

Abstract
SMT C1100 is a small molecule utrophin modulator in development to treat Duchenne muscular dystrophy. This study evaluated the safety, tolerability, and pharmacokinetics of SMT C1100 in healthy volunteers. This double‐blind, placebo‐controlled Phase 1 study comprised: Part 1, an escalating, single‐dose with/without fasting involving 50 mg/kg, 100 mg/kg, 200 mg/kg, and 400 mg/kg doses; and Part 2, a multiple 10 day dose evaluation involving 100 mg/kg bid and 200 mg/kg bid doses. Adverse events were recorded. SMT C1100 was absorbed rapidly following single and multiple oral doses, with median tmax attained within 2–3.5 hour across all doses. Considerable variability of pharmacokinetic parameters was noted among subjects. Following single doses, systemic exposure increased in a sub‐proportional manner, with the 8.0‐fold dose increment resulting in 2.7‐ and 2.4‐fold increases in AUC0‐∞ and Cmax, respectively. AUC0‐∞ and Cmax were estimated as 4.2‐ and 4.8‐fold greater, respectively, following food. Systemic exposure reduced upon repeat dosing with steady‐state concentrations achieved within 3–5 days of multiple bid dosing. No serious or severe adverse events were reported. SMT C1100 was safe and well tolerated with plasma concentrations achieved sufficient to cause a 50% increase in concentrations of utrophin in cells in vitro.
Keywords: Utrophin, Duchene muscular dystrophy, disease modifying therapy, pharmacokinetics, Phase 1
Duchenne muscular dystrophy (DMD) is a lethal muscle wasting disease characterized by a generalized weakness and progressive loss of muscle strength that ultimately leads to cardiac and respiratory difficulties leading to death in the mid‐twenties.1 The disease is an X‐linked recessive disorder resulting from mutations in the dystrophin gene.2 The major functional role of dystrophin is to provide a link between the actin cytoskeleton and the dystrophin protein complex anchored in the cell membrane enabling stabilization of the membrane during contraction and relaxation. The lack of functional dystrophin results in repeated cycles of muscle necrosis and regeneration leading to eventual replacement of muscle fibers by adipose and connective tissue.
Currently, there is no effective treatment for DMD. Various strategies developed to alleviate the symptoms include steroid treatment, anti‐inflammatory agents, growth hormone, and myostatin inhibitors.3 A limited number of disease modifying drugs are in clinical trials including: Ataluren (directed against stop‐codon mutations, PTC Therapeutics) and both drisapersen (Prosensa) and eteplirsen (Sarepta) targeting skipping of dystrophin exons 53, 51, 45, and 44.4 Each approach is aimed at a small subset of less than 13% of the DMD population.5
The potential for utrophin to functionally replace dystrophin has been recognized for some time.6, 7, 8, 9 The utrophin gene is the autosomal homolog of dystrophin; they share similar structural organizational motifs and protein binding properties. Utrophin is normally expressed in early developing skeletal muscle in the absence of dystrophin. As muscle matures toward the end of gestation, utrophin transcription is turned off and dystrophin transcription turned on. In adult muscle, utrophin expression is localized to the neuromuscular junction and myotendinous junction. In repairing muscle, utrophin expression is turned back on in the absence of dystrophin to re‐establish the continuity of the myotubes.
SMT C1100 (2‐arylbenzoxazole [5‐(ethyl sulfonyl)‐2‐(naphthalen‐2‐yl) benzo[d]oxazole]) is a small molecule utrophin modulator.10 Using SMT C1100 as treatment for DMD aims to replace the lost dystrophin with utrophin. In vitro studies have shown increased concentrations of utrophin protein and RNA in human muscle cells following dosing with SMT C100. Preclinical in vivo studies with a mouse DMD model reveal protection of dystrophin‐deficient muscle fibers and improved exercise capabilities associated with increased levels of utrophin.3
The objectives of the current study were to obtain safety and tolerability data when SMT C1100 was administered orally in single and multiple ascending doses to healthy volunteers, and to determine the single and multiple oral dose pharmacokinetics (PK) of SMT C1100, including the effect of fasting on the single dose PK of the drug.
Methods
This was a single center; Phase 1 study conducted in healthy volunteers. Prior to the start of the study, the protocol and consent form were reviewed and approved by the Ethics Committee (EC). The study commenced after receipt of a Clinical Trials Authorisation from the Medicines and Healthcare products Regulatory Agency (MHRA) and EC approval (NRES Committee South Central—Berkshire B). The study was conducted in accordance with the relevant articles of the “Declaration of Helsinki” and International Conference on Harmonisation (ICH) Good Clinical Practices (GCP) consolidated guidelines.
Study Population
The study population comprised healthy male subjects of any ethnic origin, aged between 18 and 55 years, with a body weight of 50–100 kilogram. Subjects were in good health, based on medical history, physical examination, 12‐lead electrocardiogram (ECG), and clinical laboratory evaluations. Subjects were excluded if they were unwilling to use appropriate contraception or continued to donate sperm from the time of their first dose until 3 months after the final dosing. Excluded were subjects who had received any prescribed systemic or topical medication within 7 days of first dose (excluding vitamin/mineral supplements), which were considered to potentially interfere with the study or compromise safety. Subjects who had received any medications, including St John's Wort, known to chronically alter drug absorption or elimination processes within 30 days of the first dose administration were excluded. Subjects already participating in a clinical study or who had participated within the previous 3 months were excluded. Subjects were not allowed to have donated any blood, plasma, or platelets in the 3 months prior to screening or make donations on more than two occasions within the 12 months preceding the first dose administration. Subjects with significant drug allergy or allergic disease were excluded as well as those with a supine blood pressure and supine pulse rate higher than 140/90 mm Hg and 100 beats per minute (bpm), respectively, or lower than 90/50 mm Hg and 40 bpm, respectively; who consumed >28 units of alcohol per week, had a significant history of alcoholism or drug/chemical abuse; with a positive urine drug screen or alcohol breath test result at screening or first admission; or who had smoked or used other tobacco products within 60 days of the first dose. Additional exclusion criteria for subjects: with, or with a history of, any clinically significant neurological, gastrointestinal, renal, hepatic, cardiovascular, psychiatric, respiratory, metabolic, endocrine, hematological, or other major disorders; with a clinically significant illness within 4 weeks of first dose; with serum hepatitis or carriers of the hepatitis B surface antigen (HBsAg) or hepatitis C antibody, or who have a positive result to the test for HIV antibodies; with an abnormality in the 12‐lead ECG, such as QTcB interval >450 millisecond (male), 2nd or 3rd degree atrioventricular block, complete left bundle branch block, complete right bundle branch block, or Wolff‐Parkinson‐White Syndrome.
Study Design
In preclinical toxicology with SMT C1100 in corn oil, oral doses of up to 1000 mg/kg were well tolerated in minipigs following daily dosing for 28 days. SMT C1100 has been re‐formulated as an aqueous microfluidized suspension. Non‐clinical data have shown that systemic exposure of SMT C1100 aqueous microfluidized suspension at a dose level of 100 mg/kg is similar to that of the corn oil formulation when given to male minipigs. A single dose of 50 mg/kg was selected as the starting dose in this Phase 1 study, to enable dose proportionality of SMT C1100 systemic exposure to be explored over an ascending dose range for the microfluidized formulation. Furthermore, should bioavailability of the microfluidized formulation in man be significantly greater than that seen for the corn oil formulation, systemic exposure at 50 mg/kg with the microfluidized formulation was expected to remain below levels known to be safe and well tolerated.
This double‐blind, placebo‐controlled study was conducted in 2 parts. Part 1 was an escalating, single‐dose, safety, and PK study with a fast effect evaluation. Groups A–D received 1 of the following doses in the fed state: 50 mg/kg, 100 mg/kg, 200 mg/kg, and 400 mg/kg. In each group, 6 subjects received a single dose of the study drug after food (based on lipophilic nature of SMT C1100; logP = 3.99 ± 0.01) in the morning and 2 received placebo. Food was provided 30 minutes prior to and completed approximately 10 minutes before dose administration. The high‐fat meal consisted of 2 eggs fried in blended oil, 2 rashers of grilled bacon, 1 slice of white toast with 10 gram butter, 2 hash brown potatoes and 240 milliliter full‐fat milk. The total energy count was 895 kcal and total fat content 61 gram. Doses were administered in an escalating manner following satisfactory review of safety, tolerability, and PK data (up to 48 hours postdose) from the lower doses. Group C also received a second 200 mg/kg dose after an overnight fast, 7 days after the first dose.
Part 2 of the study was a multiple‐dose, sequential safety and PK study. A total of 16 subjects were divided into 2 groups. In Group E, 6 subjects received 100 mg/kg and 2 received placebo, whereas in Group F, 6 subjects received 200 mg/kg, and 2 received placebo. The study drug doses were given bid on Days 1–9 with a 12‐hour interval, and a single dose on the morning of Day 10. The morning dose was given after a high fat breakfast (as above) and the second dose after dinner. The fat content of the dinner was the same as at breakfast. Lunch consisted of a low‐fat meal. The total dose administered did not exceed the maximum tolerated dose determined in Part 1.
SMT C1100 Formulation and Administration
SMT C1100 was given as an aqueous microfluidized suspension. Doses were given in a total volume of 240 milliliter and the appearance was identical to the placebo suspension. Subjects were dosed while standing and not allowed to lie supine for 2 hours postmorning dose, except for study procedures or if clinically indicated. With the exception of water given with the dose, subjects were not allowed any additional fluids until 2 hour after dosing. Personnel involved with the study remained blinded to the treatment randomization during the assembly and dispensing procedure
Assessment
Screening was performed within 28 days prior to first dose and included assessment of suitability for entrance to the study (demographic and medical). All subjects underwent predose assessment at Day‐1, which included providing a urine sample for a drug screen, an alcohol breath test, laboratory safety sample, and recording of body weight.
Safety
Adverse events (AEs) were monitored throughout the study. Data were collected on vital signs, blood pressure, pulse rate, body temperature, ECG, and clinical laboratory evaluations postdose for both parts of the study. The exposure to SMT C1100 in the repeated dose toxicity studies at the No Observed Adverse Effect Level (NOAEL) in the mouse (2000 mg/kg) and the minipig (1000 mg/kg) after 4 weeks daily dosing; Cmax (μg/mL) and AUC (mcg · h/mL) were 7.4, 97.9 and 6.9, 136 respectively, PK dose escalation was allowed if systemic exposure at the dose level did not exceed that seen at the NOAEL in the minipig.
Sample Collection
Two milliliter blood samples were collected from each subject at specified timepoints. For Part 1 these were predose, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, and 72 hours postdose. For Part 2, timepoints were on Day 1, predose, 0.5, 1, 2, 3, 4, 6, 8, 10, 12 hours post‐am dose, and 2, 3, and 4 hours post‐pm dose; on Day 7, pre‐am dose, 0.5, 1, 2, 3, 4, 6, 8, 10, 12 hours post‐am dose, and 3 hours post‐pm dose; on Day 10, predose, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, 48, and 72 hours postdose; and Days 2–6 and 8 and 9, pre‐am dose.
Bioanalysis
Plasma samples were prepared from the blood samples and extracted with acetonitrile. Concentrations of SMT C1100 were determined using a validated LC‐MS/MS method (range 2–2000 ng/mL; overall method accuracy and precision 85–115% and ≤15%, respectively, 80–120% and ≤20% at LLOQ). Samples were analyzed for the presence of metabolites using accurate mass LC‐MS. Analysis was performed on a Thermo LTQ Orbitrap mass spectrometer, equipped with a UPLC system and online UV detection. Data were interrogated for the presence of metabolites based on accurate masses of potential metabolites using Metworks software (version 1.0.1) in conjunction with Xcalibur 2.0. Additional LC‐MSn analyses were performed on a selected sample to elucidate the structures of the most abundant metabolites. Concentrations of metabolites were estimated by assuming equivalent detector response to parent SMT C1100.
Pharmacokinetic Analysis
PK parameters were determined from the plasma concentrations of SMT C1100 using non‐compartmental procedures. Descriptive statistics for the PK data were determined using SAS® Version 8.2. The PK analysis was conducted by Covance CRU using WinNonlin Professional Version 5.2 (Pharsight Corporation, California).
Maximum concentration of SMT C1100 (Cmax), the time of maximum concentration (tmax), apparent terminal elimination half‐life (t½) and time before the start of absorption (tlag) were determined. AUC0‐ τ (area under the plasma concentration‐time curve over a dosing interval) was calculated using the linear trapezoid method when concentrations were increasing and the logarithmic trapezoidal method when concentrations were decreasing. AUC0‐∞ (area under the plasma concentration‐time curve from time zero to infinity), %AUCextrap (the percentage of AUC0‐tlast extrapolated beyond tlast), CL/F (apparent plasma clearance), and Vz/F (apparent volume of distribution during the terminal phase) were determined using standard non‐compartmental analysis.11 The accumulation ratios based on AUC0‐τ were calculated using the formulae,
| (1) |
| (2) |
The effect of fasting on SMT C1100 AUC0‐∞ and Cmax at 200 mg/kg (Group C) was estimated, along with 95% confidence interval, by a linear mixed effect model (food status as a fixed effect and subject as a random effect) of the log‐transformed data.
Results
A total of 47 of the 48 subjects completed the study: 31 for Part 1 and 16 for Part 2. In Part 1, all subjects were white males aged 19–53 years with a BMI of 19.8–33.9 kg/m2. In Part 2, all subjects were males aged 20–47 years with a BMI of 19.3–28.0 kg/m2. All subjects were white except for one Asian male in Group F. In Part 1, 1 subject of Group C withdrew on Day −1 of Treatment Period 2 (fasted) due to a positive drugs of abuse test.
Safety
Overall, all doses of SMT C1100 were well tolerated when administered as single doses or bid doses over 10 days. Single occurrences were reported of abdominal gas (for single dose placebo fasted); bites on left ankle and palpitations (100 mg/kg dose); aching right arm near cannula site (200 mg/kg dose); and hunger and throbbing left hand (200 mg/kg dose fasted). In the multiple‐dose group: placebo (cold symptoms, sleepiness), 100 mg/kg (painful calves, tiredness, vomiting), and 200 mg/kg (loose stools, constipation, headache, leg cramps, upset stomach). The majority of AEs were mild in severity and resolved without treatment; there were no severe AEs reported. In Part 1 and Part 2, the greatest incidence was at the highest doses, 400 mg/kg and 200 mg/kg bid, respectively. The only treatment‐emergent AE related to study drug was stool coloration (discolored or pale), which was only apparent at the 200 and 400 mg/kg doses. These were not associated with any other gastrointestinal‐related AE. All subjects were within normal limits for: blood pressure; pulse rate; body temperature; 12‐lead ECG data; clinical laboratory evaluation; and physical examination.
Pharmacokinetic Evaluation
Part 1
Following a single oral dose, AUC0‐∞, and Cmax increased in a sub‐proportional manner over the 50–400 mg/kg dose range with the increase in exposure being up to 2.7‐fold for the overall 8.0‐fold increase in dose (Table 1). The sub‐proportional increase was most marked between the 100 and 200 mg/kg doses where no increase in mean systemic exposure was observed despite the 2‐fold increase in dose. CL/F and Vz/F increased as the dose of SMT C1100 increased. The sub‐proportionality across the entire dose range was confirmed by statistical analysis, with the estimates of the slopes (95%CI) from the regression analysis not spanning unity, being 0.41 (0.012–0.80) and 0.37 (0.0048–0.73) for AUC0‐∞ and Cmax, respectively. There was a rapid absorption phase (Figure 1) with median tmax increasing with dose (2.0 hours at 50 mg/kg to 3.5 hours at 400 mg/kg) (Table 1). Individual tmax values ranged from 0.5 to 4 hour across all doses, due to subject variability in the rate of absorption.
Table 1.
Summary of the Pharmacokinetic Parameters for SMT C1100 Following Single Oral Doses and Multiple Doses. Geometric Mean (Coefficient of Variation) Are Presented Unless Otherwise Stated
| Parameter | Single‐Dose Study | Multiple‐Dose Study | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 50 mg/kg (n = 6) | 100 mg/kg (n = 6) | 200 mg/kg (n = 6) | 400 mg/kg (n = 6) | 200 mg/kg Fasted (n = 5) | 100 mg/kg (n = 6) | 200 mg/kg (n = 6) | |||||
| Day 1 | Day 7 | Day 10 | Day 1 | Day 7 | Day 10 | ||||||
| AUC0‐∞ a | 1612 | 2932 | 2766 | 4215 | 492 | – | – | – | – | – | |
| (ng · h/mL) | (47.0) | (79.9) | (89.0) | (125) | (222) d | ||||||
| AUC0‐ τ | – | – | – | – | – | 1980 | 1343 | 1469 | 1390 | 1001 | 830 |
| (ng · h/mL) | (38.3) | (59.1) | (63.6) | (54.9) | (46.1) | (53.3) | |||||
| Cmax (ng/mL) | 226 (41.5) | 383 (88.2) | 373 (71.8) | 531 (103) | 73.0 (103) | 316 (40.0) | 203 (56.8) | 218 (66.8) | 255 (50.1) | 150 (45.2) | 122 (52.8) |
| tmax (hour) b | 2 (1–3) | 2.5 (0.5–3.02) | 3 (0.5–3) | 3.5 (2–4) | 2 (0.5–3) | 2.5 (1–3) | 3 (2–4) | 3 (2–4) | 2.5 (2–3) | 3 (2–4.02) | 3.53 (2–4) |
| t½ (hour) | 20.6 (42.2) | 22.6 (21.1) | 19.9 (73.6) | 19.8 (38.7) | 12.8 (246) d | – | – | 35.7 (29.8) | – | – | 29.0 (31.3) |
| CL/F (mL/min/kg) | 517 (47.0) | 569 (79.9) | 1205 (89.0) | 1582 (125) | 6768 (222) d | – | 1234 (67.3) c | 1134 (63.6) | – | 3330 (46.1) | 4014 (53.3) |
| Vz/F (L/kg) | 922 (35.3) | 1112 (73.0) | 2078 (19.6) | 2711 (139) | 7494 (37.3) d | 235 (30.7) | 475 (53.6) | 3510 (91.0) | 608 (47.3) | 1357 (32.9) | 10089 (54.1) |
| RAobs | – | – | – | – | 0.704 (25.0) | 0.770 (28.0) | – | 0.720 (29.9) | 0.597 (36.4) | ||
AUC0‐∞ = area under the plasma concentration‐time curve from time zero to infinity (percentage of AUC that is due to extrapolation from tz to infinity was <10% in all cases); AUC0− τ = area under the plasma concentration‐time curve over a dosing interval; Cmax = maximum observed plasma concentration; tmax = time of maximum observed plasma concentration; t½ = apparent plasma terminal elimination half‐life (for Day 10 of multiple‐dose study this is post drug induction); CL/F = apparent total plasma clearance; VZ/F = apparent volume of distribution during the terminal phase; RAobs = observed accumulation ratio based on AUC0‐ τ.
Arithmetic mean (CV%).
Median (min–max).
n = 5.
n = 3.
Figure 1.
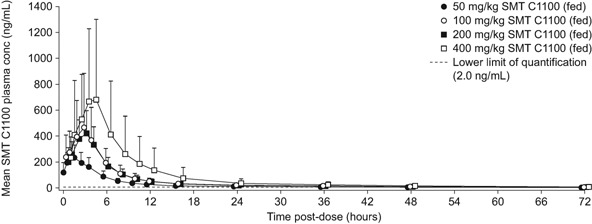
Arithmetic mean plasma concentrations of SMT C1100 following single oral dose in the fed state. The error bars represent standard deviation.
Plasma concentrations of SMT C1100 are shown in Figure 2. Following administration of SMT C1100 in the fasted state (no food for 12 hours), maximum plasma concentrations were obtained at a median tmax of 2.0 hour postdose, approximately 1 hour earlier than observed in the fed state although the range of individual tmax values was the same in both dietary states. The mean t½ under fasted conditions appeared shorter than that observed following a high fat breakfast, with mean values of 13 and 20 hours, respectively. However, the difference in mean t½ might be considered artefactual due to the SMT C1100 concentrations tending to fall below the lower limit of quantification at earlier times in fasted state, thus concealing part of the terminal elimination phase. The t½ range in individual subjects was similar between dietary states ranging from 5.7 to 34.3 hours in the fed state and 2.6 to 33.7 hours in the fasted state. Markedly lower exposure was seen for SMT C1100 under fasted conditions compared with that observed following food, with AUC0‐∞ and Cmax being 4.2 (95% CI: 2.0, 8.6)‐ and 4.8 (95% CI: 3.3, 7.0)‐fold higher, respectively, in the fed state compared with the fasted state. Hence, administration in the fed state was selected for Part 2.
Figure 2.
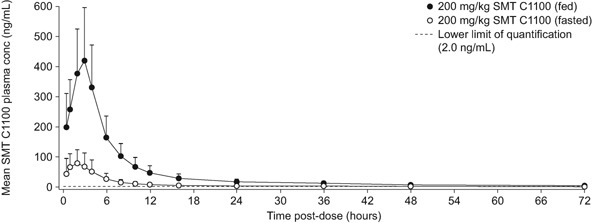
Arithmetic mean plasma concentrations of SMT C1100 following oral administration of SMT C1100 in the fed and fasted states at 200 mg/kg. The error bars represent standard deviation.
Part 2
The PK parameters of multiple‐dose study are summarized in Table 1. SMT C1100 at doses of 100 mg/kg bid (Figure 3a) and 200 mg/kg bid (Figure 3b) was rapidly absorbed, with tmax ranging from 1.0 to 4.0 hours postdose across subjects on Days 1, 7, and 10. The start of the apparent terminal elimination phase occurred between 3.0 and 6.0 hours postdose on Day 10 for both doses. The mean t½ on Day 10 was 36 and 29 hour for the 100 and 200 mg/kg bid doses, respectively, with values for individual subjects ranging from 23.6 to 52.9 hours. Steady‐state, based on plasma concentrations of drug in pre‐morning dose samples, was estimated to have been attained within 3 to 5 days of dosing (ie, on Days 4 to 6). However, systemic exposure was found to decrease with repeated doses for both groups, such that by Day 10, AUC0‐τ was approximately 23% and 40% lower than on Day 1 for the 100 and 200 mg/kg bid dose groups, respectively.
Figure 3.
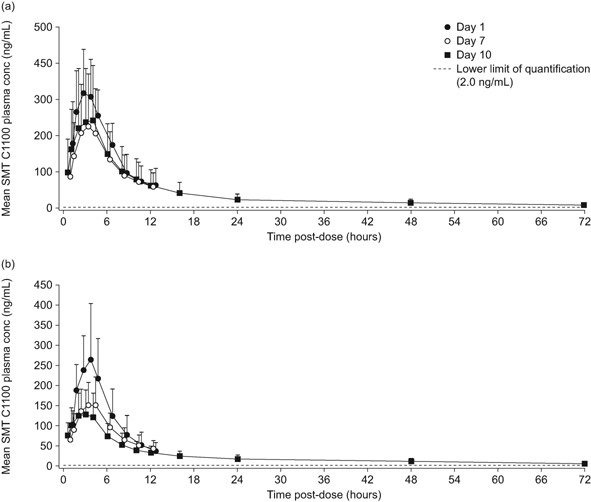
Arithmetic mean plasma concentrations of SMT C1100 at Days 1, 7, and 10 following multiple oral doses of SMT C1100 in the fed state at (a) 100 mg/kg bid and (b) 200 mg/kg bid. The error bars represent standard deviation.
Analysis of Metabolites
Plasma samples from 3 subjects in Part 1 and 3 subjects in the 100 mg/kg bid group of Part 2 were profiled for metabolites. Parent and a total of 12 metabolites were detected. Figure 4a depicts the major metabolites, and in each case the most abundant metabolites were 2 dihydrodiol metabolites, with metabolism occurring at different positions of the naphthalene moiety of SMT C1100 and the hydroxyls being trans to each other. The 2 dihydrodiol metabolites had weak in vitro utrophin modulation with EC50 > 10‐fold that of SMT C1100. Exposure to metabolites generally increased from day 1 to day 10 following multiple bid administration (see Figure 4b).
Figure 4.
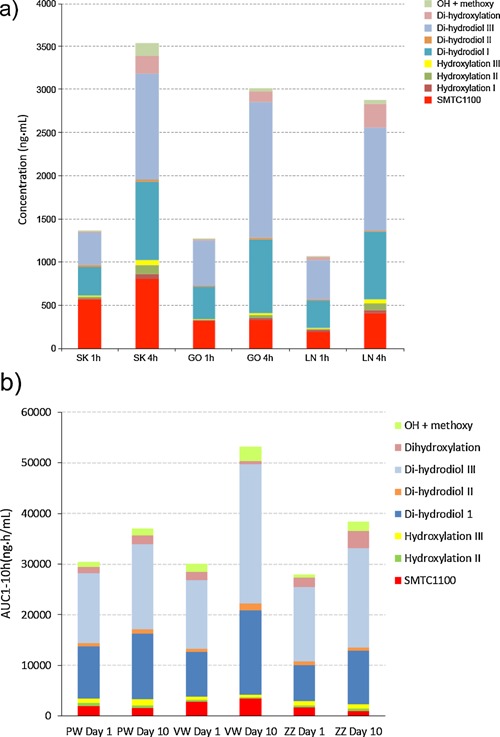
Individual healthy volunteers’ cumulative in vivo metabolism. (a) Total estimated plasma concentration of SMT C1100 parent and metabolites quantified at 1 and 4 hours after the 100 mg/kg single dose in 3 subjects. (b) Estimated plasma exposure of SMT C1100 parent and principal metabolites on Day 1 and Day 10 for 3 subjects from the 100 mg/kg bid group. Note the letter pairs indicate random blinded assignment rather than an individual subjects, initials.
Discussion
SMT C1100 has demonstrated significant disease modifying potential for DMD in non‐clinical efficacy studies. In vitro assays with human myoblasts or human DMD cells treated with SMT C1100 demonstrated a 25% increase in utrophin mRNA and a 100% increase in utrophin protein, respectively, leading to an estimated EC50 of 0.2 micromolar (67 ng/mL).3 This in vitro activity has been confirmed in a number of in vivo studies involving the dystrophin‐deficient mdx mouse model. Following daily dosing for 28 days a 2‐fold increase in utrophin mRNA was reported compared with controls.3 Importantly, increased levels of utrophin protein were reported in heart muscle (P<.01) and diaphragm (P<.05). The same study showed improvements in fatigue testing in mdx mice, which was analogous to the 6 minute distance walk test used as a primary clinical endpoint in DMD patient clinical trials. PK analysis of SMT C1100 in the mdx model confirmed plasma and muscle levels above the 0.2 micromolar concentration required to modulate utrophin transcription for several hours in a 24 hour period.3
In the current study, the most frequently reported AE was change in stool coloration. This occurred in 11 subjects predominantly at the 200 and 400 mg/kg doses and was probably a consequence of a greater proportion of unabsorbed study drug passing through the gastrointestinal tract at the higher doses. There were no treatment‐ or dose‐related trends or clinically significant findings in clinical laboratory evaluations, vital signs, 12‐lead ECG or physical examinations during this study.
The phase 1 study showed that SMT C1100 was rapidly absorbed following single and multiple oral doses of SMT C100, with median tmax being attained within 0.5–4 hours across all doses. The increase in the single oral dose from 50 mg/kg up to 400 mg/kg resulted in maximum plasma concentrations of 226–531 ng/mL, with plasma concentrations in excess of 100 ng/mL being achieved for 6 hour with the 50 mg/kg dose, 8 hours with the 100 and 200 mg/kg dose, and 12 hours with the 400 mg/kg dose (Figure 1). These plasma concentrations are above the estimated 0.2 micromolar (67 ng/mL) required for 50% increase in utrophin protein in the DMD myoblast in vitro cell assays.3
The multiple‐dose study revealed no increased plasma exposure with the 200 mg/kg bid dose compared with the 100 mg/kg bid dose; both had similar time course profiles (Figure 3). Single and multiple oral dose PK administration of SMT C1100 were characterized by sub‐proportional increases in systemic exposure with minimal increases in AUC or Cmax observed over the dose range 100–400 mg/kg. Following multiple bid dosing of SMT C1100, exposure to the drug was reduced such that systemic exposure (AUC0‐τ) was approximately 23% and 40% lower on Day 10 than on Day 1 for the 100 and 200 mg/kg bid groups, respectively (Figure 3). These data suggest that SMT C1100 causes mild auto‐induction, a finding that is similar to the mouse and female minipig.12
We believe the variation in SMT C1100 parent concentrations from individual to individual is based around differing individual activities of CYP1A1/2 (responsible for metabolism of SMT C1100). SMT C1100 metabolite profiling confirmed that although individuals had differing concentrations of SMT C1100, the total amount of parent and metabolites was very similar suggesting similar absorption followed by differing metabolism. Figure 4a illustrates this observation by comparing the total estimated amount of SMT C1100 parent and metabolites at 1 and 4 hours postdose from 3 individuals from the 100 mg/kg dose group. The total concentrations of parent and metabolites at each of the 2 time points were very similar between the 3 individuals suggesting similar absorption yet the SMT C1100 concentrations were very different. A similar finding was observed for subjects in the multiple‐dose study (Figure 4b).
Dietary state had a significant effect on systemic exposure, with AUC0‐∞ and Cmax being approximately 4.2‐ and 4.8‐fold greater, respectively, following a high fat breakfast compared with fasted conditions at the 200 mg/kg dose (Figure 2) and subsequent clinical studies will include giving drug with food. SMT C1100 exhibited bi‐phasic elimination with an apparent dose dependent terminal elimination half‐life, with mean t½ ranging from 21 to 20 hours following single doses.
As part of an expanded study to evaluate quantitatively and predict the in vivo fate of SMT C1100, and looking toward the future development of a PK‐PD model, we analyzed the concentration‐time data by compartmental analysis using SAAM II (v1.2.1, SAAM Institute, University of Washington). Individual concentration‐data, as well as group means, after oral administration of 50, 100, 200, and 400 mg/kg doses were found to fit best to a two‐compartment open model with zero‐order absorption and first‐order elimination.13 This model confirms the previously determined non‐compartmental parameters. It also explains the observed sub‐proportional relationship between AUC and dose, in so far as the absorption of SMT C1100 under the conditions of the clinical trial appears to be concentration independent (hence zero‐order) and maximal in the vicinity of the 100 mg/kg dosing regimen. The hallmark of zero‐order absorption kinetics is that the input rate into the central, rapidly perfused (plasma) compartment is constant for a fixed time and then ceases. Accordingly, doses greater than 100 mg/kg predictably yield no appreciably greater exposure.
The finding that SMT C100 follows zero‐order absorption in a determinable compartmental model further facilitates the simulation of dosing regimen outcomes, an extension of Phase 1 study information that becomes relevant in the planning of future efficacy studies. For example, as shown in Figure 5, the model, after adjustment to fit the observed increased metabolic rate (decreased AUC from day 1 to day 10) as an inverse exponential saturation function, affords comparative information in the simulation of expected peak and trough titers after bid dosing at 100 mg/kg, suggesting that near steady state, mean plasma exposure between 0.14 and 0.78 micromolar can be achieved within 1 week of dosing. Figure 5b shows the overlay of the actual 100 mg/kg bid PK data onto the predicted model demonstrating good concordance. If equilibrated with muscle, this titer range should prove sufficient to drive utrophin up‐regulation in a manner consistent with the in vitro effects observed after similar drug exposure in studies with DMD myoblasts.
Figure 5.
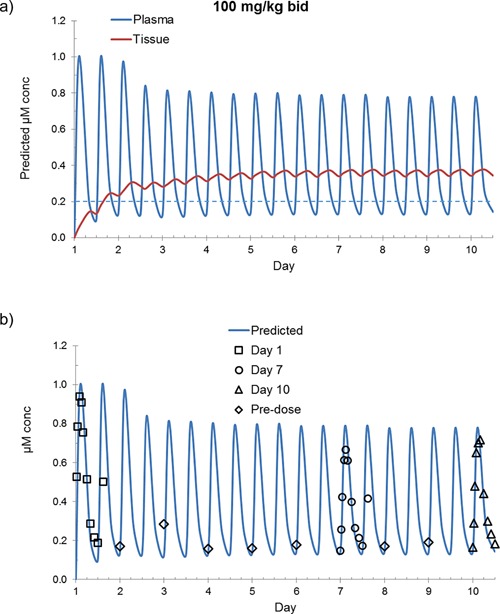
Pharmacokinetic modeling of (a) simulated concentrations of SMT C1100 in plasma and tissue and (b) predicted vs. observed concentrations in plasma, both after repeat dosing with SMT C1100 100 mg/kg bid. In (a) the predicted level required to achieve in vitro EC50 concentrations (0.2 μM/67 ng/mL) is depicted by the dashed line.
Conclusions
It was concluded that SMT C1100 was safe and well tolerated in healthy volunteers. Oral administration resulted in higher plasma concentrations than those required to cause a 50% increase in utrophin levels in cells in vitro.
Funding
We especially thank the US parent charities, Parent Project Muscular Dystrophy, Muscular Dystrophy Association USA, Cure Duchenne, Charley's Fund, Nash Avery Foundation, and Federation to Eradicate Duchenne for funding the clinical manufacture of SMT C1100 and the Phase 1 trial.
Acknowledgments
We thank Covance Clinical Research Unit (CRU) Ltd for helping design and conducting the Phase 1 study for SMT C1100 and Principal Investigator, Ashley Brooks, MBChB. We thank Dr A. Ajami for provision of the PK modeling data and critical review of the manuscript, Mr. G. Layton for critical review of the manuscript and Medscimedia Ltd for editorial support.
Declaration of Conflicting Interests
Jon Tinsley is a SUMMIT employee.
References
- 1. Bushby K, Straub V. Nonmolecular treatment for muscular dystrophies. Curr Opin Neurol. 2005; 18:511–518. [DOI] [PubMed] [Google Scholar]
- 2. Van Deutekom JC, van Ommen GJ. Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet. 2003; 4:774–784. [DOI] [PubMed] [Google Scholar]
- 3. Tinsley JM, Fairclough RJ, Storer R, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE. 2011; 6:e19189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoffman EP, Connor EM. Orphan drug development in muscular dystrophy: update on two large clinical trials of dystrophin rescue therapies. Discov Med. 2013; 16:233–239. [PubMed] [Google Scholar]
- 5. Scully MA, Pandya S, Moxley RT. Review of Phase II and Phase III clinical trials for Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2013; 1:33–46. [Google Scholar]
- 6. Squire S, Raymackers JM, Vandebrouck C, et al. Prevention of pathology in mdx mice by expression of utrophin: analysis using an inducible transgenic expression system. Hum Mol Genet. 2002; 11:3333–3344. [DOI] [PubMed] [Google Scholar]
- 7. Tinsley J, Deconinck N, Fisher R, et al. Expression of full‐length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998; 4:1441–1444. [DOI] [PubMed] [Google Scholar]
- 8. Dennis CL, Tinsley JM, Deconinck AE, Davies KE. Molecular and functional analysis of the utrophin promoter. Nucleic Acids Res. 1996; 24:1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chakkalakal JV, Stocksley MA, Harrison MA, et al. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc Natl Acad Sci USA. 2003; 100:7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chancellor DR, Davies KE, De Moor O, et al. Discovery of 2‐arylbenzoxazoles as upregulators of utrophin production for the treatment of Duchenne muscular dystrophy. J Med Chem. 2011; 54:3241–3250. [DOI] [PubMed] [Google Scholar]
- 11. Gabrielsson J, Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications. 3rd ed Stockholm, Sweden: Swedish Pharmaceutical Press; 2000:141–153. [Google Scholar]
- 12.Data on file, SUMMIT plc.
- 13. Mesnil F, Dubruc C, Mentre F, et al. Pharmacokinetic analysis of mizolastine in healthy young volunteers after single oral and intravenous doses: noncompartmental approach and compartmental modeling. J Pharmacokinet Biopharm. 1997; 25:125–147. [DOI] [PubMed] [Google Scholar]