

Abstract
Objective
Tofacitinib is an oral JAK inhibitor for the treatment of rheumatoid arthritis (RA). Systemic inflammation is proposed to play a fundamental role in the altered lipid metabolism associated with RA; however, the underlying mechanisms are unknown. We undertook this study to compare cholesterol and lipoprotein kinetics in patients with active RA with those in matched healthy volunteers.
Methods
This was a phase I open‐label mechanism‐of‐action study. Cholesterol and lipoprotein kinetics were assessed with 13C‐cholesterol and 13C‐leucine infusions. RA patients were reevaluated after receiving oral tofacitinib 10 mg twice daily for 6 weeks.
Results
Levels of high‐density lipoprotein (HDL) cholesterol, low‐density lipoprotein (LDL) cholesterol, total cholesterol, and apolipoprotein A‐I (Apo A‐I) as well as HDL cholesterol particle number were lower in RA patients (n = 36) than in healthy volunteers (n = 33). In contrast, the cholesterol ester fractional catabolic rate was higher in RA patients, but no differences were observed in cholesterol ester transfer protein, cholesterol ester production rate, HDL‐associated Apo A‐I fractional catabolic rate, or LDL‐associated Apo B fractional catabolic rate. Following tofacitinib treatment in RA patients, the cholesterol ester fractional catabolic rate decreased and cholesterol levels increased. The decrease in cholesterol ester fractional catabolic rate correlated significantly with the increase in HDL cholesterol. Additionally, HDL cholesterol particle number increased and markers of HDL cholesterol function improved.
Conclusion
This is the first study to assess cholesterol and lipoprotein kinetics in patients with active RA and matched healthy volunteers. The data suggest that low cholesterol levels in patients with active RA may be driven by increases in cholesterol ester catabolism. Tofacitinib treatment reduced cholesterol ester catabolism, thereby increasing cholesterol levels toward those in healthy volunteers, and markers of antiatherogenic HDL function improved.
Patients with rheumatoid arthritis (RA) have significantly increased cardiovascular (CV) morbidity and mortality compared with the general population 1. Paradoxically, active RA is associated with reduced levels of low‐density lipoprotein (LDL) cholesterol and total cholesterol 2. High‐density lipoprotein (HDL) cholesterol composition in patients with active RA is also altered and is associated with changes in the level and function of several HDL‐associated proteins, contributing to the formation of more pro‐oxidant, proatherogenic HDL cholesterol particles 3, 4, 5. Treatment of active RA can significantly modify lipid metabolism in patients with RA, including increases in circulating cholesterol levels 2, 6, 7. Systemic inflammation has been proposed to play a fundamental mechanistic role in this altered lipid metabolism, but no mechanistic data in humans have elucidated the potential pathways underlying this critical observation. This issue comprises a fundamental paradox in understanding lipid metabolism in the context of inflammatory diseases.
Tofacitinib is an oral JAK inhibitor which modulates intracellular signaling of multiple key cytokine receptors involved in the inflammatory cascade 8. In patients with RA, tofacitinib treatment, either as monotherapy or in combination with nonbiologic disease‐modifying antirheumatic drugs (DMARDs), resulted in significant and clinically meaningful reductions in the signs and symptoms of RA, improvements in physical function and patient‐reported outcomes, and preservation of structure, with a consistent safety profile across studies 9, 10, 11, 12, 13. During the tofacitinib clinical development program, a proportion of tofacitinib‐treated patients exhibited increases in mean HDL cholesterol, LDL cholesterol, total cholesterol, apolipoprotein A‐I (Apo A‐I), and Apo B levels, mostly occurring within the first 4–6 weeks of treatment 9, 10, 11, 12, 13. The mechanism and significance of these changes are currently unknown.
Cholesterol movement or flux through the reverse cholesterol transport pathway is critical for maintaining cellular cholesterol homeostasis, not only in the context of atherosclerotic lesion prevention, but also for the prevention of toxic levels of cholesterol in every cell. The central pathway of reverse cholesterol transport begins with the transfer of free cholesterol and phospholipid to a lipid‐poor pre–β‐HDL particle which consists principally of Apo A‐I and phospholipid (Figure 1, step 1). The free cholesterol component of the pre–β‐HDL particle is then esterified to cholesterol ester by lecithin:cholesterol acyltransferase (LCAT), generating a mature HDL particle. From this mature HDL particle, cholesterol ester may be transferred to LDL via cholesterol ester transfer protein (CETP) (Figure 1, step 2), where it can be delivered to the liver via LDL receptor uptake. Alternatively, cholesterol ester can be delivered directly to the liver from HDL through selective cholesterol ester uptake via the CD36 and LIMPII analogous‐1 receptor (also known as scavenger receptor class B type I), regenerating a lipid‐poor Apo A‐I–containing particle. Once delivered to the liver, cholesterol can leave the body via biliary secretion (Figure 1, step 3) 14, 15, 16.
Figure 1.
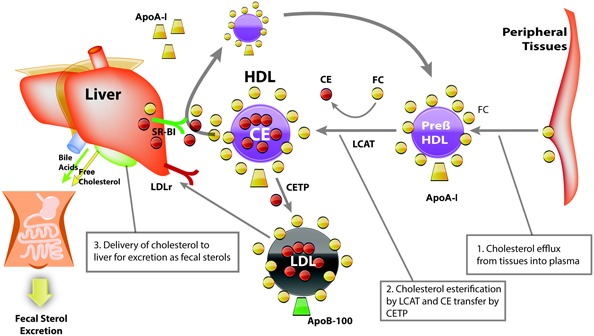
Diagram of reverse cholesterol transport. Reverse cholesterol transport starts with the transfer of free cholesterol (FC) and phospholipid to a lipid‐poor pre–β high‐density lipoprotein (pre–β‐HDL) particle. Esterification of free cholesterol to cholesterol ester (CE) by lecithin:cholesterol acyltransferase (LCAT) then generates a mature HDL particle. From this mature HDL particle, cholesterol ester may be transferred to low‐density lipoprotein (LDL) via cholesterol ester transfer protein (CETP) and then delivered to the liver via the LDL receptor (LDLr). Alternatively, selective cholesterol ester uptake via scavenger receptor class B type I (SR‐BI) can deliver cholesterol ester directly to the liver from HDL, regenerating a lipid‐poor apolipoprotein (Apo) A‐I–containing particle. Once delivered to the liver, cholesterol can leave the body via biliary secretion.
The current study used stable isotope tracers to model in vivo cholesterol and lipoprotein kinetics to evaluate potential mechanisms for the altered lipid metabolism in patients with active RA compared with healthy volunteers 16. Additionally, this study examined the effects of inflammation suppression with tofacitinib treatment on lipid metabolism parameters, as well as exploratory markers of HDL function and particle size.
SUBJECTS AND METHODS
Participants
Eligible participants were age ≥18 years with a body mass index <35 kg/m2. Lipid‐regulating agents, hormonal forms of contraception, isotretinoin, and supplements containing plant sterols/stanols or cholestin were not permitted. Participants were excluded from the study if they had triglycerides >4.52 mmoles/liter after an overnight fast or total cholesterol >3.62 mmoles/liter after an overnight fast.
Patients with RA had to meet the 1987 revised classification criteria of the American College of Rheumatology 17. Other key inclusion criteria included active RA defined by ≥4 tender/painful joints and ≥4 swollen joints and either an erythrocyte sedimentation rate (Westergren method) >28 mm/hour or a C‐reactive protein level >66.67 nmoles/liter; in addition, there had to be no evidence of active, latent, or inadequately treated Mycobacterium tuberculosis infection. Key exclusion criteria included hemoglobin <100.00 gm/liter, hematocrit <30%, white blood cell count <3.0 × 109/liter, absolute neutrophil count <1.2 × 109/liter, or platelet count <100 × 109/liter; glomerular filtration rate ≤40 ml/minute (Cockcroft‐Gault calculation [18]); and aspartate or alanine aminotransferase >1.5 times the upper limit of normal.
Use of concomitant intravenous or intramuscular corticosteroids was not permitted. Low‐dose oral corticosteroids (≤10 mg/day prednisone or equivalent) were permitted. Patients were not required to be DMARD naive; however, all biologic and nonbiologic DMARDs except methotrexate (MTX) had to have been discontinued for a predefined washout period. Patients were permitted to continue stable (≥6 weeks prior to the start of the study) background MTX (7.5–25 mg weekly) therapy. If prior MTX was discontinued, it had to have been discontinued for 4 weeks prior to the start of the study. Healthy volunteers were matched 1:1 with each patient in the RA cohort for race, age (±5 years), sex, and menopausal status (females).
Study design
This phase I open‐label mechanism‐of‐action study was conducted at 7 centers in the US and 2 centers in Hungary. The study was conducted in compliance with the Declaration of Helsinki, the International Conference on Harmonisation Guidelines for Good Clinical Practice, and local country regulations. The final protocol, any amendments, and informed consent documentation were reviewed and approved by the institutional review boards and the independent ethics committees of the investigational centers. All participants provided written informed consent.
Baseline blood samples were obtained from all participants. RA patients then received open‐label oral tofacitinib at 10 mg twice daily for 6 weeks, after which blood samples were obtained again.
The primary objective was to evaluate changes in the HDL cholesterol concentration and the cholesterol ester production rate in RA patients following 6 weeks of tofacitinib at 10 mg twice daily. Secondary objectives included the evaluation of lipid profiles and cholesterol and lipoprotein kinetics both in RA patients before and after tofacitinib treatment and in healthy volunteers. The safety profile of tofacitinib in RA patients was also assessed. Exploratory objectives were to evaluate biomarkers of HDL function and particle size in RA patients before and after tofacitinib treatment as well as to evaluate the relationship between tofacitinib treatment and changes in HDL cholesterol, LDL cholesterol, and triglyceride levels and in cholesterol ester production rate.
Collection and assessments
Lipoprotein and cholesterol concentrations and kinetics
Blood samples were collected after an overnight fast for Apo and lipoprotein cholesterol concentrations at baseline in both groups and in RA patients following tofacitinib treatment. In vivo cholesterol kinetics were assessed using blood samples obtained before and hourly throughout a 22‐hour continuous infusion (200 mg total) of labeled free cholesterol (3,4‐13C2‐cholesterol). In vivo LDL‐associated and HDL‐associated Apo kinetics were assessed using blood samples obtained before and 16, 18, and 20 hours after a 20‐hour primed continuous infusion of U‐13C6‐leucine (1.3 mg/kg body weight priming dose followed by 0.022 mg/kg body weight/minute continuous infusion). Food and calorie‐containing beverages were withheld during both infusion periods.
Blood samples of nonradioactive and isotope‐labeled cholesterol and leucine were analyzed by KineMed. Isotopic enrichments and pool sizes of plasma and red blood cell free cholesterol and cholesterol ester were measured by mass spectrometry and enzymatic colorimetric methods (Wako).
A multicompartment model was used to determine the cholesterol ester fractional catabolic rate, cholesterol ester production rate, and total cholesterol efflux rate using SAAM II software (Simulation, Analysis, and Modeling; SAAM Institute, University of Washington, Seattle). A description of the cholesterol kinetic model is detailed elsewhere 16.
HDL biomarkers
Blood samples were collected after an overnight fast for exploratory biomarkers of HDL function at baseline in both groups and following tofacitinib treatment in RA patients. Biomarkers were analyzed by Pacific Biomarkers using the Pacific Biomarkers biomarker panel. LCAT mass, CETP mass, myeloperoxidase, and total serum amyloid A (SAA) were determined by sandwich enzyme‐linked immunosorbent assay (ELISA). LCAT activity was measured using an enzymatic method with free cholesterol as the substrate. CETP activity was determined by a fluorometric method. HDL‐associated SAA was determined by isolating the HDL cholesterol particles by polyethylene glycol 8000 precipitation followed by SAA ELISA.
Lipoprotein particle number and size
Blood samples obtained after an overnight fast were analyzed by LipoScience using the LipoScience lipid profile panel to evaluate HDL cholesterol and LDL cholesterol particle size and number.
Isotope enrichments and kinetic models
A multicompartment model was used to determine plasma cholesterol ester fractional catabolic rate (%/hour), plasma cholesterol ester production rate (μmoles/kg body weight/hour), and total cholesterol efflux rate (μmoles/kg body weight/hour) using SAAM II software 16. Flux 1 is defined as the loss of free cholesterol from the rapidly exchanging free cholesterol pool in plasma and liver that does not enter into red blood cells or cholesterol ester. Plasma cholesterol esterification fractional catabolic rate (k[0,3]) and production rate (flux 3) were measured from cholesterol ester/free cholesterol enrichment time profiles and plasma cholesterol ester pool sizes. Total cholesterol efflux rate was calculated as the sum of flux 1 and flux 3 and represents the free cholesterol that was mobilized from extrahepatic tissues into the rapid‐turnover free cholesterol pool in plasma and that did not return to extrahepatic tissues during the course of the study. Isotope enrichment was measured in very low‐density lipoprotein (VLDL)–associated Apo B, LDL‐associated Apo B, and HDL‐associated Apo A‐I fractions. LDL‐associated Apo B and HDL‐associated Apo A‐I pool sizes were also measured. LDL‐associated Apo B and HDL‐associated Apo A‐I fractional catabolic rates (%/hour) were calculated from the enrichment in the Apo B and Apo A‐I fractions and using VLDL‐associated Apo B enrichments as a measure of the hepatic leucine precursor pool. LDL‐associated Apo B and HDL‐associated Apo A‐I production rates (mg/kg body weight/minute) were calculated by multiplying the fractional catabolic rates by pool sizes.
Safety
The incidence and severity of all adverse events (AEs) were recorded. Clinical laboratory tests, evaluation of vital signs, and physical examinations were performed at scheduled visits.
Statistical analysis
The full analysis set included all participants (RA patients and healthy volunteers) for whom any measurement of cholesterol ester production rate was available. The safety analysis set consisted of all enrolled participants.
Analyses of the primary end points, as well as other pharmacodynamic and kinetic end points, were based on the full analysis set. The HDL cholesterol concentration following tofacitinib treatment in RA patients was compared with baseline levels using a 1‐sample t‐test. Cholesterol ester production rate was similarly analyzed. The correlation between the change in HDL cholesterol level and the change in cholesterol ester production rate in RA patients was explored using a scatterplot. P values were calculated using Fisher's z‐transformation test of correlation coefficient. Paired t‐tests were used to analyze changes in the lipid profile, cholesterol and lipoprotein kinetics, and exploratory biomarker secondary end points versus baseline following tofacitinib treatment and between matched RA patients and healthy volunteers. No adjustment for multiple comparisons was made.
Given that this was an exploratory study, the sample size of 30 RA patients and 30 healthy volunteers was not determined by formal statistical consideration. The power calculation during protocol development was based on the assumption of a between‐subject coefficient of variation of 18.1% (calculated from a small data set in a different subject population). As initially designed, a sample size of 30 RA patients and 15 healthy volunteers would have a power of 92% to detect a 20% difference at baseline. Owing to larger‐than‐anticipated variations in the first 15 RA patients and 15 healthy volunteers, the number of healthy volunteers was increased to 30; this increased the power from 61% to 79% for the unpaired analysis and from 55% to 86% for the paired analysis. This trial (A3921130) is registered with ClinicalTrials.gov identifier NCT01262118.
RESULTS
Disposition and baseline characteristics of the subjects
The study started on May 9, 2011 and was completed on February 1, 2012. A total of 36 RA patients and 33 healthy volunteers were enrolled, and all completed the study. Patient demographics and baseline characteristics are shown in Table 1. Two healthy volunteers had no cholesterol ester production rate measurements and hence were excluded from the full analysis set. A total of 24 RA patients (66.7%) and 2 healthy volunteers (6.1%) received concomitant nonsteroidal antiinflammatory drugs during the study.
Table 1.
Demographic and baseline clinical characteristics of the participants*
| RA patients (n = 36) | Healthy volunteers (n = 33) | |
|---|---|---|
| Sex | ||
| Male | 6 | 5 |
| Female | 30 | 28 |
| Premenopausal | 10 | 8 |
| Postmenopausal | 20 | 20 |
| Age, years | ||
| 18–44 | 9 | 7 |
| 45–64 | 25 | 25 |
| ≥65 | 2 | 1 |
| Mean ± SD | 51.4 ± 9.2 | 51.3 ± 8.9 |
| Race | ||
| White | 29 | 27 |
| Black | 1 | 0 |
| Other | 6 | 6 |
| BMI, no. (%) | ||
| <25.0 kg/m2 | 13 (36.1) | 14 (42.4) |
| 25.0–29.9 kg/m2 | 14 (38.9) | 13 (39.4) |
| 30.0–39.9 kg/m2 | 9 (25.0) | 6 (18.2) |
| Weight, mean ± SD kg | 70.4 ± 14.2 | 68.3 ± 12.0 |
| Tender joint count, mean ± SD | 25.7 ± 15.8 | NA |
| Swollen joint count, mean ± SD | 14.8 ± 10.6 | NA |
Except where indicated otherwise, values are the number of subjects. RA = rheumatoid arthritis; BMI = body mass index; NA = not applicable.
Lipid and lipoprotein concentrations
At baseline, mean HDL cholesterol, LDL cholesterol, total cholesterol, and Apo A‐I concentrations were lower in RA patients than in matched healthy volunteers (Table 2). No differences in triglycerides or Apo B concentrations were noted between groups (Table 2). Following 6 weeks of tofacitinib therapy in RA patients, HDL cholesterol concentrations increased compared with baseline (P < 0.0001). Significant increases in LDL cholesterol, total cholesterol, Apo B, and Apo A‐I concentrations were also observed (Table 2).
Table 2.
Descriptive summary of lipid and lipoprotein concentrations*
| RA patients | Healthy volunteers (n = 31) | ||
|---|---|---|---|
| Baseline (n = 36) | Week 6 (n = 36) | ||
| HDL concentration, mmoles/liter | 1.41 ± 0.32 | 1.61 ± 0.39 | 1.64 ± 0.44 |
| P | – | <0.0001 | 0.0089 |
| LDL concentration, mmoles/liter | 3.23 ± 0.74 | 3.70 ± 1.01 | 3.75 ± 0.94 |
| P | – | 0.0002 | 0.0180 |
| Total cholesterol concentration, mmoles/liter | 5.02 ± 0.84 | 5.69 ± 1.07 | 5.74 ± 1.09 |
| P | – | <0.0001 | 0.0041 |
| Apo A‐I concentration, gm/litera | 1.17 ± 0.56 | 1.35 ± 0.58 | 1.28 ± 0.68 |
| P | – | 0.0022 | 0.0014 |
| Apo B concentration, gm/litera | 0.81 ± 0.42 | 0.93 ± 0.42 | 0.82 ± 0.48 |
| P | – | 0.0138 | 0.5302 |
| Triglycerides, mmoles/liter | 1.64 ± 0.75 | 1.71 ± 0.87 | 1.54 ± 1.09 |
| P | – | 0.3519 | 0.6998 |
Values are the mean ± SD. All P values are versus rheumatoid arthritis (RA) patients at baseline. HDL = high‐density lipoprotein; LDL = low‐density lipoprotein; Apo A‐I = apolipoprotein A‐I.
Data are from 35 RA patients at baseline and 32 RA patients at week 6.
Cholesterol and lipoprotein kinetics
At baseline, the cholesterol ester fractional catabolic rate was greater in RA patients than in healthy volunteers (P = 0.0085), whereas no difference in the cholesterol ester production rate or cholesterol efflux rate was observed between cohorts. Increased catabolism of cholesterol ester (greater cholesterol ester fractional catabolic rate), without a concomitant increase in the production of cholesterol ester, would be expected to result in lower circulating HDL cholesterol and LDL cholesterol levels, as noted in Table 2.
Following tofacitinib treatment, the cholesterol ester fractional catabolic rate decreased (P = 0.0014 versus baseline) in RA patients, whereas the cholesterol ester production rate and cholesterol efflux rate remained unchanged (Table 3). A significant inverse correlation (P = 0.0180) was noted between the change in cholesterol ester catabolism and the change in HDL cholesterol levels following treatment in a post hoc analysis (Figure 2A): the larger the decrease in cholesterol ester catabolism (cholesterol ester fractional catabolic rate), the larger the increase in circulating HDL cholesterol. Interestingly, an even stronger inverse correlation was observed between the decreases in cholesterol ester fractional catabolic rate and the increases in large HDL cholesterol particle numbers (P = 0.0019) (Figure 2B).
Table 3.
Descriptive summary of cholesterol and lipoprotein kinetics*
| RA patients | Healthy volunteers (n = 31) | ||
|---|---|---|---|
| Baseline (n = 34) | Week 6 (n = 33) | ||
| Cholesterol ester production rate, μmoles/kg/hour | 2.83 ± 0.62 | 2.90 ± 0.62 | 2.88 ± 0.67 |
| P | – | 0.6237 | 0.8879 |
| Cholesterol ester fractional catabolic rate, %/hour | 2.43 ± 0.39 | 2.23 ± 0.31 | 2.17 ± 0.36 |
| P | – | 0.0014 | 0.0085 |
| Cholesterol efflux rate, μmoles/kg/hour | 10.39 ± 2.48 | 10.45 ± 2.79 | 11.25 ± 3.96 |
| P | – | 0.5987 | 0.2542 |
| LDL‐associated Apo B production rate, mg/kg/houra | 0.49 ± 0.12 | 0.52 ± 0.12 | 0.50 ± 0.13 |
| P | – | 0.3604 | 0.9097 |
| LDL‐associated Apo B fractional catabolic rate, %/houra | 1.61 ± 0.37 | 1.57 ± 0.41 | 1.50 ± 0.35 |
| P | – | 0.5680 | 0.2699 |
| HDL‐associated Apo A‐I production rate, mg/kg/houra | 0.57 ± 0.11 | 0.65 ± 0.15 | 0.59 ± 0.16 |
| P | – | 0.0017 | 0.4684 |
| HDL‐associated Apo A‐I fractional catabolic rate, %/houra | 1.08 ± 0.22 | 1.11 ± 0.28 | 1.02 ± 0.22 |
| P | – | 0.3031 | 0.6718 |
Values are the mean ± SD. All P values are versus RA patients at baseline. See Table 2 for definitions.
Data are from 32 RA patients at baseline, 32 RA patients at week 6, and 30 healthy volunteers.
Figure 2.

Correlation of change in cholesterol ester fractional catabolic rate (FCR) with change in HDL cholesterol levels (A) and change in large HDL cholesterol particle levels (B) following treatment. See Figure 1 for other definitions.
At baseline, mean LDL‐associated Apo B and HDL‐associated Apo A‐I production rates and their respective fractional catabolic rates were similar between RA patients and healthy volunteers. Following tofacitinib treatment, only the HDL‐associated Apo A‐I production rate had changed (an increase) (P = 0.0017) versus baseline in RA patients (Table 3).
Exploratory analyses
HDL biomarker findings
Patients with active RA had higher levels of HDL‐associated SAA (P = 0.0084), SAA (P = 0.0079), and myeloperoxidase (P = 0.0221) and lower LCAT activity (P = 0.0302) and mass (P = 0.0013) compared with healthy volunteers (Table 4). CETP activity and mass were similar between cohorts.
Table 4.
Descriptive summary of exploratory biomarkers and particle size*
| RA patients | Healthy volunteers (n = 31) | ||
|---|---|---|---|
| Baseline (n = 36) | Week 6 (n = 36) | ||
| LCAT activity, nmoles/ml/hour | 596.03 ± 138.58 | 642.47 ± 133.57 | 687.81 ± 120.52 |
| P | – | 0.0184 | 0.0302 |
| LCAT mass, μg/liter | 8,200 ± 2,020 | 8,980 ± 2,320 | 9,810 ± 1,540 |
| P | – | 0.0023 | 0.0013 |
| CETP activity, pmoles/ml/minute | 60.41 ± 8.87 | 58.87 ± 7.25 | 58.25 ± 6.58 |
| P | – | 0.2472 | 0.3165 |
| CETP mass, μg/liter | 1,960 ± 520 | 1,970 ± 450 | 1,920 ± 570 |
| P | – | 0.9011 | 0.6687 |
| SAA concentration, mg/liter | 51.93 ± 95.61 | 24.97 ± 48.96 | 3.47 ± 2.71 |
| P | – | 0.0588 | 0.0079 |
| HDL‐associated SAA concentration, mg/liter | 34.76 ± 62.83 | 17.79 ± 34.45 | 2.98 ± 2.33 |
| P | – | 0.0647 | 0.0084 |
| Myeloperoxidase concentration, pmoles/liter | 1,092.00 ± 1,017.05 | 942.86 ± 942.64 | 736.39 ± 344.79 |
| P | – | 0.4755 | 0.0221 |
| HDL particles (total), μmoles/liter | 30.66 ± 4.80 | 33.85 ± 5.51 | 35.00 ± 5.92 |
| P | – | <0.0001 | 0.0020 |
| HDL particle size, nm | 9.10 ± 0.40 | 9.09 ± 0.51 | 9.20 ± 0.39 |
| P | – | 0.8573 | 0.3209 |
| LDL particles (total), nmoles/liter | 1,276.19 ± 391.50 | 1,352.67 ± 498.79 | 1,357.87 ± 540.11 |
| P | – | 0.1382 | 0.3448 |
| LDL particle size, nm | 21.01 ± 0.85 | 21.20 ± 0.97 | 21.36 ± 0.89 |
| P | – | 0.0379 | 0.0639 |
Values are the mean ± SD. All P values are versus RA patients at baseline. LCAT = lecithin:cholesterol acyltransferase; CETP = cholesterol ester transfer protein; SAA = serum amyloid A (see Table 2 for other definitions).
Following tofacitinib therapy, markers of HDL cholesterol function improved versus baseline, including increases in LCAT mass and activity (P = 0.0023 and P = 0.0184, respectively) and decreases in total SAA (P = 0.0588) and HDL‐associated SAA (P = 0.0647). No differences were noted in myeloperoxidase levels or CETP activity/mass (Table 4).
Number and size of lipoprotein particles
The total HDL cholesterol particle number was decreased in RA patients at baseline compared with healthy volunteers and increased (P < 0.0001) following treatment with tofacitinib. HDL cholesterol size, LDL cholesterol particle number, and LDL cholesterol size were not significantly different in RA patients compared with healthy volunteers. LDL cholesterol size increased following tofacitinib treatment (P = 0.0379) (Table 4).
Safety findings
Forty‐four AEs were reported by 19 RA patients, and 6 AEs were reported by 6 healthy volunteers. The most common AEs in RA patients were upper respiratory tract infection (n = 7), headache (n = 4), urinary tract infection (n = 3), abdominal distension (n = 2), and dyspepsia (n = 2); the most common AE in healthy volunteers was headache (n = 4).
There were no serious AEs, permanent discontinuations, or withdrawals due to AEs. Two RA patients discontinued tofacitinib temporarily due to AEs of moderate severity: one had dizziness, headache, and anxiety, and the other had a respiratory tract infection. These AEs resolved before the end of the study.
DISCUSSION
Suppression of circulating cholesterol levels in the setting of active systemic inflammation has long been recognized 2, 4, 19, 20. Hudgins et al demonstrated that endotoxin rapidly and significantly decreased LDL cholesterol and total cholesterol levels in healthy volunteers concomitant with onset of the inflammatory response 20. The suppression of cholesterol levels in patients with active RA has also been increasingly recognized 2, 4, 19. When RA therapy is associated with significant improvement in clinical parameters, it is also often associated with an increase in circulating cholesterol levels 2, 6, 7, 19. However, the fundamental mechanisms driving the increase in circulating cholesterol levels in effectively treated RA patients remain largely unknown.
We used a recently described novel in vivo assay to measure cholesterol and lipoprotein kinetics in patients with active RA and matched healthy volunteers. We also investigated the effects of tofacitinib, a JAK inhibitor used in the treatment of RA, on cholesterol and lipoprotein kinetics. The results reported here suggest for the first time a potential mechanism that may explain the cholesterol changes in patients with active RA through changes in cholesterol ester metabolism. Moreover, the results of the exploratory analyses suggest potential beneficial effects of tofacitinib treatment in RA patients on markers of the antiatherogenic function of HDL.
HDL cholesterol, LDL cholesterol, and total cholesterol levels in patients with active RA at baseline were lower than those in healthy volunteers. Following 6 weeks of tofacitinib treatment in RA patients, these cholesterol levels increased and approached the baseline measures of the healthy volunteers. RA patients also had a higher cholesterol ester fractional catabolic rate compared with healthy volunteers, without any differences in cholesterol ester production rate. Cholesterol ester is carried by mature HDL particles and may be transferred to LDL particles via CETP. Therefore, increased cholesterol ester catabolism, without a concomitant increase in cholesterol ester production, would be expected to result in lower circulating levels of HDL cholesterol and LDL cholesterol, as was observed in patients with active RA in the current study. Following 6 weeks of tofacitinib treatment in RA patients, the cholesterol ester fractional catabolic rate decreased and HDL cholesterol and LDL cholesterol levels increased. A significant inverse correlation was noted between the change in cholesterol ester catabolism and the change in HDL cholesterol levels following treatment: the larger the decrease in cholesterol ester catabolism, the larger the increase in circulating HDL cholesterol. Increased cholesterol ester fractional catabolic rate in the absence of changes in HDL‐associated Apo A‐I or LDL‐associated Apo B fractional catabolic rates may reflect increased selective cholesterol ester uptake by scavenger receptor class B type I in patients with active RA, which could have been normalized by tofacitinib.
Accumulating data suggest that the particle function of HDL may be an important component of the protective capacity of HDL in preventing CV disease 21, 22, 23. Data in the non‐RA population indicate impairment in the antioxidant and efflux functions of HDL in patients with coronary heart disease (CHD) compared with healthy volunteers 22, 23. Khera et al reported that the efflux capacity of HDL was a strong inverse predictor of both coronary artery disease (CAD) and carotid intima‐media thickness in non‐RA patients, even after adjustment for HDL cholesterol levels and traditional CV risk factors 23. Li et al found a similar cross‐sectional association of HDL's efflux capacity with prevalent CAD status; however, they also demonstrated a “paradoxical” association of efflux capacity with future CV events in a prospective study 24. The latter work has raised important questions regarding the HDL “specificity” of a commonly used ex vivo cholesterol efflux assay 24.
In RA patients, HDL function is abnormal and has previously been associated with high levels of systemic inflammation—a known link to CV risk in the RA population 3, 25. HDL normally carries a “cargo” of multiple associated proteins, which may be integral to its antiatherogenic functions 26. In the setting of active RA, this protein cargo changes, including the accumulation of several acute‐phase proteins and the displacement and/or functional inhibition of normally protective proteins and enzymes 3, 5, 26. In the current study, select protein biomarkers related to HDL function were examined in exploratory analyses of patients with active RA (before and after tofacitinib treatment) and healthy volunteers. HDL‐associated SAA levels were higher in patients with active RA than in healthy volunteers and decreased following treatment with tofacitinib. These data are consistent with a prior study by Raterman et al which showed changes in the HDL protein composition from proatherogenic to less atherogenic, with specific decreases in HDL‐associated SAA, in RA patients responding clinically to rituximab therapy 27.
Higher HDL‐associated SAA levels have previously been proposed to contribute to the generation of a proatherogenic HDL particle, in part through displacement of HDL's major protein, Apo A‐I 21, 26, 28. Apo A‐I is required for activation of LCAT—an HDL‐associated enzyme that is integral to HDL's efflux capacity 21, 28. In the current study, LCAT activity and mass were lower at baseline in patients with active RA compared with healthy volunteers and increased following tofacitinib treatment. While the current work did not demonstrate differences in the cholesterol efflux rate between healthy controls and RA patients before and after therapy, it should be noted that the current methodology may be insensitive to detecting changes in efflux mediated by reverse cholesterol transport at the cellular level in cells such as macrophages. In addition, MTX was allowed in the current study, and it has previously been shown to abrogate interferon‐γ–induced down‐regulation of ATP‐binding cassette transporter protein, member 1, in THP‐1 macrophages 29.
Exploratory biomarker analyses demonstrated that the total HDL particle number was lower in patients with active RA than in healthy volunteers and that it increased following tofacitinib therapy. Smaller LDL particles were also noted in patients with active RA compared with healthy volunteers at baseline, and LDL particle size increased significantly after 6 weeks of tofacitinib therapy. Lipoprotein particle size and number have previously been implicated in CV risk across several studies. The presence of small dense LDL particles has been strongly associated with increased CHD risk in the general population, and increased levels of small dense LDL particles have previously been reported in drug‐naive patients with early RA 30. Low concentrations of small HDL particles have also been reported in RA patients and have been linked to increased coronary atherosclerosis 31. Although the current data suggest potentially beneficial CV effects with tofacitinib treatment on LDL and HDL particle size and number, these data must be considered preliminary in nature, with further larger studies warranted.
In summary, this is the first study to assess cholesterol and lipoprotein kinetics in patients with active RA and matched healthy volunteers. Our findings suggest a novel mechanism that may explain the suppression of circulating cholesterol levels in patients with active RA, in part through increases in cholesterol ester catabolism. These data may have wider implications across a range of inflammatory diseases in which lipid metabolic disturbances are reported and in which vascular comorbidity is described. Tofacitinib treatment in RA patients was associated with decreases in cholesterol ester catabolism and increases in cholesterol levels toward the range found in healthy volunteers. Additionally, improvements in markers of antiatherogenic HDL function were observed, as well as potentially beneficial effects on lipoprotein particle size and number. Further investigation of these findings with direct correlation to CV risk in the RA population is warranted.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Charles‐Schoeman had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Turner, Beysen, Milad, Hellerstein, Luo, Kaplan, Riese, Zuckerman, McInnes.
Acquisition of data. Charles‐Schoeman, Fleischmann, Davignon, Schwartz, Turner, Beysen, Milad, Luo, Kaplan, Riese, Zuckerman.
Analysis and interpretation of data. Charles‐Schoeman, Fleischmann, Davignon, Schwartz, Turner, Beysen, Milad, Hellerstein, Luo, Kaplan, Riese, Zuckerman, McInnes.
ROLE OF THE STUDY SPONSOR
This study was supported by Pfizer Inc. Both Pfizer and non‐Pfizer authors participated in the study design, data collection, data analysis, open scientific discussion of the data, interpretation of the data, and development of the associated manuscript. Atrium Research & Consulting LLC were paid contractors to Pfizer in the design, conduct, and data analysis for the study and development of the manuscript. Editorial support was provided by Complete Medical Communications and funded by Pfizer Inc.
ADDITIONAL DISCLOSURES
Authors Turner, Beysen, and Hellerstein are employees of KineMed, Inc. Author Milad is an employee of Atrium Research & Consulting LLC.
Acknowledgments
We thank the study investigators and participants. We also thank Anne Marie Reid, PhD (Complete Medical Communications), for editorial support, which was funded by Pfizer Inc. Finally, we thank John Bradley, Birgitta Benda, and Tamas Koncz (all employees of Pfizer Inc) for providing intellectual input during manuscript development.
ClinicalTrials.gov identifier: NCT01262118.
The views and opinions expressed within the manuscript are those of all authors and do not necessarily represent those of the funding organization.
REFERENCES
- 1. Maradit‐Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, et al.Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population‐based cohort study.Arthritis Rheum 2005;52:402–11. [DOI] [PubMed] [Google Scholar]
- 2. Boers M, Nurmohamed MT, Doelman CJ, Lard LR, Verhoeven AC, Voskuyl AE, et al.Influence of glucocorticoids and disease activity on total and high density lipoprotein cholesterol in patients with rheumatoid arthritis.Ann Rheum Dis 2003;62:842–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Charles‐Schoeman C, Watanabe J, Lee YY, Furst DE, Amjadi S, Elashoff D, et al.Abnormal function of high‐density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis.Arthritis Rheum 2009;60:2870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kitas GD, Gabriel SE.Cardiovascular disease in rheumatoid arthritis: state of the art and future perspectives.Ann Rheum Dis 2011;70:8–14. [DOI] [PubMed] [Google Scholar]
- 5. Watanabe J, Charles‐Schoeman C, Miao Y, Elashoff D, Lee YY, Katselis G, et al.Proteomic profiling following immunoaffinity capture of high‐density lipoprotein: association of acute‐phase proteins and complement factors with proinflammatory high‐density lipoprotein in rheumatoid arthritis.Arthritis Rheum 2012;64:1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Curtis JR, John A, Baser O.Dyslipidemia and changes in lipid profiles associated with rheumatoid arthritis and initiation of anti–tumor necrosis factor therapy.Arthritis Care Res (Hoboken) 2012;64:1282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jamnitski A, Visman IM, Peters MJ, Dijkmans BA, Voskuyl AE, Nurmohamed MT.Beneficial effect of 1‐year etanercept treatment on the lipid profile in responding patients with rheumatoid arthritis: the ETRA study.Ann Rheum Dis 2010;69:1929–33. [DOI] [PubMed] [Google Scholar]
- 8. Meyer DM, Jesson MI, Li X, Elrick MM, Funckes‐Shippy CL, Warner JD, et al.Anti‐inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP‐690,550, in rat adjuvant‐induced arthritis.J Inflamm (Lond) 2010;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burmester GR, Blanco R, Charles‐Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al.Tofacitinib (CP‐690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial.Lancet 2013;381:451–60. [DOI] [PubMed] [Google Scholar]
- 10. Fleischmann R, Kremer J, Cush J, Schulze‐Koops H, Connell CA, Bradley JD, et al.Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis.N Engl J Med 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- 11. Van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al, and the ORAL Scan Investigators .Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve‐month data from a twenty‐four–month phase III randomized radiographic study.Arthritis Rheum 2013;65:559–70. [DOI] [PubMed] [Google Scholar]
- 12. Van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, Garcia Meijide JA, Wagner S, et al, for the ORAL Standard Investigators .Tofacitinib or adalimumab versus placebo in rheumatoid arthritis [published erratum appears in N Engl J Med 2013;369:293].N Engl J Med 2012;367:508–19. [DOI] [PubMed] [Google Scholar]
- 13. Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin‐Mola E, et al.Tofacitinib in combination with nonbiologic DMARDs in patients with active rheumatoid arthritis: a randomized trial.Ann Intern Med 2013;159:253–61. [DOI] [PubMed] [Google Scholar]
- 14. Cohen DE. Lipoprotein metabolism and cholesterol balance In: Arias IM, editor.The liver: biology and pathobiology.Hoboken (NJ):John Wiley & Sons;2009. p.271–85. [Google Scholar]
- 15. Dikkers A, Tietge UJ.Biliary cholesterol secretion: more than a simple ABC.World J Gastroenterol 2010;16:5936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turner S, Voogt J, Davidson M, Glass A, Killion S, Decaris J, et al.Measurement of reverse cholesterol transport pathways in humans: in vivo rates of free cholesterol efflux, esterification, and excretion.J Am Heart Assoc 2012;1:e001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al.The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis.Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 18. Cockcroft DW, Gault MH.Prediction of creatinine clearance from serum creatinine.Nephron 1976;16:31–41. [DOI] [PubMed] [Google Scholar]
- 19. Toms TE, Panoulas VF, Kitas GD.Dyslipidaemia in rheumatological autoimmune diseases.Open Cardiovasc Med J 2011;5:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hudgins LC, Parker TS, Levine DM, Gordon BR, Saal SD, Jiang XC, et al.A single intravenous dose of endotoxin rapidly alters serum lipoproteins and lipid transfer proteins in normal volunteers.J Lipid Res 2003;44:1489–98. [DOI] [PubMed] [Google Scholar]
- 21. Navab M, Berliner JA, Subbanagounder G, Hama S, Lusis AJ, Castellani LW, et al.HDL and the inflammatory response induced by LDL‐derived oxidized phospholipids.Arterioscler Thromb Vasc Biol 2001;21:481–8. [DOI] [PubMed] [Google Scholar]
- 22. Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, et al.Inflammatory/antiinflammatory properties of high‐density lipoprotein distinguish patients from control subjects better than high‐density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment.Circulation 2003;108:2751–6. [DOI] [PubMed] [Google Scholar]
- 23. Khera AV, Cuchel M, de la Llera‐Moya M, Rodrigues A, Burke MF, Jafri K, et al.Cholesterol efflux capacity, high‐density lipoprotein function, and atherosclerosis.N Engl J Med 2011;364:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li XM, Tang WH, Mosior MK, Huang Y, Wu Y, Matter W, et al.Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks.Arterioscler Thromb Vasc Biol 2013;33:1696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maradit‐Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE.Cardiovascular death in rheumatoid arthritis: a population‐based study.Arthritis Rheum 2005;52:722–32. [DOI] [PubMed] [Google Scholar]
- 26. Van der Westhuyzen DR, de Beer FC, Webb NR.HDL cholesterol transport during inflammation.Curr Opin Lipidol 2007;18:147–51. [DOI] [PubMed] [Google Scholar]
- 27. Raterman HG, Levels H, Voskuyl AE, Lems WF, Dijkmans BA, Nurmohamed MT.HDL protein composition alters from proatherogenic into less atherogenic and proinflammatory in rheumatoid arthritis patients responding to rituximab.Ann Rheum Dis 2012;72:560–5. [DOI] [PubMed] [Google Scholar]
- 28. Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, et al.Anti‐inflammatory HDL becomes pro‐inflammatory during the acute phase response: loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures.J Clin Invest 1995;96:2758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reiss AB, Carsons SE, Anwar K, Rao S, Edelman SD, Zhang H, et al.Atheroprotective effects of methotrexate on reverse cholesterol transport proteins and foam cell transformation in human THP‐1 monocyte/macrophages.Arthritis Rheum 2008;58:3675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rizzo M, Spinas GA, Cesur M, Ozbalkan Z, Rini GB, Berneis K.Atherogenic lipoprotein phenotype and LDL size and subclasses in drug‐naive patients with early rheumatoid arthritis.Atherosclerosis 2009;207:502–6. [DOI] [PubMed] [Google Scholar]
- 31. Chung CP, Oeser A, Raggi P, Sokka T, Pincus T, Solus JF, et al.Lipoprotein subclasses determined by nuclear magnetic resonance spectroscopy and coronary atherosclerosis in patients with rheumatoid arthritis.J Rheumatol 2010;37:1633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]