

Abstract
Background
Recently, it was shown that a knock‐out (KO) of the polycomb histone methyltransferase Ezh2 leads to cardiac hypertrophy in mice, which was driven by the homeodomain transcription factor Six1. Here, we analyzed the expression of Six1 and its regulating factor Ezh2 in cardiac tissue of patients with end‐stage dilative cardiomyopathy (DCM).
Methods
Tissue samples of patients with end‐stage DCM (n = 35) were compared with controls (n = 12) for the protein expression of Ezh1, Ezh2, Six1, and a marker of protein expression p70S6K.
Results
Contrary to the Ezh2‐KO mouse model, we found a down‐regulation of Six1 (26%) and an up‐regulation of Ezh2 (76%) in DCM hearts, (both P < 0.05). Expression of Ezh2 and Six1 did not correlate in human tissue (DCM: r 2: 0.03, P = 0.31 and donor: r 2: 0.05, P = 0.45). Expression of Six1 weakly correlated with left ventricular end‐systolic diameter and fractional shortening. In DCM, Six1 also showed a positive correlation to the expression of the ribosomal protein p70S6K (r: 0.39, P = 0.029), which is involved in protein synthesis. This correlation was not seen in donor tissue, which showed a trend for a negative correlation (r: −0.49, P = 0.08).
Conclusion
Our data indicate that the Ezh2/Six1 axis might be involved in human DCM. However, Six1 expression may be regulated by factors other than Ezh2, and more research is needed to determine the precise role of Ezh2/Six1 in human DCM.
Keywords: DCM, Cardiomyopathy, Signalling
Introduction
Recently, a novel insight in a possible contribution of epigenetic factors towards a pathophysiological remodelling of the heart has been described by Delgado‐Olguín and colleagues.1 They showed that the knock‐out of the polycomb histone methyltransferase Ezh2 leads to cardiac hypertrophy in mice, which was effectively driven by the homeodomain transcription factor Six1.1 Six1 is normally active during embryogenesis and plays an important role for normal cardiovascular development.2 It is repressed by Ezh2's modification of histones in the postnatal heart. Non‐physiological remodelling of the heart, i.e. pathological hypertrophy as described by Delgado‐Olguín et al. is likely to progress to dilative cardiomyopathy (DCM) in humans. In human DCM, a myosin heavy chain (MHC) isoform switch from alpha to beta MHC is observed; the latter is normally repressed during postnatal development.3, 4 This switch to a foetal contractile protein gene program is directly associated with the DCM phenotype and involves multiple signalling pathways, including modification of chromatin structure by regulation of histone deacetylases.5 Interestingly, Six1 over‐expression in the slow fiber‐type soleus muscle has been associated with the induction of glycolytic, fast fibre‐type muscular proteins.6 Therefore, the Ezh2/Six1 axis may be of relevance in development of human DCM.
Here, we assessed the protein expression of Ezh2 and the related Ezh1, which have been shown to be expressed in cardiomyocytes,7 as well as Six1 in left ventricular (LV) tissue samples of patients with end‐stage DCM in relation to the expression in healthy controls.
Methods
Human tissue
Samples of human LV tissue were taken immediately after explantation of the heart of patients with end‐stage DCM (n = 35) and stored at −80°C until further use. Tissue used in this study was taken from a slice in the area of the papillary muscle in order to ensure that all samples were taken from similar regions. Samples of rejected donor hearts (control, n = 12) were obtained and stored the same way as soon as the heart had been found unsuitable for transplantation by expert cardiac surgeons. All surgeries were performed 2000/2001. All procedures have been approved by the local ethics committee.
Western blotting
Approximately 50 mg of pulverized tissue was put into 500 μL lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Trition‐X100, 2.5 mM Na4P2O7, 1% phosphatase inhibitor (P2850 Sigma), 20 mM NaF, 1 mM DTT, 1 mM Na3VO4, 1 mM β‐glycerophosphate). Electrophoresis was performed using a 12% separating gel (12% polyacrylamid, 0.38 M Tris pH 8.8, 0.1% SDS, 0.1% APS, 0.04% TEMED).
For protein transfer, a standard BioRad Trans‐Blot® semi‐dry electrophoretic transfer cell was used overnight at 5 V. The protein was transferred from the gel to PVDF membrane.
The membrane was blocked with 5% BSA/Tris‐buffered saline with Tween 20 (TBST) buffer, washed with TBST three times and incubated with protein‐specific primary antibodies, Ezh1 (SAB2100737, Sigma), Ezh2 (AV38470, Sigma), Six1 (SAB2102157, Sigma), p70S6K (9202, Cell Signalling), and p‐p70S6K (9205, Cell Signalling) (diluted in 5% BSA/TBST buffer, 1:1000) overnight. The following day, the antibody was removed and membrane was washed three times with TBST. Membranes were incubated with appropriate secondary antibodies (diluted in 5% BSA/TBST buffer, 1:1000) at room temperature for 1 h. Membranes were washed three times in TBST.
For visualization, the membranes were incubated with chemiluminescent signal CDP‐star®‐assay buffer (0.2 mM CDP‐star®, 20 mM Tris (pH 9.8), 1 mM MgCl2, H2O) for 5 min. Signal was detected using chemiluminescent films (Amersham Hyperfilm ECL, GE Healthcare), developed by an automatic autoradiography processor (Siemens, AGFA Curix 60). The density of bands was analysed with ImageJ (Image Processing and Analysis in Java). Results were normalized to comparison glyceraldehyde‐3‐phosphate dehydrogenase (G9545, Sigma). All expression data are given in % values compared with the mean of the donor group for the relevant proteins.
Statistics
Data were analyzed using GraphPad PRISM 5.0 (GraphPad Software, Inc, La Jolla, CA, USA). Results are shown as mean ± SEM. Data were tested for normal distribution using the Kolmogorov Smirnov test. Data with normal distribution were analyzed by Student's t‐test, while data without normal distribution were analyzed by Mann Whitney U‐test. Correlation analysis was performed using Spearman's signed rank test. A P‐value <0.05 was considered significant.
Results
Recipients had a mean age of 44.9 ± 2.1 years, mean body weight of 74.2 ± 3.1 kg, and a mean BMI of 24.3 ± 0.7. Four recipients were female and 31 male. Donors mean age was 44.7 ± 3.2 years, mean body weight of 78.1 ± 2.1 kg, and a mean BMI of 25.4 ± 0.7. Five donors were female, and 7 were male. Data on cardiac function and medication were only available for patients with DCM (Table 1).
Table 1.
Patient's characteristics, cardiac function, and medication for patients and donors with DCM
| DCM | Donor | |
|---|---|---|
| n [male/female] | 35 [31/4] | 12 [7/5] |
| Age [y] | 44.9 ± 2.1 | 44.7 ± 3.2 |
| Weight [kg] | 74.2 ± 3.1 | 78.1 ± 2.1 |
| BMI [kg/m2] | 24.3 ± 0.7 | 25.4 ± 0.7 |
| LVEF [%] | 18.5 ± 1.7 | NA |
| LVFS [%] | 10.5 ± 1.4 | NA |
| LVEDD [mm] | 72.4 ± 2.3 | NA |
| Heart rate [bpm] | 81 ± 4 | NA |
| Beta‐blocker [n] | 23 | NA |
| ACE inhibitor [n] | 19 | NA |
| Aldosterone antagonist [n] | 20 | NA |
| Diuretics [n] | 32 | NA |
| Digitalis [n] | 28 | NA |
| Anticoagulant [n] | 28 | NA |
NA, data not available.
Contrary to the findings of Delgado‐Olguín et al., we found a 76% up‐regulation of the Ezh2 protein expression (Figure 1 B) and a down‐regulation of Six1 (Figure 1 C) by 26% in cardiac tissue of patients with end‐stage DCM compared with donor tissue. Ezh1 expression was down‐regulated by 17% vs donors (Figure 1 A; for protein bands refer to Figure 1 D). While the absolute data suggest a direct regulation of Six1 expression by Ezh2, no correlation between the two proteins was seen in either group, DCM: r 2 = 0.03, P = 0.31 and donor: r 2 = 0.05, P = 0.45 (Figure 1 E). Moreover, no correlation was seen between Ezh1/Ezh2 (DCM: r 2 = 0.11, P = 0.19 and donor: r 2 = 0.04, P = 0.57) or Ezh1/Six1 (DCM: r 2 = 0.04, P = 0.44 and donor: r 2 = 0.16, P = 0.43; data not shown).
Figure 1.
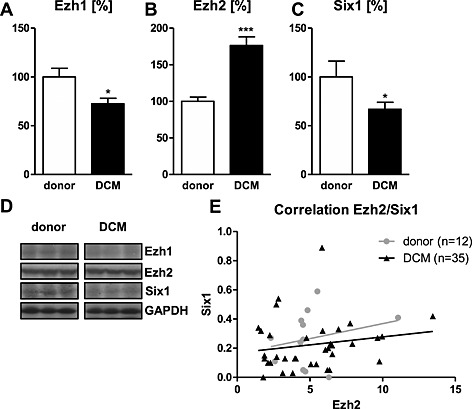
A down‐regulation of Ezh1 (A) and Six1 (C) protein levels was seen in left ventricular tissue samples of patients with end‐stage dilated cardiomyopathy (DCM) compared with control. Expression of Ezh2 was up‐regulated compared with donor tissue (B). Data are shown in percent compared with the mean value of donors. (D) Representative western blot bands for the respective targets. No correlation was found between the expression of Ezh2 and Six1 (E) in either donor or DCM group. *P < 0.05, ***P < 0.001 vs donor.
Interestingly, expression of Six1 correlated with a lower LV end‐systolic diameter (r 2: 0.21, P = 0.038; Figure 2A) but not with the LV end‐diastolic diameter in patients with end‐stage DCM (r 2: 0.02, P = 0.24; Figure 2B). LV fractional shortening was higher in patients with a higher Six1 expression (r: 0.43, P = 0.043; Figure 2C), while LV ejection fraction showed no correlation (r: 0.10, P = 0.58; Figure 2D).
Figure 2.

Six1 showed a negative correlation with left ventricular (LV) end‐systolic diameter (ESD; A) and LV fractional shortening (FS; C), while LV end‐diastolic diameter (EDD; B) and LV ejection fraction (EF; D) were independent of Six1 expression. No data on cardiac dimension and function were available on donor hearts.
Overall expression and also phosphorylation (activation) of the S6 ribosomal protein p70S6K was down‐regulated in the DCM group by 29% (P = 0.0029) and 22% (P = 0.04), respectively (Figure 3A, B). However, the ratio of activated and total p70S6K was not changed between groups (P > 0.3; Figure 3C; for protein bands refer to Figure 1D). In DCM, a higher expression of Six1 correlates with a higher expression of p70S6K (r: 0.39, P = 0.029; Figure 3 E), while a trend for a negative association was observed in donor hearts (r: −0.49, P = 0.08; Figure 3 E). Phosphorylation of p70S6K showed no correlation in either group (DCM: r: 0.11, P = 0.57; donor: r: 0.19, P = 0.53, data not shown).
Figure 3.
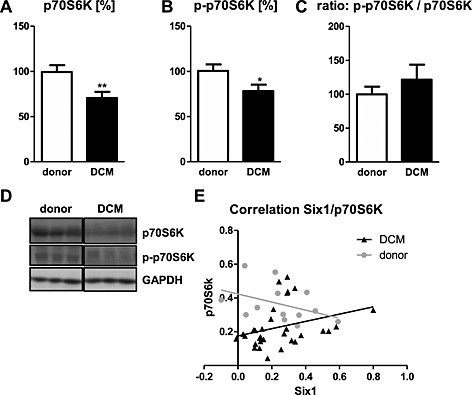
The overall expression level (A) and the phosphorylation (B) of the ribosomal protein p70S6K were lower in DCM compared with donors. Data are shown in percent compared with the mean value of donors. (D) Representative western blot bands for the respective targets. (E) In DCM, Six1 showed a positive correlation to p70S6K expression, while donors showed a trend for a negative correlation. *P < 0.05, **P < 0.01 vs donor.
Discussion
The main finding of this study is a down‐regulation of Six1 expression in the heart of patients with DCM compared with samples obtained from controls, while Ezh2 shows the opposite regulation. However, the expression of these two proteins seems to be unrelated in human tissue. This contradicts a recent report that showed an up‐regulation of Six1 by a knock‐out of Ezh2 in a mouse model.1
Delgado‐Olguín et al. reported that a knock‐out of Ezh2 induced hypertrophy by deregulation of Six1, but they did not look at a later stage of pathological remoulding that would be similar to the end‐stage DCM tissue used in our study. Hence, one possible explanation is the spatial difference between the two studies, in which we may have observed an endogenous (over‐) compensation of Ezh2 expression after or during the transition from the earlier cardiac hypertrophy to dilatation. A differential and spatial expression of beta‐actin isoforms in cardiac hypertrophy and subsequent decompensation has been described.8 A fibre‐type switch associated with chronic heart failure is also observed in skeletal muscle, which may be influenced by drug treatment, e.g. beta‐blockers.9 Therefore, a further reason for the differences to the mouse study may be that interventional drug treatment of the patients with DCM may have had direct or indirect effects on the expression Ezh2 and Six1. A direct, e.g. aldosterone antagonism and reduction of fibrosis, and/or indirect regulation, e.g. reduction of fibrosis by diuretics as a result of reduced volume load, of numerous proteins involved in ventricular remodelling in heart failure by chronic heart failure medication has been described.10 However, in experimental heart failure, a positive effect of carvedilol on remodelling was shown but without affecting the foetal gene re‐expression.11
In the present study, a lower expression of the ribosomal protein p70S6K was seen in the DCM group. When phosphorylated, p70S6K induces protein synthesis at the ribosome.12 While the level of phosphorylation was not different between groups, the overall lower expression of p70S6K suggests a reduced protein synthesis in DCM, as elevated p70S6K expression has been shown to be related to hypertrophy of cardiomyocytes.13 The positive correlation of Six1 and p70S6K in the DCM group of our study may point towards a role of Six1 in the control of translational machinery associated with the ribosome. Interestingly, in healthy cardiac tissue, a trend for a negative correlation of Six1 and p70S6K was seen suggesting that Six1 mainly plays a role in the heart under pathophysiological conditions. Patients with DCM with higher Six1 had a somewhat better preserved contractile function as LVFS as well as LVESD correlated with Six1. This may be because of its role in protein synthesis and may reflect different stages of DCM.
In conclusion, the Ezh2/Six1 axis can be considered as an interesting concept of how epigenetic factors may contribute to the development of DCM, and while the mouse data are certainly quite compelling, human samples show a contrary regulation of Ezh2 and Sixt1, which also lack a correlated expression in our experiments. Therefore, more work is needed to clarify the precise role of Ezh2/Six1 in human cardiomyopathy.
Declaration of interest
None declared.
Tschirner A., Palus S., Hetzer R., Meyer R., Anker S. D., and Springer J. (2014) Six1 is down‐regulated in end‐stage human dilated cardiomyopathy independently of Ezh2, ESC Heart Failure, 1, 154–159, doi: 10.1002/ehf2.12017.
References
- 1. Delgado‐Olguin P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet 2012;44: 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A, Li X. A Tbx1‐Six1/Eya1‐Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Invest 2011;121: 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yazaki Y, Tsuchimochi H, Kurabayashi M, Kawana M, Kimata S. Distribution of cardiac myosin isozymes in cardiomyopathy: immunohistochemical and gene analysis. Jpn Circ J 1987;51: 676–681. [DOI] [PubMed] [Google Scholar]
- 4. LeWinter MM. Functional consequences of sarcomeric protein abnormalities in failing myocardium. Heart Fail Rev 2005;10: 249–257. [DOI] [PubMed] [Google Scholar]
- 5. Calalb MB, McKinsey TA, Newkirk S, Huynh K, Sucharov CC, Bristow MR. Increased phosphorylation‐dependent nuclear export of class II histone deacetylases in failing human heart. Clin Transl Sci 2009;2: 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grifone R, Laclef C, Spitz F, Lopez S, Demignon J, Guidotti JE, Kawakami K, Xu PX, Kelly R, Petrof BJ, Daegelen D, Concordet JP, Maire P. Six1 and Eya1 expression can reprogram adult muscle from the slow‐twitch phenotype into the fast‐twitch phenotype. Mol Cell Biol 2004;24: 6253–6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008;32: 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berni R, Savi M, Bocchi L, Delucchi F, Musso E, Chaponnier C, Gabbiani G, Clement S, Stilli D. Modulation of actin isoform expression before the transition from experimental compensated pressure‐overload cardiac hypertrophy to decompensation. Am J Physiol Heart Circ Physiol 2009;296: H1625–H1632. [DOI] [PubMed] [Google Scholar]
- 9. Dalla Libera L, Ravara B, Gobbo V, Betto DD, Germinario E, Angelini A, Evangelista S, Vescovo G. Skeletal muscle proteins oxidation in chronic right heart failure in rats: can different beta‐blockers prevent it to the same degree? Int J Cardiol 2010;143: 192–199. [DOI] [PubMed] [Google Scholar]
- 10. Barry SP, Townsend PA. What causes a broken heart‐‐molecular insights into heart failure. Int Rev Cell Mol Biol 2010;284:113–179. [DOI] [PubMed] [Google Scholar]
- 11. Li B, Liao YH, Cheng X, Ge H, Guo H, Wang M. Effects of carvedilol on cardiac cytokines expression and remodeling in rat with acute myocardial infarction. Int J Cardiol 2006;111:247–255. [DOI] [PubMed] [Google Scholar]
- 12. Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin‐FKBP specifically blocks growth‐dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992;69:1227–1236. [DOI] [PubMed] [Google Scholar]
- 13. Koda M, Takemura G, Okada H, Kanoh M, Maruyama R, Esaki M, Li Y, Miyata S, Kanamori H, Li L, Ogino A, Kondo T, Minatoguchi S, Fujiwara T, Fujiwara H. Nuclear hypertrophy reflects increased biosynthetic activities in myocytes of human hypertrophic hearts. Circ J 2006;70: 710–718. [DOI] [PubMed] [Google Scholar]