increase in HMGB1 may be responsible for the HSC activation and collagen deposition in ALF. During ALF, HMGB1 primarily released from macrophage and breakage of hepatocytes, the activation of HSC by HMGB1 represents a crosstalk between parenchyma cell and non‐parenchyma cell.
Recently, autophagy has been implicated in HSC activation 18, 31, 32, 33. To further explore the mechanism of HSC activation induced by HMGB1, we examined if HMGB1 induce autophagy in HSC. Firstly, we found an elevated serum HMGB1 level and induced autophagy by LC3‐II staining in explanted livers tissue. Next, our in vitro study clearly showed that rHMGB induced an increase in LC3 lipidation in time‐ and dose‐dependent manner in primary rat HSCs. Consistently, autophagic flux also increased when treated with rHMGB1. Moreover, GFP‐LC3 puncta also significantly increased by dynamic observation in rHMGB1 treated HSC‐T6 cell. Several previous investigations have suggested that autophagy provides energy that is essential to support HSC activation. Mitochondrial uncouplers inhibit hepatic stellate cell activation. Basal autophagy is present in all cell types and is rapidly up‐regulated as an adaptive response under conditions of cellular stress as a means to generate intracellular nutrients and energy. In normal liver, lysosomal activity is reported to be very low. Here, we found autophagy was induced during ALF. More important, we identified autophagy is triggered by HMGB1 released from hepatocytes and macrophage trigger during ALF. To our knowledge, this is the first time to detect autophagy during HMGB1 induced HSC activation.
Our study identified ongoing fibrosis in progression stage of acute liver failure, increased serum HMGB1 activates HSCs via autophagy. The findings integrate HMGB1, HSCs activation, autophagy into a common framework for understanding the regulation of stellate cell activation and thus fibrosis in ALF.
Supporting information
Fig. S1. Representative Light Photomicrographs of liver Tissue Showing Collagen Deposition. Original Magnification 100(A) or 200 (B).
Fig. S2. Serum HMGB1 level in patients with ALF.
Fig. S3. HMGB1 induces autophagy.
Table S1 Demographic data and clinical characteristics of controls* and ALF patients.
Acknowledgements
Funding: The Fundamental Research Funds for the Central Universities, National 11th (China, No. 2008ZX10002‐007) and 12th (No.2012ZX10002007) Five‐Year Special Grand Project for Infectious Diseases; National Nature and Science Foundation of China (30700712).
Conflict of interest: The authors do not have any disclosures to report.
Liver Int. 2015; 35: 1877–1885
Handling Editor: Frank Tacke
References
- 1. Wu ZG, Han MF, Chen T, Yan WM, Ning Q. Acute liver failure: mechanisms of immune‐mediated liver injury. Liver International 2010; 30: 782–94. [DOI] [PubMed] [Google Scholar]
- 2. Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41: 1179–97. [DOI] [PubMed] [Google Scholar]
- 3. Su TH, Kao JH, Liu CJ. Molecular mechanism and treatment of viral hepatitis‐related liver fibrosis. Int J Mol Sci 2014; 15: 10578–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Comprehensive Physiology 2013; 3: 1473–92. [DOI] [PubMed] [Google Scholar]
- 5. Dechene A, Sowa JP, Gieseler RK, et al Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology 2010; 52: 1008–16. [DOI] [PubMed] [Google Scholar]
- 6. Yin CY, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. Journal of Clinical Investigation 2013; 123: 1902–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen KT, Chang WJ, Gao XD, et al Depletion of activated hepatic stellate cell correlates with severe liver damage and abnormal liver regeneration in acetaminophen‐induced liver injury. Acta Biochim Biophys Sin 2011; 43: 307–15. [DOI] [PubMed] [Google Scholar]
- 8. Issa R, Zhou XY, Trim N, et al Mutation in collagen‐I that confers resistance to the action of collagenase results in failure of recovery from CCl4‐induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. Faseb Journal 2002; 16: 47–9. [DOI] [PubMed] [Google Scholar]
- 9. Bianchi ME, Scaffidi P, Misteli T. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–5. [DOI] [PubMed] [Google Scholar]
- 10. Tracey KJ, Lotze MT. High‐mobility group box 1 protein (HMGB): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005; 5: 331–42. [DOI] [PubMed] [Google Scholar]
- 11. Adebayo D, Mookerjee RP, Jalan R. Mechanistic biomarkers in acute liver injury: are we there yet? J Hepatol 2012; 56: 1003–5. [DOI] [PubMed] [Google Scholar]
- 12. Bechmann LP, Jochum C, Kocabayoglu P, et al Cytokeratin 18‐based modification of the MELD score improves prediction of spontaneous survival after acute liver injury. J Hepatol 2010; 53: 639–47. [DOI] [PubMed] [Google Scholar]
- 13. Tracey KJ, Yang H, Ochani M, et al Reversing established sepsis with antagonists of endogenous high‐mobility group box 1. Proc Natl Acad Sci USA 2004; 101: 296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lowenstein PR, Curtin JF, Liu NY, et al HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 2009; 6: 83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laskin DL, Dragomir AC, Laskin JD. Macrophage activation by factors released from acetaminophen‐injured hepatocytes: potential role of HMGB1. Toxicol Appl Pharmacol 2011; 253: 170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang H, Nace GW, Mcdonald KA, et al Hepatocyte‐specific high‐mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high‐mobility group box 1 in cellular protection. Hepatology 2014; 59: 1984–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamada N, Maeyama T, Kawaguchi T, et al The role of high mobility group box1 in pulmonary fibrosis. Am J Respir Cell Mol Biol 2008; 39: 440–7. [DOI] [PubMed] [Google Scholar]
- 18. Van De Graaf S, Beuers U. Autophagy ‐ another piece of the puzzle towards understanding primary biliary cirrhosis? Liver International 2014; 34: 481–3. [DOI] [PubMed] [Google Scholar]
- 19. Hernandez‐Gea V, Friedman SL. Autophagy fuels tissue fibrogenesis. Autophagy 2012; 8: 849–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thoen LF, Guimaraes EL, Grunsven LA. Autophagy: a new player in hepatic stellate cell activation. Autophagy 2012; 8: 126–8. [DOI] [PubMed] [Google Scholar]
- 21. Hernandez‐Gea V, Ghiassi‐Nejad Z, Rozenfeld R, et al Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012; 142: 938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moles A, Tarrats N, Fernandez‐Checa JC, Mari M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology 2009; 49: 1297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang R, Livesey KM, Zeh HJ 3rd, Lotze MT, Tang D. Metabolic regulation by HMGB1‐mediated autophagy and mitophagy. Autophagy 2011; 7: 1256–8. [DOI] [PubMed] [Google Scholar]
- 24. Chen T, He Y, Liu X, et al Nucleoside analogues improve the short‐term and long‐term prognosis of patients with hepatitis B virus‐related acute‐on‐chronic liver failure. Clinical and Experimental Medicine 2012; 12: 159–64. [DOI] [PubMed] [Google Scholar]
- 25. Tandon B, Bernauau J, O'grady J, et al Recommendations of the International Association for the Study of the Liver Subcommittee on nomenclature of acute and subacute liver failure. J Gastroenterol Hepatol 1999; 14: 403–4. [DOI] [PubMed] [Google Scholar]
- 26. Wang X, Tang X, Gong X, et al Regulation of hepatic stellate cell activation and growth by transcription factor myocyte enhancer factor 2. Gastroenterology 2004; 127: 1174–88. [DOI] [PubMed] [Google Scholar]
- 27. Albayrak A, Uyanik MH, Cerrah S, et al Is HMGB1 a new indirect marker for revealing fibrosis in chronic hepatitis and a new therapeutic target in treatment? Viral Immunol 2010; 23: 633–8. [DOI] [PubMed] [Google Scholar]
- 28. Wang J, Huang WC, Xu RX, et al MicroRNA‐24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med 2012; 16: 2150–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gobe GC, Bennett NC, West M, et al Increased progression to kidney fibrosis after erythropoietin is used as a treatment for acute kidney injury. Am J Physiol Renal Physiol 2014; 306: F681–92. [DOI] [PubMed] [Google Scholar]
- 30. Qiao J, Zhang MJ, Bi JM, et al Pulmonary fibrosis induced by H5N1 viral infection in mice. Respir Res 2009; 10: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cardinal J, Pan P, Tsung A. Protective role of cisplatin in ischemic liver injury through induction of autophagy. Autophagy 2009; 5: 1211–2. [DOI] [PubMed] [Google Scholar]
- 32. Rautou PE, Mansouri A, Lebrec D, et al Autophagy in liver diseases. J Hepatol 2010; 53: 1123–34. [DOI] [PubMed] [Google Scholar]
- 33. Ni HM, Williams JA, Yang H, et al Targeting autophagy for the treatment of liver diseases. Pharmacological Research: The Official Journal of the Italian Pharmacological Society 2012; 66: 463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative Light Photomicrographs of liver Tissue Showing Collagen Deposition. Original Magnification 100(A) or 200 (B).
Fig. S2. Serum HMGB1 level in patients with ALF.
Fig. S3. HMGB1 induces autophagy.
Table S1 Demographic data and clinical characteristics of controls* and ALF patients.