

Abstract
Background and purpose
Tauroursodeoxycholic acid (TUDCA) is a hydrophilic bile acid that is produced in the liver and used for treatment of chronic cholestatic liver diseases. Experimental studies suggest that TUDCA may have cytoprotective and anti‐apoptotic action, with potential neuroprotective activity. A proof of principle approach was adopted to provide preliminary data regarding the efficacy and tolerability of TUDCA in a series of patients with amyotrophic lateral sclerosis (ALS).
Methods
As a proof of principle, using a double‐blind placebo controlled design, 34 ALS patients under treatment with riluzole who were randomized to placebo or TUDCA (1 g twice daily for 54 weeks) were evaluated after a lead‐in period of 3 months. The patients were examined every 6 weeks. The primary outcome was the proportion of responders [those subjects with improvement of at least 15% in the Amyotrophic Lateral Sclerosis Functional Rating Scale Revised (ALSFRS‐R) slope during the treatment period compared to the lead‐in phase]. Secondary outcomes included between‐treatment comparison of ALSFRS‐R at study end, comparison of the linear regression slopes for ALSFFRS‐R mean scores and the occurrence of adverse events.
Results
Tauroursodeoxycholic acid was well tolerated; there were no between‐group differences for adverse events. The proportion of responders was higher under TUDCA (87%) than under placebo (P = 0.021; 43%). At study end baseline‐adjusted ALSFRS‐R was significantly higher (P = 0.007) in TUDCA than in placebo groups. Comparison of the slopes of regression analysis showed slower progression in the TUDCA than in the placebo group (P < 0.01).
Conclusions
This pilot study provides preliminary clinical data indicating that TUDCA is safe and may be effective in ALS.
Keywords: amyotrophic lateral sclerosis, cholic acids, tauroursodeoxycholic acid
Short abstract
Click here to view the accompanying paper in this issue.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease characterized by impairment of upper and lower motor neurons 1. Degeneration of lower motor neurons leads to progressive muscular atrophy. A number of complex biochemical and regulatory pathways are probably involved in the pathogenesis of ALS. These different pathways interact with each other, eventually leading to selective cell death or apoptosis 2. In addition, recent evidence suggests a role for mitochondrial dysfunction 3.
The development of a disease‐modifying therapy that may reverse the progression of disability is a high priority in ALS research. Thus far, only riluzole has shown a modest disease‐modifying efficacy, as it prolonged survival by 3 months after 18 months of treatment 4. Unfortunately, the benefit is modest leading to an increase in survival of perhaps 10%–20% 5.
Tauroursodeoxycholic acid (TUDCA) is a hydrophilic bile acid that is normally produced endogenously in humans in the liver, by conjugation of taurine to ursodeoxycholic acid (UDCA). It is commonly used for treatment of chronic cholestatic liver diseases and for gallstone 6. TUDCA possesses many additional ancillary features, including the inhibition of mitochondrial‐associated apoptosis through different mechanisms 7. Results of experimental studies suggest that the cytoprotective and anti‐apoptotic action of TUDCA may be responsible for potential neuroprotective activity for a variety of chronic neurodegenerative conditions 8.
Ursodeoxycholic acid and TUDCA have been shown to strongly inhibit apoptosis in different types of cells, by either stabilizing the mitochondrial membrane or modulating the expression of specific upstream targets of apoptosis 9. It has recently been shown that, in a cellular model of superoxide dismutase 1 neurodegeneration, glycine‐conjugated UDCA inhibits nitrite production and prevents matrix metallopeptidase 9 activation 10. These recent data have high relevance for human ALS.
In this scenario, TUDCA may be a potential therapeutic candidate in ALS, due to its multiple mechanisms of cytoprotective actions which may include anti‐apoptotic, immunomodulatory and antioxidant effects.
This study was performed on early‐stage non‐severely disabled patients whose disease progression could be monitored. It was designed as a proof of principle 11, i.e. with the aims of collecting cost‐efficient data regarding the tolerability of TUDCA in patients with ALS and providing indications whether the potential neuroprotective effects of TUDCA in animal models might be replicated in human individuals with ALS.
It was intended as a pilot study in order to obtain preliminary information on the possible therapeutic efficacy of TUDCA as an add‐on treatment in ALS patients taking riluzole. A lead‐in trial design as suggested by a consensus on designing clinical trials in ALS 12 was used. In order to evaluate a homogeneous group of patients only spinal‐onset ALS patients with <18 months' disease duration were included.
Methods
This study was designed to collect preliminary safety and efficacy data regarding the long‐term biological effects of TUDCA in patients with treated ALS. It was an investigator‐led, charity‐funded project without commercial sponsorship and was a phase II, multicenter (three Italian centers), randomized (1:1), double‐blind, placebo controlled, parallel group study. The licensed product of TUDCA consists of 250 mg of tauroursodeoxycholic acid dihydrate; the placebo was excipient lactose. Both treatment groups received identical tablets containing either 250 mg of TUDCA or 250 mg of placebo. Both TUDCA and placebo were kindly provided by Bruschettini Srl (Genoa, Italy).
The duration of study treatment was 54 weeks with either placebo or TUDCA (1 g twice daily by mouth), preceded by a 12‐week lead‐in period with riluzole only (50 mg twice daily). A parallel group, randomized, controlled design was chosen, with follow‐up over a 54‐week period. The patient group was chosen to represent typical spinal ALS forms; eligible patients were aged 18–75 years and had clinically probable or definite ALS disease, as defined by the revised El Escorial diagnostic criteria 13, with spinal onset and a disease duration of <18 months at study entry.
Patients were required to have (i) a forced vital capacity (FVC) ≥75% of the predicted one, (ii) a steady treatment regimen with riluzole for at least 3 months according to current European guidelines 14 and (iii) evidence of disease progression over the last 3 months assessed by worsening of self‐reported disability scales and Amyotrophic Lateral Sclerosis Functional Rating Scale Revised (ALSFRS‐R) scores 15. The following patients were excluded: those submitted to tracheostomy or to gallbladder resection, those with electromyographic evidence of motor or sensory nerve conduction blocks, with dementia, active peptic ulcer or active malignancy. Patients with bulbar onset were excluded. Pregnant women and breast‐feeding mothers or women with child bearing potential who did not practise an effective method of birth control were also excluded.
Randomization was independently performed by generating a randomized list that was kept blinded to patients, investigators and statisticians. All patients were followed for the 12‐week lead‐in period, during which they underwent two in‐person visits, to assess medication for concomitant conditions, occurrence of adverse events, compliance to current treatment and level of disability, evaluated by ALSFRS‐R 15. Disease progression at time of enrolment was measured using the ΔFS score 16. At the end of the lead‐in phase, the eligible patients entered a 54‐week randomized treatment period, with in‐person visits every 6 weeks. Blood samples for biological examinations were taken at randomization and during each follow‐up visit. Patients underwent disease progression monitoring with the ALSFRS‐R scale; concurrent clinical conditions, occurrence of adverse events and compliance to treatment were evaluated. Furthermore, at randomization, at week 24 and at the end of the treatment period, the following assessments were performed: neurological examination, respiratory function, FVC, muscle strength, and the Medical Research Council (MRC) Scale. Quality of life was assessed by administering the short form 36 (SF‐36) questionnaire 17. At each visit, safety was assessed by monitoring adverse events, vital signs and laboratory test data for complete blood count and basic chemistry panel, including liver function and creatine kinase serum dosage. Safety was evaluated by the incidence and severity of adverse events and their relationship to treatment which were based on the results of laboratory tests, patients' reports and the investigator's judgments.
The proportion of responder patients in the two treatment groups was the primary outcome measure of the study. Responder patients were defined as those showing an improvement of at least 15% in the ALSFRS‐R slope during the treatment period compared to the lead‐in period. This threshold was chosen based on indications from the ALS consensus committee to ensure that a small, but useful, effect is not missed 12. Other efficacy outcomes included between‐treatment comparison of (i) ALSFRS‐R at study end; (ii) the slopes of the two linear regression analyses of ALSFRS‐R mean scores during the treatment period; (iii) survival time; (iv) FVC at the end of the study; (v) physical component summary and mental component summary scores of SF‐36; and (vi) MRC scores for right and left muscle groups 18. A pre‐specified analysis was performed for the primary end‐point of the study; post hoc analyses were performed in the evaluation of other outcome measures.
Intention‐to‐treat analysis was performed, including all randomized patients receiving at least one dose of the study medication and having at least one primary efficacy assessment after randomization. The last observation carried forward imputation method was used to replace missing values at study end for patients prematurely leaving the study. Homogeneity of clinical characteristics and efficacy variables at baseline between the two randomization groups (between‐group baseline differences) was assessed by analysis of variance for continuous variables and by a chi‐squared test for discrete variables. All efficacy end‐points were compared between the two randomization groups at study end (between‐group differences at study end) by means of analysis of covariance for continuous variables, adjusting for baseline value and for center effect, and by a chi‐squared test for discrete variables. Survival time was compared between treatments by a Kaplan–Meier survival analysis. The slopes of the two linear regression equations of ALSFRS‐R scores during the treatment period were compared by linear regression and generalized linear model analysis. The level of statistical significance was kept at 0.05 throughout the study and all the results are reported with two‐sided P values. Data are shown as mean ±SD or as mean and 95% confidence interval (CI) (continuous variables) or as absolute (n) and relative frequency (categorical variables). A two‐group chi‐squared test with a 0.05 two‐sided significance level will have 80% power to detect the difference between a group 1 proportion of responders of 0.30 and a group 2 proportion of responders of 0.80 (odds ratio 9.33) when the sample size in each group is at least 15 subjects included in the study. Statistical analysis was performed using SAS Statistical Analysis System (version 9.2; Cary, NC, USA).
The study was conducted according to good clinical practice guidelines and the Declaration of Helsinki and was approved by the Carlo Besta Institute internal review board (coordinating center, registered at the Office for Human Research Protection as IORG0006168) and by the review boards of the other two centers involved. All eligible patients gave written informed consent to participate in this study. The trial was registered at clinicaltrials.gov as EudraCT No: 2007‐001592‐10/NCT00877604.
Results
A total of 34 patients were randomized, 17 to placebo and 17 to TUDCA treatment. The first patient was enrolled on 8 October 2008. Of the 34 patients, five dropped out before the second visit, i.e. before starting treatment. Four patients withdrew consent and one was lost to follow‐up. The full analysis set included 29 randomized patients, four of whom died during the study period, one in the TUDCA group and three in the placebo group (Fig. 1), due to respiratory failure in all instances. The patients' baseline characteristics are reported in Table 1. Disease progression at time of enrolment was equally distributed between the two groups (ΔFS score of 1.05 for placebo and 1.45 for TUDCA).
Figure 1.
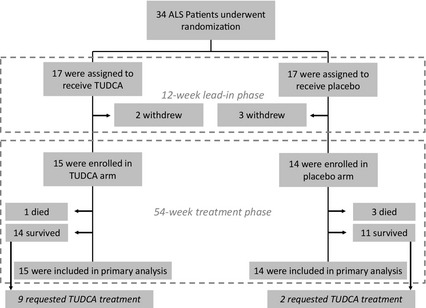
Random assignments to treatment, withdrawals during the lead‐in phase, completion of the trial, and requests for open‐label treatment. The number of patients who died during the treatment phase is also reported.
Table 1.
Demographic and clinical characteristics of the study population at the time of treatment phase. Data are shown as means ± SD or as absolute value (n); P values refer to the statistical significance of the between‐group differences (t test for continuous variables, chi‐squared test for discrete variables)
| Placebo (n = 14) | TUDCA (n = 15) | P value | |
|---|---|---|---|
| Age (years) | 58.2 ± 12.9 | 54.0 ± 12.2 | 0.377 |
| Disease duration (years) | 1.0 ± 0.4 | 1.1 ± 0.7 | 0.814 |
| Gender | |||
| Men (n) | 9 | 10 | 0.893 |
| Women (n) | 5 | 5 | |
| ALSFRS‐R scale | 38.4 ± 6.4 | 38.7 ± 4.9 | 0.887 |
| ΔFS | 1.04 ± 0.6 | 1.45 ± 0.8 | 0.605 |
| FVC (%) | 96.1 ± 7.9 | 94.9 ± 12.2 | 0.735 |
| SF‐36 questionnaire | |||
| PCS | 38.0 ± 7.0 | 39.3 ± 9.9 | 0.695 |
| MCS | 45.4 ± 13.0 | 50.9 ± 11.8 | 0.263 |
| MRC scale | |||
| Right muscle group | 56.9 ± 7.6 | 58.2 ± 3.9 | 0.626 |
| Left muscle group | 54.9 ± 9.0 | 55.6 ± 8.9 | 0.851 |
TUDCA, tauroursodeoxycholic acid; ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale Revised; ΔFS, progression rate calculated as per 16; FVC, forced vital capacity; SF‐36, short form 36; PCS, physical component summary; MCS, mental component summary; MRC, Medical Research Council.
Thirteen patients (87%) were classified as responders in the TUDCA group, whereas six (43%) were responders in the placebo group (P = 0.021). At study end (54 weeks) the baseline‐adjusted ALSFRS‐R score was significantly higher in TUDCA‐treated than in placebo‐treated patients [mean value and 95% CI: 23.3 (19.9–26.6) vs. 16.3 (12.9–19.7); P = 0.007]. Also, the ALSFRS‐R bulbar subscore was significantly higher in TUDCA‐treated than in placebo‐treated patients (Tables 2 and S1). At the end of the study period the ALSFRS‐R mean score in the TUDCA group corresponded to the mean value reached by the placebo group at week 36, i.e. 18 weeks earlier; the slopes of the two regression lines were different (−0.262 for the TUDCA group, −0.388 for the placebo group; P < 0.01) (Fig. 2).
Table 2.
Other secondary outcome measures, data are shown as relative percentages (%) or baseline‐adjusted mean values and 95% confidence intervals; P values refer to the statistical significance of the between‐group differences (54 weeks, anova analysis)
| Placebo (baseline) | Placebo (54 weeks) | TUDCA (baseline) | TUDCA (54 weeks) | P value | |
|---|---|---|---|---|---|
| FVC (%) | 96.1 (90.0–102.2) | 87.7 (80.9–95.3) | 94.9 (86.8–101.4) | 89.1 (81.4–96.7) | 0.778 |
| SF‐36 questionnaire | |||||
| PCS | 38.0 (33.8–42.2) | 35.0 (30.4–39.6) | 39.3 (33.3–45.3) | 34.8 (30.2–39.4) | 0.951 |
| MCS | 45.4 (37.5–53.2) | 42.3 (35.5–49.2) | 50.9 (43.8–58.1) | 49.0 (42.1–55.8) | 0.173 |
| MRC scale | |||||
| Right muscle group | 56.9 (51.8–62.0) | 47.0 (35.6–58.5) | 58.2 (55.6–60.8) | 49.2 (44.9–53.4) | 0.695 |
| Left muscle group | 54.9 (48.8–61.0) | 43.7 (32.9–54.6) | 55.6 (49.6–61.6) | 47.0 (41.6–52.4) | 0.553 |
| ALSFRS‐R bulbar subscore | 11.3 (10.8–11.8) | 1.9 (0.8–2.9) | 10.8 (10.1–11.5) | 3.8 (2.2–5.4) | 0.037 |
| ALSFRS‐R upper limbs subscore | 11.6 (9.6–13.6) | 8.1 (5.9–10.4) | 11.4 (9.7–13.2) | 11.1 (8.4–13.8) | 0.087 |
| ALSFRS‐R lower limbs subscore | 4.4 (3.3–5.4) | 4.9 (3.7–6.1) | 5.1 (3.8–6.4) | 4.8 (3.4–6.2) | 0.947 |
| ALSFRS‐R respiratory subscore | 11.2 (10.4–12.0) | 2.2 (1.2–3.3) | 11.5 (11.0–12.0) | 3.6 (2.1–5.1) | 0.125 |
TUDCA, tauroursodeoxycholic acid; FVC, forced vital capacity; SF‐36, short form 36; PCS, physical component summary; MCS, mental component summary; MRC, Medical Research Council; ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale Revised.
Figure 2.
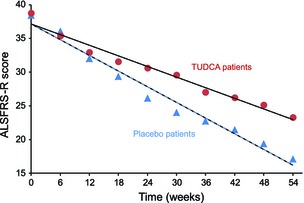
Linear regression analysis of ALSFFRS‐R mean scores over time for the TUDCA (circles, slope −0.388) and placebo groups (triangles, slope −0.262).
The other secondary outcomes did not differ between treatment groups (Table 2). Cumulative incidence of death in the course of the observation period (median follow‐up of 66 weeks) was three deaths in the placebo group and one death in the TUDCA group (NS, P = 0.092, Fig. 3). Accordingly, the average survival time in TUDCA treatment was 65.7 weeks and in the placebo group 61.1 weeks [mean value and 95% CI: 65.7 (65.2–66.3) weeks in TUDCA vs. 61.1 (55.3–66.9) weeks in placebo].
Figure 3.
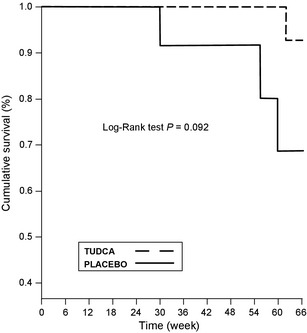
Kaplan–Meier estimates of the probability of survival in the placebo and TUDCA groups (P = 0.092; log‐rank test).
The population for safety analysis consisted of 15 patients who took TUDCA and 14 patients who took placebo. The treatment was well tolerated in all patients. Laboratory parameters did not change in either treatment group during the course of the study. Except for the expected complications related to ALS, no changes in vital signs and laboratory values that could possibly be attributed to the study drug or placebo were recorded. Overall, five adverse events were considered by the investigators to be study related based on the patients' descriptions. Two events were reported in the 15 TUDCA‐treated patients (13.3%); three events occurred in the 14 placebo‐treated patients (21.4%). Mild diarrhea occurred in two patients treated with TUDCA and in two treated with placebo; anorexia was reported in a placebo‐treated patient. Four patients died during the study period, one in the TUDCA group and three in the placebo group.
At the end of the study and before breaking the treatment code, each patient was allowed to decide whether to receive TUDCA on a compassionate basis: 64% of the patients in the treatment group and 20% of those in the placebo group opted for open‐label TUDCA treatment.
Discussion
This trial is the first to report tolerability and pilot data of the effects of TUDCA in patients with a neurodegenerative disease. It was observed by using a double‐blind controlled design that treatment with TUDCA was associated with a slower deterioration of function in ALS patients. The study was performed in a homogeneous population of patients with overlapping phenomenology, the spinal‐onset phenotype, and disease duration at study entry of <18 months.
These findings demonstrate that TUDCA is well tolerated by ALS patients and that clinically relevant differences emerged between the two groups and persisted for the 12‐month exposure to the study drug. Considering that TUDCA has no known symptomatic effect on motor function or muscle strength, this observation may potentially reflect a disease‐modifying effect.
This pilot study was designed to allow the collection of data in a cost‐efficient manner to provide first data of a possible effect of TUDCA in ALS. A proof of principle approach was adopted to provide preliminary data regarding its tolerability and possible biological effects.
The inclusion and exclusion criteria for patient selection were defined on a pragmatic basis to minimize the risk of including patients with advanced ALS, considering that patients with advanced disease would be likely to have fewer salvageable motor neurons.
The design of this trial included a lead‐in period with riluzole. In the absence of a surrogate, assessments made during the lead‐in period provide the best prediction of disease course for each patient and reduce inter‐patient variability 19. This strategy was used to determine a slope indicative of the rate of disease progression. A lead‐in design is more suited to linear efficacy measures, such as the ALSFRS for which the entry level and slope are predictive of survival 15.
The present data are of particular interest in view of the recent observation that glycine‐conjugated UDCA has anti‐apoptotic properties in a cellular model of ALS 10. Taking together pre‐clinical and clinical observations it may be argued that a biological effect of TUDCA deserves to be further explored in patients with ALS and other neurodegenerative diseases.
No important safety concerns emerged from this study; gastrointestinal symptoms are a common side effect of TUDCA, as confirmed in this ALS population, but did not compromise trial participation in any patient. The frequency of adverse events in the TUDCA group was similar to that seen in the previous clinical trials in primary biliary cirrhosis patients 20, 21.
The findings reported here are to be considered preliminary, particularly in view of the limited sample size. In our study there were no significant differences between the two groups at baseline; however, despite randomization, minor differences in the baseline characteristics of the treated and control groups can influence subsequent disease progression. It is also possible that observed results are related to a subgroup of patients. The last observation carried forward imputation method and the high death rate in the treatment phase are other possible contributing factors which might have amplified the transient supplemental effect of TUDCA.
Bearing in mind these limitations, our data provide preliminary information about TUDCA tolerability and efficacy in ALS and allow consideration of the size of the biological effects seen in comparison to previous trials of patients with disease‐modifying medications. Previous trials demonstrated a small, albeit statistically significant, prolongation of survival in participants receiving the intermediate and high dose of riluzole compared to placebo controls 4, 22. In these studies, there was a small positive benefit on limb function 23, with a per‐year rate of decline of the Norris scale of about 4 points (on a 0–63 score) in the riluzole group compared to placebo. In this study functional assessment was performed using the ALSFRS‐R, which measures the physical functional status with strong internal consistency and construct validity, and is a good predictor of survival time 24. The per‐year decline rate of ALSDRS‐R was found to be about 7 points smaller (on a 0–48 score) in the TUDCA group compared to placebo.
At variance with the primary outcome, no between‐group differences were observed in FVC, MRC scale or in the ALSFRS‐R limbs and respiratory subscores. This could be due to the limited sample size of the study that it is not powered enough to detect differences in secondary outcome measures. A lesser decline in lung function was observed in TUDCA‐treated patients, indicating a possible positive impact of treatment on respiratory involvement, since a progressive reduction of FVC is associated with poor prognosis 25. TUDCA treatment was also associated with a less pronounced deterioration in muscle strength and quality of life. Whether these measures will prove significant in larger studies with longer follow‐up is unknown.
These data support planning new double‐blind trials of TUDCA for a potential disease‐modifying effect in ALS, which will incur substantially greater costs than were required for this study. It is arguable that prevention of deterioration is more achievable in the earlier stages of ALS, when a greater number of motor neurons are still viable. This innovative phase 2 study provides the premises for larger phase 3 trials, that should be designed taking into account reasons of disappointment that have led to negative results in recent studies 26.
This study shows that treatment with TUDCA for 1 year at a dose of 2 g daily was associated with a slower deterioration of function in ALS patients. This effect was additional to that of riluzole and almost 90% of patients treated with TUDCA had a ≥15% improvement in the ALSFRS‐R slope. This could represent a clinically significant reduction of function deterioration, as a slowing of the decline in ALSFRS‐R by 15% is estimated to be equal to a prolongation of median survival by 4–5 months 27. At the end of the 54‐week treatment period, patients in the TUDCA group had a mean ALSFRS‐R score corresponding to that of the placebo group at week 36. This suggests that a 1‐year TUDCA treatment may slow ALS deterioration by 18 weeks and leads us to suppose that a longer duration of treatment may produce an even more accentuated between‐group divergence.
Our findings are consistent with an earlier observation hinting at a potential neuroprotective efficacy of biliary acids in ALS. In a recent publication, an oral soluble UDCA formula was tested for 3 months in ALS patients with a cross‐over, randomized, placebo controlled design 28. A relative slowing of the rate of progression was observed in the treatment group compared to placebo. These short‐term data are in keeping with the present longer‐term results using TUDCA, the taurine conjugated derivative of UDCA. This is in line with pre‐clinical studies demonstrating that, both in vitro and in vivo, TUDCA benefits neurodegenerative disorders 29, 30.
Disclosure of conflicts of interest
The authors declare no financial or other conflicts of interest.
Trial registration
The trial was registered at clinicaltrials.gov as NCT00877604.
Supporting information
Table S1. Individual ALSFRS‐R subscores for each patient. Bulbar, upper limb, lower limb and respiratory scores are reported at baseline and at the end of the study.
Acknowledgements
This study was supported by an unrestricted educational grant from Bruschettini Srl, who did not play any role in the study design and conduct.
See editorial by A. C. Ludolph on page 11.
References
- 1. Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol 1998; 24: 104–117. [DOI] [PubMed] [Google Scholar]
- 2. Turner MR, Bowser R, Bruijn L, et al Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2013; 14(Suppl. 1): 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cozzolino M, Ferri A, Valle C, Carri MT. Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci 2013; 55: 44–49. [DOI] [PubMed] [Google Scholar]
- 4. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose‐ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996; 347: 1425–1431. [DOI] [PubMed] [Google Scholar]
- 5. Sreedharan J, Brown RH Jr. Amyotrophic lateral sclerosis: problems and prospects. Ann Neurol 2013; 74: 309–316. [DOI] [PubMed] [Google Scholar]
- 6. Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 1999; 159: 2647–2658. [DOI] [PubMed] [Google Scholar]
- 7. Amaral JD, Xavier JM, Steer CJ, Rodrigues CM. The role of p53 in apoptosis. Discov Med 2010; 9: 145–152. [PubMed] [Google Scholar]
- 8. Amaral JD, Viana RJ, Ramalho RM, Steer CJ, Rodrigues CM. Bile acids: regulation of apoptosis by ursodeoxycholic acid. J Lipid Res 2009; 50: 1721–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodrigues CM, Steer CJ. The therapeutic effects of ursodeoxycholic acid as an anti‐apoptotic agent. Expert Opin Investig Drugs 2001; 10: 1243–1253. [DOI] [PubMed] [Google Scholar]
- 10. Vaz AR, Cunha C, Gomes C, Schmucki N, Barbosa M, Brites D. Glycoursodeoxycholic acid reduces matrix metalloproteinase‐9 and caspase‐9 activation in a cellular model of superoxide dismutase‐1 neurodegeneration. Mol Neurobiol 2014; PMID: 24848512 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 11. Schmidt B. Proof of principle studies. Epilepsy Res 2006; 68: 48–52. [DOI] [PubMed] [Google Scholar]
- 12. Leigh PN, Swash M, Iwasaki Y, et al Amyotrophic lateral sclerosis: a consensus viewpoint on designing and implementing a clinical trial. Amyotroph Lateral Scler Other Motor Neuron Disord 2004; 5: 84–98. [DOI] [PubMed] [Google Scholar]
- 13. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 293–299. [DOI] [PubMed] [Google Scholar]
- 14. Andersen PM, Abrahams S, Borasio GD, et al EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS) – revised report of an EFNS task force. Eur J Neurol 2012; 19: 360–375. [DOI] [PubMed] [Google Scholar]
- 15. Gordon PH, Miller RG, Moore DH. ALSFRS‐R. Amyotroph Lateral Scler Other Motor Neuron Disord 2004; 5(Suppl. 1): 90–93. [DOI] [PubMed] [Google Scholar]
- 16. Kimura F, Fujimura C, Ishida S, et al Progression rate of ALSFRS‐R at time of diagnosis predicts survival time in ALS. Neurology 2006; 66: 265–267. [DOI] [PubMed] [Google Scholar]
- 17. Jenkinson C, Hobart J, Chandola T, Fitzpatrick R, Peto V, Swash M. Use of the short form health survey (SF‐36) in patients with amyotrophic lateral sclerosis: tests of data quality, score reliability, response rate and scaling assumptions. J Neurol 2002; 249: 178–183. [DOI] [PubMed] [Google Scholar]
- 18. Winhammar JM, Rowe DB, Henderson RD, Kiernan MC. Assessment of disease progression in motor neuron disease. Lancet Neurol 2005; 4: 229–238. [DOI] [PubMed] [Google Scholar]
- 19. Magnus T, Beck M, Giess R, Puls I, Naumann M, Toyka KV. Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve 2002; 25: 709–714. [DOI] [PubMed] [Google Scholar]
- 20. Crosignani A, Battezzati PM, Setchell KD, et al Tauroursodeoxycholic acid for treatment of primary biliary cirrhosis. A dose−response study. Dig Dis Sci 1996; 41: 809–815. [DOI] [PubMed] [Google Scholar]
- 21. Larghi A, Crosignani A, Battezzati PM, et al Ursodeoxycholic and tauro‐ursodeoxycholic acids for the treatment of primary biliary cirrhosis: a pilot crossover study. Aliment Pharmacol Ther 1997; 11: 409–414. [DOI] [PubMed] [Google Scholar]
- 22. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994; 330: 585–591. [DOI] [PubMed] [Google Scholar]
- 23. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 2012; 3: CD001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cedarbaum JM, Stambler N, Malta E, et al The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 1999; 169: 13–21. [DOI] [PubMed] [Google Scholar]
- 25. Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry 2006; 77: 390–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 2014; 13: 1127–1138. [DOI] [PubMed] [Google Scholar]
- 27. Miller RG, Moore DH, Jackson CE. Western ALS Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord 2004; 5(Suppl. 1): 121–124. [DOI] [PubMed] [Google Scholar]
- 28. Min JH, Hong YH, Sung JJ, Kim SM, Lee JB, Lee KW. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: a randomized cross‐over trial. J Korean Med Sci 2012; 27: 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramalho RM, Viana RJ, Low WC, Steer CJ, Rodrigues CM. Bile acids and apoptosis modulation: an emerging role in experimental Alzheimer's disease. Trends Mol Med 2008; 14: 54–62. [DOI] [PubMed] [Google Scholar]
- 30. Shacka JJ, Roth KA. Regulation of neuronal cell death and neurodegeneration by members of the Bcl‐2 family: therapeutic implications. Curr Drug Targets CNS Neurol Disord 2005; 4: 25–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Individual ALSFRS‐R subscores for each patient. Bulbar, upper limb, lower limb and respiratory scores are reported at baseline and at the end of the study.