

Abstract
Alkaptonuria (AKU) is a rare genetic disease that affects the entire joint. Current standard of treatment is palliative and little is known about AKU physiopathology. Chondroptosis, a peculiar type of cell death in cartilage, has been so far reported to occur in osteoarthritis, a rheumatic disease that shares some features with AKU. In the present work, we wanted to assess if chondroptosis might also occur in AKU. Electron microscopy was used to detect the morphological changes of chondrocytes in damaged cartilage distinguishing apoptosis from its variant termed chondroptosis. We adopted histological observation together with Scanning Electron Microscopy and Transmission Electron Microscopy to evaluate morphological cell changes in AKU chondrocytes. Lipid peroxidation in AKU cartilage was detected by fluorescence microscopy. Using the above‐mentioned techniques, we performed a morphological analysis and assessed that AKU chondrocytes undergo phenotypic changes and lipid oxidation, resulting in a progressive loss of articular cartilage structure and function, showing typical features of chondroptosis. To the best of our knowledge, AKU is the second chronic pathology, following osteoarthritis, where chondroptosis has been documented. Our results indicate that Golgi complex plays an important role in the apoptotic process of AKU chondrocytes and suggest a contribution of chondroptosis in AKU pathogenesis. These findings also confirm a similarity between osteoarthritis and AKU. J. Cell. Physiol. 230: 1148–1157, 2015. © 2014 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
Alkaptonuria (AKU) is an ultra‐rare metabolic disease due to a deficient activity of the enzyme homogentisate 1,2‐dioxygenase (HGD) leading to accumulation of homogentisic acid (HGA). HGA‐oxidized derivative benzoquinone acetic acid (BQA) forms a melanin‐like pigmentation known as “ochronosis,” causing dramatic tissue degeneration. A severe form of arthropathy, with articular cartilage degeneration, is the most common clinical presentation of AKU. Focal degeneration of the articular cartilage, associated with altered subchondral bone remodeling, sclerosis, bone hypertrophy, and the presence of osteophytes are main AKU features (Taylor et al., 2011a).
Over the last two decades, there has been increasing evidence showing association between cartilage degradation and chondrocyte death in rheumatologic diseases (Hayami et al., 2004; Li et al., 2013), and different types of cell death in cartilage have been reported including apoptosis and chondroptosis. The term chondroptosis indicates that cells undergo apoptosis in a non‐classical manner that appears to be typical in chondrocytes death in vivo (Roach et al., 2004; Sitte et al., 2009). Chondroptosis is nothing else than a special form of apoptosis of chondrocytes, which resides in cartilage matrix so that the apoptotic rests cannot be handled as in vascularized tissue. Although chondroptosis has some features in common with classical apoptosis and other types of cell death such as cell shrinkage, chromatin condensation and the probable involvement of caspases, other features are different such as a cytoplasmic vacuolization without nuclear fragmentation and prominent Golgi and rough endoplasmic reticulum (RER). Chondroptosis has been reported to play an important role in osteoarthritis (OA) (Roach et al., 2004; Pérez et al., 2005; Almonte‐Becerril et al., 2010; Zamli and Sharif, 2011).
Recent advances on the study of articular tissue degeneration in AKU have been related to cartilage and extracellular matrix (ECM) structural and biochemical characterization (Taylor et al., 2011b), describing different zones in the tissue (Taylor et al., 2010). AKU histopathology has been assessed by the degenerative stages of different zones of the articular human cartilage (Taylor et al., 2011a).
Our previous work (Braconi et al., 2010,b; Tinti et al., 2010; Tinti et al., 2011a,2011b; Braconi et al., 2012; Laschi et al., 2012; Millucci et al., 2012; Braconi et al., 2013; Spreafico et al., 2013) suggested that morphological changes of chondrocytes in AKU cartilage may be attributed to apoptosis, but until now no in depth study exists to elucidate if chondroptosis may occur in AKU. Moreover, ultra‐structural observations were complemented with biochemical and proteomic characterization of chondrocytes isolated from the ochronotic cartilage of AKU patients, indicating that AKU chondrocytes are characterized by HGA‐induced apoptosis, protein aggregation, nitric oxide release, and oxidative stress (Tinti et al., 2011a; Laschi et al., 2012; Millucci et al., 2012; Braconi et al., 2013; Spreafico et al., 2013). AKU patients have high levels of plasma serum amyloid A and pro‐inflammatory cytokines (Tinti et al., 2011b; Laschi et al., 2012; Millucci et al., 2012; Braconi et al., 2013; Millucci et al., 2014). The local expression of HGD in human osteoarticular system (Laschi et al., 2012) dramatically increases the effects in AKU cartilage degeneration. We also reported that plasma from AKU patients and an AKU human plasma model contains inflammatory cytokines, free radicals and oxidants acting as cytotoxic agents for chondrocytes (Tinti et al., 2011a; Braconi et al., 2012; Laschi et al., 2012; Braconi et al., 2013; Spreafico et al., 2013; Millucci et al., 2014).
In the present work, we performed a morphological analysis and assessed that AKU chondrocytes undergo phenotypic changes and lipid oxidation, resulting in a progressive loss of articular cartilage structure and function, showing typical features of chondroptosis and suggesting its role in AKU pathogenesis, similarly to what observed in OA.
Materials and Methods
Ethical approval
The whole study was conducted following the approval of Siena University Hospital Ethics Committee. Patients gave a written informed consent prior to inclusion in the study. The informed consent conformed to the standards set by the latest revision of the Declaration of Helsinki containing few substantive changes relative to the April 2013 iteration.
Histology
AKU, OA, and healthy cartilage fragments were placed in 5% formaldehyde. Samples were dehydrated with increasing concentrations of alcohol and embedded in paraffin. Sections were then stained with hematoxylin‐eosin (H/E).
Histological analyses of proteoglycans
Sections were stained with Safranin‐O and fast green.
AKU and healthy chondrocytes
Alkaptonuric chondrocytes were obtained from articular cartilage of AKU patients and cultured, as described (Tinti et al., 2010). Healthy chondrocytes were from articular cartilage of a healthy young male subject who underwent surgery for a traumatic accident (Table I).
Table I.
Patients and control features
| Features | Control 1 | Control 2 | Control 3 | Control 4 | Control 5 | Control 6 | Control 7 | Control 8 | Control 9 | Control 10 | Control 11 | Control 12 | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 71 | 68 | 63 | 67 | 72 | 70 | 60 | 59 | 65 | 80 | 84 | 30 | 71 | 63 | 60 | 65 |
| Sex | F | F | F | F | M | M | M | F | F | F | M | M | F | F | F | M |
| Pathology | OA | OA | OA | OA | OA | OA | OA | OA | OA | OA | OA | Healthy | AKU | AKU | AKU | AKU |
| OARSI grade | 4 | 5 | 5 | 3 | 6 | 4 | 4 | 3 | 4 | 6 | 5 | 0 | 6 | 6 | 5 | 5 |
| Cartilage sample | Knee | Knee | Shoulder | Knee | Shoulder | Shoulder | Hip | Hip | Knee | Hip | Hip | Knee | Hip | Hip | Shoulder | Knee |
| Sample analysis | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM | H | H, IF, TEM | H, IF, TEM | H, IF, TEM | H, IF, TEM |
F, female; M, male; OA, osteoarthritis; AKU, alkaptonuria; H, histology; IF, immunofluorescence; TEM, transmission electron microscopy.
Fluorescence microscopy
In order to evaluate the presence of major products of lipid peroxidation, 3 µm unstained slices of AKU cartilage were used for immunofluorescence staining with anti‐4‐hydroxy‐2‐nonenal (4‐HNE) mouse monoclonal antibody (Percipio Biosciences, Inc., Burlingame, CA). A secondary FITC‐conjugated rabbit anti‐mouse IgG (Santa Cruz, CA) was adopted for final detection. The nuclei were counter stained with blue DAPI DNA stain (Abcam, Cambridge, UK). 4‐HNE immunofluorescence was also performed on AKU chondrocytes obtained from all AKU patients (Table I) and from a healthy subject used as control. Apoptosis was determined by staining cells with DAPI DNA stain (Abcam, Cambrige, UK).
The activation of tissue transglutaminase (Tgase; also called transglutaminase type 2 [TGase 2]) as hallmark of apoptosis was studied by a cytochemical assay based on the use of fluoresceinated cadaverine (Sigma–Aldrich, Saint Louis, MO). AKU chondrocytes were grown on coverslips for 24 h labeled with 0.05 mM mono‐dansylcadaverine in PBS at 37°C for 10 min. After incubation, cells were washed four times with PBS and analyzed using an Axiovert 405 M, inverted microscope equipped with epi‐fluorescence.
Statistical analysis
Multiple linear regression analysis was used to model the percentage of cells that were apoptotic.
Scanning electron microscopy
Scanning electron microscopy (SEM) observations and chemical microanalysis were carried out using a Philips® XL30 device operated at 20 kV equipped with an EDAX energy‐dispersive (EDS) X‐ray. The volume of sample analyzed with EDAX, at the actual operating condition, was estimated to have a diameter of ca. 3 µm; hence areas smaller than ca. 5 µm had chemical analyses mixed with adjacent phases. Initially, the cartilage was analyzed without carbon coating to check the presence of C in the sample itself. Once verified that C was a component of the ochronotic pigment, the fragment was carbon coated in order to obtain good quality images under SEM.
Transmission electron microscopy (TEM)
Tissue was fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4) containing 2% tannin and post‐fixed in 1% osmium tetroxide. Tissues were dehydrated in ethanol and embedded in epoxy resin. Ultrathin sections were contrasted with uranyl acetate and lead citrate. Specimens were examined in a transmission electron microscope (100CX II, JEOL, Peabody, MA) operated at 80 kV. Samples were also analyzed by an electron dispersive X‐ray (EDX) microanalysis system using Quantex software (Kevex Corp., San Carlos, CA).
Results
Histology—analysis of cartilage in AKU joints
AKU knees with significant cartilage pathology contained degenerated cartilage severely pigmented. Calcium deposition on roughened and fibrillated surfaces was often present. Tears were frequently observed (Fig. 1b,c). No meniscal ganglions or external cysts were observed. The intensity of Safranin‐O staining was directly proportional to the proteoglycan content in normal cartilage (Fig. 1a). Safranin‐O has thus been used to demonstrate any changes that occurred in articular AKU cartilage. Representative images of Safranin‐O staining are shown in Figure 1. In AKU specimens, hardly a feeble Safranin‐O staining was observed. The Safranin‐O staining in AKU cartilage was significantly weaker than that in normal cartilage (Fig. 1b–d), while it was more prominent in the surface zone than that in the deep zone (Fig. 1c,d). Histopathological evaluation of cartilage from AKU joints revealed a more severe fibrocartilaginous disruption. The extracellular matrix features fine fibrillations and a loss of structure in most cases, most likely due to an edematous swelling of the tissue. There was an extensive variability in the pattern of proteoglycan staining with areas with more appreciable intensity as well as areas nearly devoid of staining (Fig. 1b,d). We observed large variation in cell distribution with hypercellular, hypocellular, and acellular areas as well as areas containing large and abundant cell clusters. Abnormal cell clusters were found close to the articular surface, typically associated with tears of the meniscus (Fig. 1c). These cell clusters were seen in both positive and negative Safranin‐O stained matrix. Cells around frayed regions and tears were often larger compared to cells in normal regions.
Figure 1.
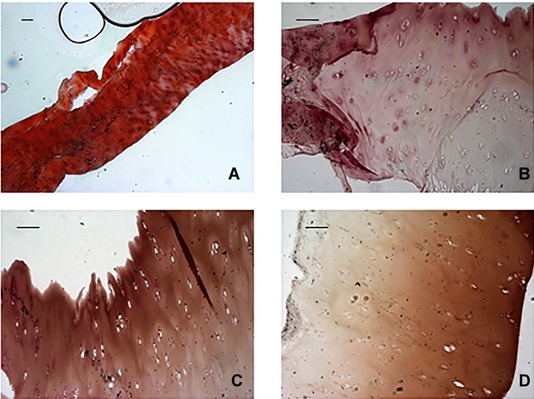
Representative images of Safranin‐O staining. (A) Normal cartilage (horizontal sections of the central portion of menisci) displayed strong orange‐red staining, indicating a completely preserved cartilage. In contrast (B, C, D), AKU menisci (horizontal sections of the central portion of menisci) displayed moderate orange‐red staining, indicating advanced cartilage destruction. Scale bar = 100 μm.
Morphological changes in AKU cartilage were indicative of phenotypic variability (Fig. 2A). The ultrastructural characteristics of AKU chondrocytes let us to classify them into two types according to their location and phenotypic changes: (1) normal chondrocytes, observed in non‐fibrillated regions distant from the damaged cartilage (Fig. 2A,b); (2) chondrocytes with secretory phenotype, clustered (groups of 2–4 cells, “clones”) flattened or ellipsoidal chondrocytes within the most damaged region of AKU cartilage (Fig. 2A,c,d). Normal and chondrocytes with secretory phenotype were not the same as the different types typically seen in normal cartilage. Chondrocytic death in AKU cartilage was witnessed by the presence of large number of empty lacunae (Fig. 2A,b–d) and confirmed previous data related to mechanical injury, increased production of reactive oxygen species (ROS) and disruption of extracellular matrix integrity (Braconi et al., 2010a,b; Tinti et al., 2010). The clusters of chondrocytes with secretory phenotype were isogenous groups when located in the lower area of alkaptonuric cartilage, as visible in Figure 2B,c. On the contrary, chondrocytes with secretory phenotype located in the superficial zone of AKU cartilage (Fig. 2B,a) were not isogenous, but had the same chondroptotic phenotype, as also confirmed by TEM analysis (Fig. 6A,B).
Figure 2.

Histological anatomo‐pathological analysis of hematoxylin and eosin stained AKU cartilage. (A,a) Healthy cartilage from the knee of a healthy young male subject who underwent surgery for a traumatic accident (Control 12, Table I). (b) AKU cartilage in non‐fibrillated regions, distant from the damaged cartilage. Hypocellularity and empty lacunae (arrow) are showed. Specimen was from Patient 1 (Table I). (c) Image from the most damaged region of AKU cartilage. Different grading of ochronotic pigmentation was clearly visible, being more pronounced in the subchondral zone. Specimen was from Patient 2 (Table I). (d) Chondrocyte aggregates or “clones” were found in great matrix lacunae. Clustered chondrocytes in the pigmented areas organized in clones are shown (arrows). Specimen was from Patient 3. Scale bar = 100 μm. (B) Photomicrograph showing the different patterns of ochronotic pigmentation in AKU cartilage. Ochronotic pigmentation was intense at the deep and middle cartilage areas. Scale bar = 100 μm. (a) Superficial zone showing the articular surface with matrix scarcely pigmented but presenting a defined area more ochronotic where dismorphic pigmented chondrocytes were random positioned; (b) Middle hypertrophic zone with dense ochronotic pigment in the extracellular matrix. Chondrocytes showed intracellular deposits and appeared phenotipically normal; (c) Deep zone showing intense pigment deposition in the extracellular matrix. Chondrocytes showed intracellular pigmentation, and some of them were chondroptotic. Their dimension was larger in volume than chondrocytes present in the hypertrophic zone. Resorption of the subchondral plate was also appreciable. (C) 4‐HNE detection in AKU, OA, and normal cartilage. (a) Healthy cartilage, as a negative control, from Control 12 (Table I); (b–d) AKU cartilage from Patient 1 (Table I); (e–g) AKU cartilage from Patient 2 (Table I); (h) OA cartilage, as a positive and reference control. Immuno‐fluorescence to 4‐HNE was relevant in all AKU cartilage specimens (b–g). Immuno‐specificity of the staining was assessed by the absence of fluorescence in the superficial zones of AKU cartilage (e–g) evidenced only by DAPI fluorescence (blue). The deep zone of cartilage surface, the pericellular matrix, the intracellular matrix, and the matrix around the cells, all showed positive staining which suggested the presence of strong lipid peroxidation in these areas. Scale bar = 100 μm.
Figure 6.
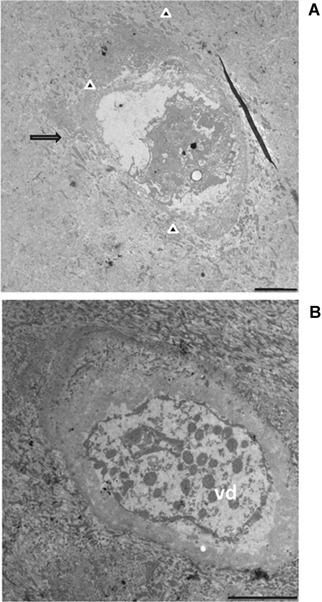
TEM observation of AKU cartilage. Samples from Patient 1 (Table I) were from the superficial zone of cartilage (see Fig. 1B,a). Chondrocytes were phenotypically ascribed to those with phenotype, though being located in a non‐pigmented area. Extrusion of cellular material into the lacunae into extracellular space, typical feature in chondroptosis, was evident (A, arrow). The disintegration of cell in chondroptosis is a combination of digestion in autophagic vacuoles and of the release of resultant cell fragment into the extracellular space, as observable in part A (arrow heads). In this terminal stage of cell death, cytoplasm organelles were not recognizable and only some nuclear remnant is often visible (A and B). Chondroptotic remnants were disintegrated into vesicular detritus (B, vd). Bars A, B: 25 μm.
4‐HNE detection
Lipid peroxidation (LPO) was clearly evident in AKU cartilage from different patients, compared to control (Fig. 2C). HNE is specific major aldehydic product of lipid peroxidation. It is believed to be largely responsible for the cytopathologic effects observed during the oxidative stress of lipids (Pompella et al., 2002). In AKU samples, fluorescence is very specific for pigmented and degraded zones of cartilage. In Figure 2C, d–g, it is appreciable that HNE fluorescence is intensely diffuse only in hypertrophic and deep zone of cartilage, corresponding to pigmented areas visible in Figure 1B and hosting chondrocytes with secretory phenotype. On the contrary, superficial AKU cartilage zone that is scarcely pigmented is not reactive for HNE and evidenced only by DAPI fluorescence. In contrast, normal cartilage (Fig. 2C,a) showed only a very faint staining of the cartilage surface. In OA sample (Fig. 2C,h), the positivity for HNE fluorescence was evident as well as for AKU cartilage. Moreover, LPO was observed also in primary AKU chondrocytes (Fig. 3). DAPI staining allowed observe apoptotic nuclei and apoptotic bodies in primary AKU chondrocytes (Fig. 3B). Taken together, our observations suggest a role of lipid peroxidation in peculiar chondrocytes death in OA and AKU.
Figure 3.
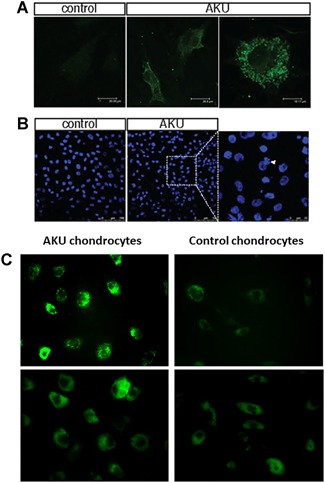
4‐HNE detection in AKU and normal chondrocytes. (A) 4HNE‐positive staining was found in AKU chondrocyte cytoplasm as assessed by confocal microscopy. (B) Nuclear fragmentation. The assessment of nuclear damage in AKU and control chondrocytes using nuclear stain DAPI was observed on confocal microscopy. AKU cell showed DNA aggregation (arrow) and more frequent smeared, aggregated, and condensed cellular DNA (arrow). (C) Visualization of fluoresceinated cadaverine indicative of apoptosis in AKU primary chondrocytes and control.
Activation of tissue transglutaminase
Extensive protein cross‐linking takes place during apoptosis. The ubiquitous TGase was identified as the enzyme responsible for this reaction. Because protein cross‐linking makes them less soluble, it is presumed that activation of TGase during apoptosis prevents a release of soluble and immunogenic proteins from dying cells and thereby decreases the possibility of inducing an autoimmune reaction. Further, protein packaging into apoptotic bodies may be facilitated when they remain in solid state rather than in solution. Here, we monitor fluoresceinated cadaverine becoming attached in situ to the protein in live AKU cells by TGase. As shown in Figure 3C, in AKU cells the accumulation of monodansylcadaverine was very evident whereas in control cells, monodansylcadaverine was faintly detected indicating that apoptosis occurs mainly in AKU cells.
SEM
SEM analysis of AKU cartilage revealed extensive fissuring, fibrillation, and clustering of cells (Fig. 4) in the deep zone of AKU cartilage.
Figure 4.

SEM image of AKU cartilage. Ultrastructural features of most damaged region of AKU cartilage showing extensive fissuring of tissue (asterisk) and evident clustering of cells with numerous clones ellipsoidal in shape (arrow). s.i.: superficial interface. Electron Dispersive X‐ray microanalysis (EDX) of mineralized areas visible in AKU cartilage are shown. Calcium and phosphate peaks were prominent and indicated that these areas were composed of calcium phosphate crystals. Table reports element composition of pigmented areas and analysis for the X‐ray spectrum of the pigment. The analysis gives the percentage by weight (wt%) and the percentage by number of atoms (at%) of each of the elements identified. The letter (K) after each element (e.g., CrK) refers to the characteristic X‐ray wavelength due to the energy released by an electron moving from one shell to an inner shell of the atom. The acronym 'ZAF' describes a procedure in which matrix corrections for atomic number effects (Z), absorption (A), and fluorescence (F), are calculated separately from suitable physical models. (The atomic number correction encompasses both the stopping power and backscattering factors).
EDS microanalyses
To get light on the composition of AKU cartilage, we performed EDS microanalyses on the surface of samples from patients 2 and 4 (Fig. 4). Results were very reproducible and revealed that pigmented areas were composed by C, O, N, S, Na, and variable amount of Ca. This mineral phase can indicate the presence of hydroxyapatite suggesting that in AKU ochronotic cartilage an active process of calcification was occurring.
TEM
To observe the ultrastructural features of chondroptotic chondrocytes and focusing on AKU chondrocytes with secretory phenotype, we performed TEM analysis using a positive OA (Fig. 5A) control and AKU cartilage arising from all AKU patients listed in Table I (Figs. 5B, 6, and 7). Figure 5A shows typical chondroptotic chondrocytes: intense vacuolization, anular chromatin condensation, organelle destruction, multiple protrusions of cell membrane, and self‐destruction through autophagic vacuoles.
Figure 5.
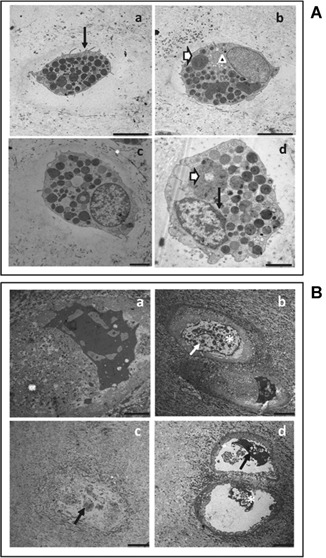
TEM observation of OA and AKU cartilage. (A) OA cartilage was adopted as positive control (Control 11, Table I). Images show chondroptotic chondrocytes, characterized by evident intensive vacuolization, multiple protrusions of cell membrane (part a, arrow), organelle destruction (parts b and d, white arrow), clusters of swollen mitochondria (part b, arrowhead), and anular chromatin condensation (part d, black arrow). Self‐destruction through autophagic vacuoles and blebbing was evident in parts a–d. Analogous results were obtained from other OA controls. (B) AKU cartilage from Patient 1 (Table I). Images show chondrocytes with secretory phenotype arising from the deep zone of pigmented cartilage (see Fig. 1B,c). Chondroptosis features were more pronounced respecting OA cartilage, suggesting a key role of HGA in the degeneration of articular tissues and cell death in AKU. Fragmented nuclear condensation was always visible with convoluted nuclei (parts b and d, asterisk), and abundant autophagic vacuoles (parts c and d, arrows). Analogous results were obtained from Patients 2, 3, 4. Bars: A,a: 25 μm; A,b,c: 10 μm; A,d: 15 μm; B,a: 10 μm; B,b,c,d: 25 μm.
Figure 7.
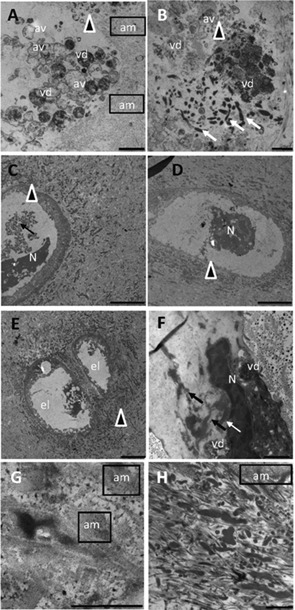
TEM observation of AKU cartilage. Images offer an overview of AKU chondroptotic chondrocytes with secretory phenotype located in the deep zone and an ultrastructural analysis of extracellular matrix where collagen disaggregation (G, H) and diffuse pigment deposition (C–H) were evident. Cellular disintegration as the final stage of chondroptosis was evident in parts A, B, and E. The cell was totally fragmented into vesicles (vd: vesicular detritus) accumulated in the extracellular matrix, and the nucleus was barely visible (A, B). Cell remnants (arrowheads) were observable in parts C–F, where glycogen was absent, nucleus (N) was still visible but as a dense central region with some vacuoles and no trace of cytoplasm and organelles. In parts B and F condensed endoplasmic reticulum and Golgi were still visible (arrows), although very deteriorated, often forming compartments within which organelles were sequestered and digested (vd). This phenomenon of degradation also took place in autophagic vacuoles (A, B, av) and was followed by the expulsion of blebs into the lacunar space. In the nucleus, chromatin was patchy condensed and spread throughout the nucleus (E, F). Finally, the end stage of chondroptosis was an empty lacuna (C, el). Amyloid (am) was clearly visible in different images of AKU cartilage, as well as ochronotic pigment patchy dispersed in the degraded cartilage matrix (G, H). Samples were from Patient 2 (A, B), Patient 3 (C, D, H), and Patient 4 (E, F, G). Bars: A: 5 μm; B: 10 μm ; C, D, E: 25 μm; F: 10 μm; G: 2.5 μm; H: 5 μm.
AKU chondrocytes with secretory phenotype were irregular and dismorphic, electron dense and characterized by an abundant RER and by prominent Golgi complexes (Fig. 5B,b–d), showing typical features of chondroptosis. Chondroptotic nuclei showed irregular outlines and contained very‐condensed chromatin (Fig. 5B,a–d), sometimes scattered within the nucleous (Fig. 6A,B). Chondroptotic AKU chondrocytes showed characteristic signs of degeneration and organelle destruction: increased amount of condensed heterochromatin in the nuclei (Fig. 7C–F), anular chromatin condensation at the nuclear envelope (Figs. 5B,b and 6B), budding of the nuclear envelope (Fig. 7B), clusters of swollen mitochondria (Fig. 7A,B), and multiple protrusions of cell membrane (Figs. 5B,c and 6A). Some chondrocytes showed shrinking and self‐destruction through autophagic vacuoles and blebbing with disruption of the cellular membrane (Fig. 7A,B). Prominent RER, fragmented and expanded Golgi membranes (Fig. 7B), dense chromatin (Fig. 7C–F), cell fragmentation and disintegration (Fig. 7A,B,E). The ultrastructural evidence of different morphological phenotypes, including chondroptosis, within clustered chondrocytes suggested that cell modifications were asynchronic and not a synchronic event. Moreover, in Figure 7G,H the ultrastructural appearence of AKU ECM is shown: patchy pigment deposition is visible and disruption of collagen fibers is evident and may be a possible precursor of osteoarthritic changes. The remarkable sparse dotted pigmentation distributed within the tissue appears to be responsible of the collagen fibers fragmentation and of the lack of their periodicity. We were also able to find trace of amyloid fibrils thinly interconnected to ECM weave (Fig. 7A,G,H), randomly diffused and interposed with pigment droplets in the cluttered and disorganized net of broken collagen fibers. The presence of amyloid in all AKU patients analyzed demonstrates the fundamental role of amyloid deposits in the triggering and worsening osteoarthrosic degeneration in AKU.
Discussion
Apoptosis is a physiological process for maintaining homeostasis in both embryogenic and adult tissue but it may also have a role in diseases involving articular cartilage degeneration, such as OA and AKU. In the past years, a different ultra‐structural pattern for dying chondrocytes articular cartilage has been documented, displaying characteristics different from conventional apoptosis and defined as chondroptosis (Roach et al., 2004; Pérez et al., 2005; Almonte‐Becerril et al., 2010).
In this work, typical chondroptotic features, like prominent Golgi and RER and autophagic destruction (Figs. 6 and 7), were observed in human chondrocytes from damaged AKU cartilage, suggesting that in AKU the development of the secretory pathway may be involved with apoptosis. This result agrees with the present tendency to emphasize the role of stress of the cell secretory machinery as an important apoptosis inductor (Ahmed et al., 2007) and suggests that Golgi and rough endoplasmic reticulum might have a key role in AKU. To the best of our knowledge, this is the first report on the occurrence of chondroptosis in AKU and the second report on the occurrence of this type of cell death after osteoarthritis (Roach et al., 2004; Pérez et al., 2005; Almonte‐Becerril et al., 2010) and one case report of traumatic injury (Sitte et al., 2009).
Chondrocyte death results in cell disintegration, associated with cytoplasmic budding, release of membrane‐bounded vesicles and, finally, fragmentation of dead cells, with some remnants containing portions of the nucleus. These cell‐derived fragments might be considered apoptotic bodies, like as those observed also in AKU chondrocytes (Figs. 6 and 7).
The remarkable morphological changes observed in AKU chondrocytes found in the most damaged tissue region might correspond to different physio‐pathological stages of AKU cartilage degeneration, shifting from a reparative to a degradation pattern, related to ECM disruption and cell death. Based on these findings, we hypothesize that normal chondrocytes (still presents in not fissuring cartilage) undergo “activation” and go through major changes leading to cells with a secretory phenotype that triggers a recovery mechanism followed by ECM degradation and chondroptosis when repair is unsuccessful.
Safranin‐O staining indicated that proteoglycan content decreased in AKU cartilage when compared to normal cartilage. Taken together, our findings indicate that cartilage degeneration in AKU follow different pathways. In AKU cartilage both severe collagen loss and proteoglycan loss occur, indicating that both collagen‐degrading enzymes and proteoglycan‐degrading enzymes are actively involved in the degenerative process.
The relationship between chondrocyte ultrastructure and the synthesis of extracellular matrix molecules has been determined. Tseche and Miosge (Tesche and Miosge, 2004) reported that fibroblast‐like chondrocytes, synthesize perlecan (a molecule involved in cartilage development and maintenance), decorin and biglycan, though not collagen type II, in areas adjacent to the main defect of the cartilage. These authors suggested that fibroblast‐like chondrocytes, which might correspond to chondrocytes with secretory phenotype reported among upper zone OA chondrocytes (Kouri et al., 1996) are involved in the regeneration of the extracellular matrix of the damaged tissue. Interestingly, upper zone chondrocytes have also been described to synthesize MMP‐3 over their inhibitor (Pelletier and Martel‐Pelletier, 1994), a cysteine protease that disrupts the ECM. This strongly suggests that during the pathogenesis of osteoarthritis, chondrocytes shift their synthesis pattern from reparative to degradative mode, which leads to cartilage breakdown. Also, apoptosis might be mediated by a reorganization of the cytoskeleton, which leads to the release of the pro‐apoptotic signal. Actually, we previously observed two different chondrocytic populations isolated from ochronotic AKU cartilage and showing distinct proteomic profiles (Braconi et al., 2012) that could be related to normal and chondrocytes with secretory phenotype here described. In agreement with chondroptosis here reported, proteomics of AKU chondrocytes revealed an over‐expression of programmed cell death 6‐interacting protein (PDC6I) and a profound alteration in the levels of proteins involved in protein folding and cell organization, cytoskeleton integrity, RER‐associated protein catabolic process, RER‐unfolded protein response, retrograde protein transport (RER to cytosol), and protein ubiquitination (Braconi et al., 2012).
Oxidative stress is at the center of AKU pathophysiological scene and ROS injury is crucial in many degenerative diseases (Bocci and Valacchi, 2013; Signorini et al., 2014); oxidative stress is also related to HGA‐induced AKU chondrocyte apoptosis and cartilage degeneration, as we recently reported (Braconi et al., 2010a,b; Tinti et al., 2010, 2011a,2011b; Braconi et al., 2012; Laschi et al., 2012; Millucci et al., 2012; Braconi et al., 2013; Spreafico et al., 2013). There is growing evidence (Armstrong, 2002; Poli et al., 2008) that HNE, generated during the LPO process and here reported to be present in AKU chondroptotic cartilage (Fig. 2), is a key mediator of oxidative stress‐induced pathophysiological effects. HNE plays a role in cartilage degradation in OA via its ability to alter cellular viability and the metabolic activity of chondrocytes (Silberberg et al., 1965; Bentz et al., 2012). All these data indicate a pivotal role of lipid peroxidation in AKU and suggest HNE implication in the induction of chondroptosis, similarly to what already observed for apoptosis in osteoarthritic cartilage (Vaillancourt et al., 2008).
Progressive loss of articular cartilage, inflammation, matrix degradation, pain, and subchondral bone remodeling are shared features between OA and AKU, whose similarity is further documented by occurrence of chondroptosis in both diseases.
Chondroptosis clearly involves autophagy, as suggested by the massive presence of autophagic vacuoles in both OA and AKU chondrocytes. At this proposal it is interesting to note that the existence of a kind of cellular death featured by autophagocytosis was known in insect metamorphosis and in ovarian atrophy (Bowen et al., 1996; D'Herde et al., 1996) and this can be the clue that autophagic cell death is an alternative to normal apoptosis when environmental clues change. This can be the case of AKU where HGA and amyloid represent a peculiar and surely abnormal milieu for the cells.
Finally, since we reported that AKU is an amyloidogenic disease (Millucci et al., 2012), at the best of our knowledge, the present work is the first associating chondroptosis and amyloidosis.
Authors' Contribution
Lia Millucci: acquisition of data, collection and assembly of data, analysis and interpretation of data, drafting the article, final approval of the version to be submitted. Alessandro Paffetti: acquisition of data, final approval of the version to be submitted. Giovanna Giorgetti: acquisition of data, final approval of the version to be submitted. Cecilia Viti: acquisition of data, final approval of the version to be submitted. Lorenzo Ghezzi: acquisition of data, final approval of the version to be submitted. Barbara Marzocchi: acquisition of data, final approval of the version to be submitted. Silvia Gambassi: acquisition of data, final approval of the version to be submitted. Daniela Braconi: acquisition of data, final approval of the version to be submitted. Giulia Bernardini: acquisition of data, final approval of the version to be submitted. Pietro Lupetti: acquisition of data, final approval of the version to be submitted. Maurizio Orlandini: acquisition of data, drafting the article, final approval of the version to be submitted. Annalisa Santucci: conception and design of the study, acquisition of data, analysis and interpretation of data, drafting the article, revising it critically for important intellectual content, obtaining of funding, final approval of the version to be submitted.
Acknowledgements
The authors thank alkaptonuric patients who generously donated samples for the present study, AimAKU (Associazione Italiana Malati di Alcaptonuria, ORPHA263402), Toscana Life Sciences Orphan_1 project and Fondazione Monte dei Paschi di Siena. The authors also thank E. Vannuccini and E. Paccagnini for their technical assistance. This work has been supported by Telethon Italy grant GGP10058. Telethon Italy had no involvement in the study design, collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
Conflicts of interest: None.
Literature Cited
- Ahmed YA, Tatarczuch L, Pagel CN, Davies HM, Mirams M, Mackie EJ. 2007. Physiological death of hypertrophic chondrocytes. Osteoarthritis Cartilage 15:575–586. [DOI] [PubMed] [Google Scholar]
- Almonte‐Becerril M, Navarro‐Garcia F, Gonzalez‐Robles A, Vega‐Lopez MA, Lavalle C, Kouri JB. 2010. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of osteoarthritis within an experimental model. Apoptosis 15:631–638. [DOI] [PubMed] [Google Scholar]
- Armstrong D. 2002. Methods in molecular biology. Florida: Humana Press; 356 p. [Google Scholar]
- Bentz M, Zaouter C, Shi Q, Fahmi H, Moldovan F, Fernandes JC, Benderdour M. 2012. Inhibition of inducible nitric oxide synthase prevents lipid peroxidation in osteoarthritic chondrocytes. J Cell Biochem 113:2256–2267. [DOI] [PubMed] [Google Scholar]
- Bocci V, Valacchi G. 2013. Free radicals and antioxidants: How to reestablish redox homeostasis in chronic diseases. Curr Med Chem. 20:3397–3415. [DOI] [PubMed] [Google Scholar]
- Bowen ID, Mullarkey K, Morgan SM. 1996. Programmed cell death during metamorphosis in the blow‐fly Calliphora vomitoria. Microsc Res Tech 34:202–217. [DOI] [PubMed] [Google Scholar]
- Braconi D, Bernardini G, Bianchini C, Laschi M, Millucci L, Amato L, Tinti L, Serchi T, Chellini F, Spreafico A, Santucci A. 2012. Biochemical and proteomic characterization of alkaptonuric chondrocytes. J Cell Physiol 227:3333–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braconi D, Laschi M, Amato L, Bernardini G, Millucci L, Marcolongo R, Cavallo G, Spreafico A, Santucci A. 2010. Evaluation of anti‐oxidant treatments in an in vitro model of alkaptonuric ochronosis. Rheumatology 49:1975–1983. [DOI] [PubMed] [Google Scholar]
- Braconi D, Laschi M, Taylor AM, Bernardini G, Spreafico A, Tinti L, Gallagher JA, Santucci A. 2010. Proteomic and redox‐proteomic evaluation of homogentisic acid and ascorbic acid effects on human articular chondrocytes. J Cell Biochem 111:922–932. [DOI] [PubMed] [Google Scholar]
- Braconi D, Millucci L, Ghezzi L, Santucci A. 2013. Redox proteomics gives insights into the role of oxidative stress in alkaptonuria. Expert Rev Proteomics 10:521–535. [DOI] [PubMed] [Google Scholar]
- D'Herde K, De Prest B, Roels F. 1996. Subtypes of active cell death in the granulosa of ovarian atretic follicles in the quail (Coturnix coturnix japonica). Reprod Nutr Dev 36:175–189. [DOI] [PubMed] [Google Scholar]
- Hayami T, Pickarski M, Wesolowski GA, McLane J, Bone A, Destefano J, Rodan GA, Duong le T. 2004. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum 50:1193–1206. [DOI] [PubMed] [Google Scholar]
- Kouri JB, Argüello C, Quintero M, Chico A, Ramos ME. 1996. Variability in the cell phenotype of aggregates or “clones” of human osteoarthritic cartilage. A case report. Biocell 3:191–200. [PubMed] [Google Scholar]
- Laschi L, Tinti L, Braconi D, Millucci L, Ghezzi L, Amato L. 2012. Homogentisate 1,2 dioxygenase is expressed in human osteoarticular cells: Implications in alkaptonuria. J Cell Physiol 227:3254–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Yin J, Gao J, Cheng TS, Pavlos NJ, Zhang C, Zheng MH. 2013. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res Ther 15:‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millucci L, Ghezzi L, Braconi D, Laschi M, Geminiani M, Amato L, Orlandini M, Benvenuti C, Bernardini G, Santucci A. 2014. Secondary amyloidosis in an alkaptonuric aortic valve. Int J Cardiol 172:e121–e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millucci L, Ghezzi L, Paccagnini E, Giorgetti G, Viti C, Braconi D, Laschi M, Geminiani M, Soldani P, Lupetti P, Orlandini M, Benevenuti C, Perfetto F, Spreafico A, Bernardini G, Santucci A. 2014. Amyloidosis, inflammation, and oxidative stress in the heart of an alkaptonuric patient. Med Inflamm 2014:258471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millucci L, Spreafico A, Tinti L, Braconi D, Ghezzi L, Paccagnini E, Bernardini G, Amato L, Laschi M, Selvi E, Galeazzi M, Mannoni A, Benucci M, Lupetti P, Chellini F, Orlandini M, Santucci A. 2012. Alkaptonuria is a novel human secondary amyloidogenic disease. Biochim Biophys Acta 1822:1682–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JP, Martel‐Pelletier J. 1994. Role of synovial inflammation, cytokines and IGF‐1 in the physiopathology of osteoarthritis. Rev Rhum Ed Fr 61:103S–108S. [PubMed] [Google Scholar]
- Pérez HE, Luna MJ, Rojas ML, Kouri JB. 2005. Chondroptosis: An immunohistochemical study of apoptosis and Golgi complex in chondrocytes from human osteoarthritic cartilage. Apoptosis 10:1105–1110. [DOI] [PubMed] [Google Scholar]
- Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 2008. 4‐Hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med Res Rev 28:569–631. [DOI] [PubMed] [Google Scholar]
- Pompella A, Dominici S, Frank J, Biesalski HK. 2002. Indirect immunofluorescence detection of protein‐bound 4‐hydroxynonenal in tissue sections and isolated cells. Methods Mol Biol 196:41–46. [DOI] [PubMed] [Google Scholar]
- Roach HI, Aigner T, Kouri JB. 2004. Chondroptosis: A variant of apoptotic cell death in chondrocytes. Apoptosis 9:265–277. [DOI] [PubMed] [Google Scholar]
- Signorini C, Leoncini S, De Felice C, Pecorelli A, Meloni I, Ariani F, Mari F, Amabile S, Paccagnini E, Gentile M, Belmonte G, Zollo G, Valacchi G, Durand T, Galano JM, Ciccoli L, Renieri A, Hayek J. 2014. Redox imbalance and morphological changes in skin fibroblasts in typical Rett syndrome. Oxid Med Cell Longev 2014:195935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg M, Silberberg R, Orcutt B. 1965. Modifying effect of linoleic acid on articular aging and osteoarthrosis in lard‐fed mice. Gerontologia 11:179–187. [DOI] [PubMed] [Google Scholar]
- Sitte I, Kathrein A, Pfaller K, Pedross F, Roberts S. 2009. Intervertebral disc cell death in the porcine and human injured cervical spine after trauma: A histological and ultrastructural study. Spine (Phila Pa 1976) 34:131–140. [DOI] [PubMed] [Google Scholar]
- Spreafico A, Millucci L, Ghezzi L, Geminiani M, Braconi D, Amato L, Chellini F, Frediani B, Moretti E, Collodel G, Bernardini G, Santucci A. 2013. Antioxidants inhibit SAA formation and pro‐inflammatory cytokine release in a human cell model of alkaptonuria. Rheumatology 52:1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Batchelor TJ, Adams VL, Helliwell TR, Gallagher JA, Ranganath LR. 2011b. Ochronosis and calcification in the mediastinal mass of a patient with alkaptonuria. J Clin Pathol 64:935–936. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Boyde A, Wilson PJ, Jarvis JC, Davidson JS, Hunt JA, Ranganath LR, Gallagher JA. 2011a. The role of calcified cartilage and subchondral bone in the initiation and progression of ochronotic arthropathy in alkaptonuria. Arthritis Rheum 12:3887–3896. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Wlodarski B, Prior IA, Wilson PJ, Jarvis JC, Ranganath LR, Gallagher JA. 2010. Ultrastructural examination of tissue in a patient with alkaptonuric arthropathy reveals a distinct pattern of binding of ochronotic pigment. Rheumatology 49:1412–1414. [DOI] [PubMed] [Google Scholar]
- Tesche F, Miosge N. 2004. Perlecan in late stages of osteoarthritis of the human knee joint. Osteoarthritis Cartilage 12:852–862. [DOI] [PubMed] [Google Scholar]
- Tinti L, Spreafico A, Braconi D, Millucci L, Bernardini G, Chellini F, Cavallo G, Selvi E, Galeazzi M, Marcolongo R, Gallagher JA, Santucci A. 2010. Evaluation of antioxidant drugs for the treatment of ochronotic alkaptonuria in an in vitro human cell model. J Cell Physiol 225:84–91. [DOI] [PubMed] [Google Scholar]
- Tinti L, Spreafico A, Chellini F, Galeazzi M, Santucci A. 2011a. A novel ex vivo organotypic culture model of alkaptonuria‐ochronosis. Clin Exp Rheumatol 29:693–696. [PubMed] [Google Scholar]
- Tinti L, Taylor AM, Santucci A, Wlodarski B, Wilson PJ, Jarvis JC, Fraser WD, Davidson JS, Ranganath LR, Gallagher JA. 2011b. Development of an in vitro model to investigate joint ochronosis in alkaptonuria. Rheumatology 50:271–277. [DOI] [PubMed] [Google Scholar]
- Vaillancourt F, Fahmi H, Shi Q, Lavigne P, Ranger P, Fernandes JC, Benderdour M. 2008. 4‐Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: The protective role of glutathione‐S‐transferase. Arthritis Res Ther 10:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamli Z, Sharif M. 2011. Chondrocyte apoptosis: A cause or consequence of osteoarthritis. Int J Rheum Dis 14:159–166. [DOI] [PubMed] [Google Scholar]