

Abstract
Tumor relapse after chemotherapy-induced regression is a major clinical problem, because it often involves inoperable metastatic disease. Tumor-associated macrophages (TAM) are known to limit the cytotoxic effects of chemotherapy in preclinical models of cancer. Here, we report that an alternatively activated (M2) subpopulation of TAMs (MRC1+TIE2HiCXCR4Hi) accumulate around blood vessels in tumors after chemotherapy, where they promote tumor revascularization and relapse, in part, via VEGF-A release. A similar perivascular, M2-related TAM subset was present in human breast carcinomas and bone metastases after chemotherapy. Although a small proportion of M2 TAMs were also present in hypoxic tumor areas, when we genetically ablated their ability to respond to hypoxia via hypoxia-inducible factors 1 and 2, tumor relapse was unaffected. TAMs were the predominant cells expressing immunoreactive CXCR4 in chemotherapy-treated mouse tumors, with the highest levels expressed by MRC1+ TAMs clustering around the tumor vasculature. Furthermore, the primary CXCR4 ligand, CXCL12, was upregulated in these perivascular sites after chemotherapy, where it was selectively chemotactic for MRC1+ TAMs. Interestingly, HMOX-1, a marker of oxidative stress, was also upregulated in perivascular areas after chemotherapy. This enzyme generates carbon monoxide from the breakdown of heme, a gas known to upregulate CXCL12. Finally, pharmacologic blockade of CXCR4 selectively reduced M2-related TAMs after chemotherapy, especially those in direct contact with blood vessels, thereby reducing tumor revascularization and regrowth. Our studies rationalize a strategy to leverage chemotherapeutic efficacy by selectively targeting this perivascular, relapse-promoting M2-related TAM cell population.
Introduction
The regrowth of tumors after treatment with cytotoxic agents poses a major threat to survival in cancer patients, particularly those with inoperable primary and/or metastatic tumors as they often rely heavily on chemotherapy to slow tumor growth and reduce its burden. The development of resistance results in poor survival rates for patients with, for example, inoperable pancreatic or lung cancer, who often survive for less than 12 months after diagnosis (1, 2). New therapeutic strategies are, therefore, urgently needed to delay or prevent tumor regrowth after early cycles of chemotherapy as these would extend life.
Malignant tumors contain various CD11b+ myeloid cells, including granulocytes, myeloid-derived suppressor cells (MDSC), and tumor-associated macrophages (TAM; ref. 3). The latter are recruited as monocytes from the peripheral blood, which are themselves derived from progenitor cells in the bone marrow (4, 5). After entry into tumors, monocytes differentiate into macrophages (6) and promote tumor progression by stimulating tumor invasion, neovascularization and metastasis, and suppressing antitumor immunity (5, 7). TAMs express a broad spectrum of activation states between the two extreme forms of “classical” (M1) and “alternative”’ (M2) activation, with a tendency toward the latter (8). M2-skewed TAMs are characterized by their upregulation of various receptors, including the mannose receptor C-type lectin (MRC1/CD206) and the angiopoietin receptor, TIE2 (9). Indeed, TAMs expressing high levels of these two receptors have been shown to play an essential role in promoting angiogenesis in untreated mouse tumors (10).
A variety of anticancer therapies have been shown to stimulate the recruitment of CD11b+ myeloid cells by mouse tumors (11, 12). For example, TAMs accumulate in mouse tumors after chemotherapy (13-15), ionizing radiation (16-18), and the vascular disrupting agent combretastatin-A4-P (CA-4-P; ref. 19). Importantly, the mononuclear phagocyte growth factor CSF1 (13) and chemokines CCL2 (14) and CXCL12 (16, 19) are increased in tumors after such anticancer therapies, and can trigger monocyte recruitment (4, 5). These cells then reduce the efficacy of chemotherapy by limiting vascular permeability via their expression of MMP9 (14), promoting resistance to therapy-induced death via their expression of cathepsin serine proteases (15) and by suppressing the recruitment/activation of cytotoxic T cells (12, 13).
Considerable evidence has emerged recently for M2-activated macrophages playing an important role in repair and remodeling after tissue injury. For example, they are prominent in diseased tissues in spinal cord injury, myocardial infarction, and various forms of renal disease, (20). Furthermore, TAMs with similar phenotypes have been implicated in tumor relapse after therapies like irradiation and CA-4-P (17, 19). Although MRC1+ TAMs are increased in MMTV-PyMT mammary tumors after doxorubicin treatment (14), the role of M2 TAMs in tumor relapse after chemotherapy has not been defined.
Our studies show that MRC1+ TAMs are elevated in mouse tumors after treatment with various chemotherapeutic agents. Moreover, this TAM subset was further defined as MRC1HiTIE2HiCXCR4HiVEGFA+ and shown to accumulate preferentially in vascularized, CXCL12-rich regions of tumors after chemotherapy. Blockade of CXCR4 signaling prevented this close association with the tumor vasculature after chemotherapy, resulting in a marked delay in subsequent tumor revascularization and relapse. These findings suggest that selective targeting of vessel-associated, M2-skewed TAMs after chemotherapy could increase the relapse-free survival of cancer patients.
Materials and Methods
Mouse studies
To investigate the mechanisms regulating tumor relapse after chemotherapy, we primarily used the Lewis lung carcinoma model (21) and the MMTV-PyMT model of breast cancer (15). These syngeneic tumor models respond to chemotherapy with an initial phase of tumor growth inhibition, followed by a distinct regrowth phase. Transgenic tumor models were not considered suitable as their responses to some cytotoxic agents can be so minimal that a relapse phase is not evident (13, 22). Furthermore, tumors in these models are often multifocal making the kinetics of tumor relapse difficult to assess accurately.
Our mouse studies were conducted in accordance with either UK Home Office regulations (C.E. Lewis/M. Muthana/R. Hughes), the Veterinary Authorities of the Canton Vaud (M. De Palma), the institutional standards of the Research Animals Resource Centre at Memorial Sloan Kettering Cancer Centre (J.A. Joyce; New York, NY), and the Albert Einstein College of Medicine Animal Use Committee (J.W. Pollard). LLC1/cyclophosphamide studies using 8-week-old C57BL/6 mice were performed as described previously (21). For LLC1/AMD3100 studies, AMD3100 was given concurrently (10 mg/kg) with cyclophosphamide, i.p. every day for 7 days. Tumors and organs were harvested at the indicated times. Mice were treated i.p. with pimonidazole (60 mg/kg) and bromodeoxyuridine (BrdUrd; 100 mg/kg) 2 hours before sacrifice to label hypoxia and proliferating cells, respectively.
4T1/paclitaxel studies
Female Balb/c mice (>8 weeks) were orthotopically implanted with 1 × 106 4T1 murine mammary adenocarcinoma cells. Tumors were grown for 10 days before mice were injected i.p. with either cremaphore or cremophore + paclitaxel (10 mg/kg) every 5 days for a total of three doses, the mice were sacrificed and tissues harvested 4 days after the last treatment. Orthotopic MMTV-PyMT/doxorubicin studies were performed as previously described (15). Mice bearing implanted tumors were injected i.v. with a single dose of doxorubicin (5 mg/kg) when at 250 mm3 and sacrificed 7 days after therapy.
Monocyte/macrophage-specific ablation of Vegfa studies
Female Tg(Csf1r-Mer-iCre-Mer)1jwp; Vegfafl/fl) mice (>8 weeks), shown to specifically lack the expression of VEGFA in the monocyte/macrophage lineage after treatment with tamoxifen (23), or Cre-negative litter mates (ages 8 weeks+), were orthotopically implanted with the syngeneic MMTV-PyMT cell line F246-6. Vegfa floxed mice were the kind gift of Dr. Naploeone Ferrar, Genentech. Mice with established tumors were treated with a single i.v. injection of either vehicle (PBS) or doxorubicin (5 mg/kg) and 24 hours later with tamoxifen via their food (3 mg/20 g body weight/day). Treatment with tamoxifen was then maintained throughout tumor regrowth. Tumor growth was monitored by caliper measurements. In the LLC1/TAM adoptive transfer studies, C57BL/6 mice implanted with LLCs and treated with cyclophosphamide as outline above, 48 hours after the last dose of cyclophosphamide, donor mice were sacrificed, their tumors removed, enzymatically dissociated, and MRC1Hi and MRC1Lo TAMs were purified by flow cytometry. Recipient mice were randomized into three groups and received either 50 × 103 MRC1Hi TAMs, 50 × 103 MRC1Lo TAMs, or saline via a 25 μL intratumoral injection. After injection of these cells or saline, tumor regrowth was then monitored.
Human breast carcinomas and metastatic bone lesions
Four patients with newly diagnosed breast cancer underwent neoadjuvant chemotherapy as previously described (24). Residual tumor tissues were collected at the time of surgery (which took place 21–28 days after the last dose of paclitaxel). Biopsies of bone metastatic lesions were also obtained from five advanced breast cancer patients. All biopsies were obtained under informed consent following procedures approved by the Ottawa Health Sciences Research Ethics Board. Samples were collected as previously described (25). All biopsies were taken within 6 to 15 days of treatment with either chemotherapy alone (paclitaxel, docetaxel, or 5-fluoruracil/doxorubicin/cyclophosphamide) or without pamidronate.
Immunofluorescence and IHC studies
Frozen tumor sections were blocked with 1% BSA and 5% goat serum for 30 minutes and incubated with various primary antibodies (Supplementary Table S1), for 1 hour at room temperature. Alexa fluor–conjugated anti-rabbit or anti-rat secondary antibodies were used with unconjugated primary antibodies. For BrdUrd staining, DNA in frozen tissue sections was denatured with 2NHCl for 15 minutes before immunostaining, as described above. Nuclei in all tumor sections were counterstained with DAPI. Formalin-fixed, paraffin-embedded tissues were rehydrated, peroxidase blocked, then antigen retrieved, serum blocked, and incubated with primary antibodies for 1 to 2 hours. Primary antibodies were detected with appropriate ABC or Polymer detection kits followed by chromagen staining with DAB.
Distribution of MRC1+ TAMs relative to blood vessels in human primary breast tumors
The number of MRC1+ TAMs present within a 250-μm radius of a given blood vessel was counted in six randomly selected areas per tumor section. Then, within this 250 μm radius, the distance of each MRC1+ TAM was measured to the nearest vessel and the number of MRC1+ TAMs within or beyond 150 μm from that vessel was expressed as a% MRC1+ TAMs within the region being analyzed. These were defined as “perivascular” or “avascular” MRC1+ TAM subsets, respectively.
Tumor dissociation/flow-cytometric studies
Tumors were enzymatically dissociated and labeled with antibodies as described previously (9). All antibody incubations were performed for 1 hour at 4°C. Cell staining analyses and cell sorting experiments were performed using BD LSRII and BD FACSAria, respectively.
VEGFA ELISA studies
CD45+CD11b+Ly6G−F4/80+ TAMs were isolated from three pooled, dissociated control, and cyclophosphamide-treated LLC1 tumors. One hundred thousand sorted cells were seeded in 100 μL of medium and cultured overnight. Conditioned medium was examined for VEFGA release using a mouse VEGFA Quantikine Sandwich ELISA (R&D Systems).
Chemotaxis assay
MRC1+/Hi (CD11b+Ly6G−F4/80+MRC1Hi) and MRC1−/Lo (CD11b+Ly6G−F4/80+MRC1Hi) TAMs were FACSorted from dissociated cyclophosphamide-treated LLC1 tumors and seeded into Transwell inserts (4 μm pores, VWR International), 50 × 103 cell per well. Lower chambers contained either medium alone, medium supplemented with 10 nmol/L BSA or 10 nmol/L recombinant murine CXCL12 (100 ng/mL). After an incubation of 6 hours at 37°C, the chambers were dissassembled and the membranes stained with crystal violet. The upper surface of each Transwell insert was scraped to remove non-migrated cells before quantification.
Quantification of spontaneous LLC1 lung metastases
Sections (4 μm thick) were cut from formalin-fixed, paraffin-embedded lungs and stained with haemotoxylin and eosin. These stained sections were imaged using the Aperio slide scanner (Leica Biosystems), and the number of metastases per section and total metastatic area quantified using ImageScope analysis software (Leica Biosystems).
Statistical analysis
All data represent mean values ± SEM. P values of less than 0.05 were considered to be significant. All statistical comparisons were made using the nonparametric Mann–Whitney U test (paired or unpaired as appropriate).
Results
M2-skewed (MRC1+TIE2HIVEGFA+) TAMs are abundant in mouse tumors after chemotherapy and promote their relapse
Three i.p. injections of the cytotoxic agent, cyclophosphamide, resulted in the complete cessation of LLC1 tumor growth, followed by regrowth beginning 7 days after treatment stops (Fig. 1A, left). Forty-eight hours after the last cyclophosphamide injection (day 6), LLC1s contained significantly (P < 0.05) shorter blood vessels and more hypoxia than size-matched controls (Supplementary Fig. S1A). Consistent with earlier observations for MMTV-PyMT implants treated with paclitaxel (13), TAMs were enriched among those leukocytes present in LLC1s 48 hours after cyclophosphamide (Supplementary Fig. S1B). In addition, there was a significant (P < 0.05) increase in the overall number of F4/80+ TAMs in cyclophosphamide-treated LLCs compared with size-matched controls. This was also seen in paclitaxel-treated, orthotopic 4T1 tumors, and doxorubicin-treated ortho-topic MMTV-PyMT implants (Supplementary Fig. S1C).
Figure 1.
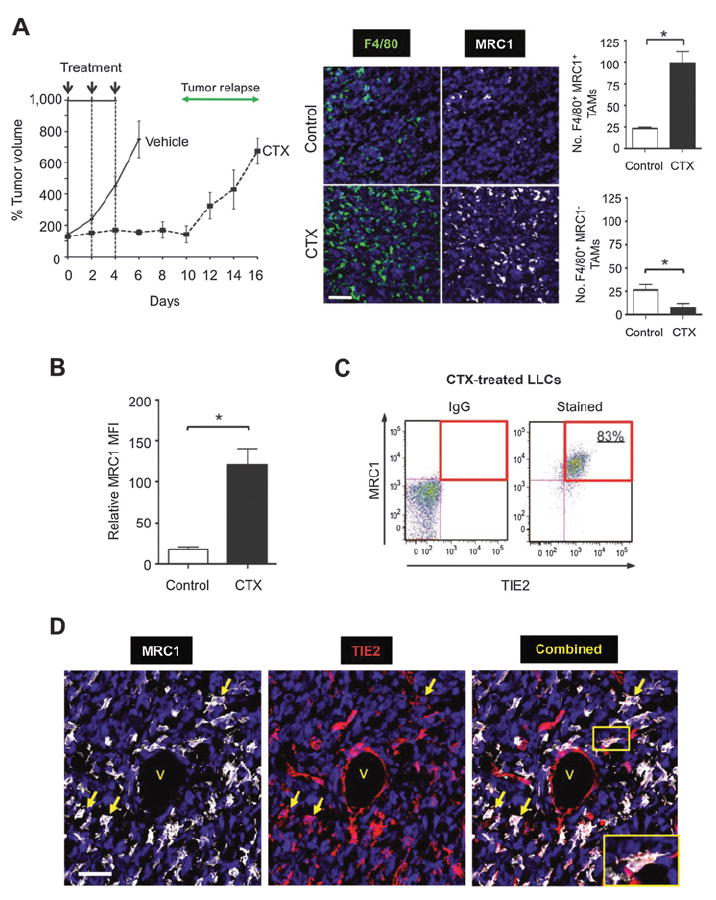
Effects of cyclophosphamide (CTX) on tumor growth and accumulation of M2 TAMs in LLC1 tumors. A and B, growth kinetics of LLC1s after three i.p. injections with either 150 mg/kg cyclophosphamide or PBS (left; A), the number of MRC1+ or MRC1− F4/80+ TAMs (right; n = 7–8/group), and their cell-surface expression of MRC1 (B), as detected by flow cytometry (n = 5–6/group), 2 days after the last dose of cyclophosphamide (day 6, i.e., before the regrowth phase). C, flow-cytometric analysis of TIE2 and MRC1 on TAMs from cyclophosphamide-treated tumors. D, colocalization of MRC1 and TIE2 on TAMs in cyclophosphamide-treated tumors (see yellow arrows and inset for dual-stained cells). V, vessel. Bars, 50 μm. *, P < 0.05.
Cyclophosphamide treatment of LLC1s resulted in a significant (P < 0.05) increase in the number of F4/80+ TAMs expressing the M2-marker, MRC1, relative to size-matched controls. In contrast, there was a significant (P < 0.05) drop in F4/80+/MRC1− TAMs after cyclophosphamide (Fig. 1A, right). Also, TAMs from cyclophosphamide-treated LLC1s expressed higher surface MRC1 than those from size-matched controls (Fig. 1B). Consistent with previous findings in untreated mouse tumors (9, 10), MRC1Hi TAMs in cyclophosphamide-treated LLC1s coexpressed elevated TIE2 (Fig. 1C and D). A similar increase in MRC1+ TAMs was also seen in orthotopic 4T1 and MMTV-PyMT implants after paclitaxel and doxorubicin, respectively (Supplementary Fig. S1D). In all three models, the vast majority of MRC1+ cells F4/80+ TAMs, and the small number of F4/80−MRC1+ (presumably dendritic) cells were not significantly increased after treatment with cyclophosphamide, paclitaxel, or doxorubicin (Supplementary Fig. S2A).
BrdUrd uptake was negligible in MRC1+ TAMs in both size-matched control and cyclophosphamide-treated LLC1 tumors, indicating their nonproliferative status (Supplementary Fig. S2B). As chemotherapy-induced changes in circulating hematopoietic stem and progenitor cells (HS/PCs) could, in theory, contribute to the increase of TAM numbers after therapy, we also performed dual immunofluorescence labeling for HS/PC markers, c-Kit and Sca1. Although c-Kit+Sca1+ cells were detected in the spleens of LLC1 tumor-bearing mice (positive control for the staining), no such cells were detected in either control or cyclophosphamide-treated LLC1s (Supplementary Fig. S2C). These data indicate that the chemotherapy-induced increase in TAMs is most likely to be due to increased monocyte recruitment rather than the proliferation of existing TAMs or their differentiation from recruited HS/PCs.
We then isolated F4/80+MRC1+/Hi and F4/80+MRC1−/Lo TAMs from cyclophosphamide-treated LLC1 tumors by FACS and injected them into cyclophosphamide-treated LLC1 tumors in littermates (Fig. 2A). MRC1+/Hi TAMs, but not MRC1−/Lo TAMs from treated tumors or TAMs from vehicle-treated tumors, accelerated tumor regrowth after cyclophosphamide. This was accompanied by a significant (P < 0.05) increase in the number of MRC1+ TAMs, CD31+ blood vessels, and a moderate increase in BrdUrd+ (proliferating) cells in cyclophosphamide-treated LLC1s receiving MRC1+/Hi TAMs. Very few (< 1%) of CD45+ leukocytes contained immunodetectable BrdUrd (Fig. 2B–E).
Figure 2.
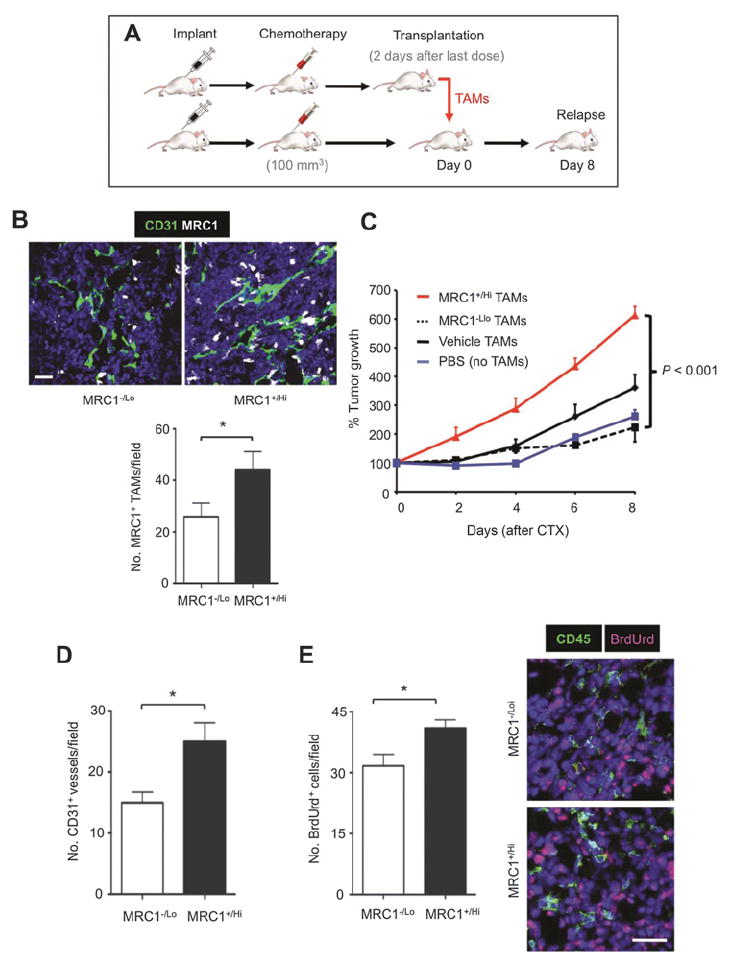
Effects of adoptive transfer of MRC1Hi vs MRC1Lo TAMs on LLC1 relapse after cyclophosphamide (CTX). A, design schematic of the TAM transfer experiment. B, quantification of MRC1+ TAMs in tumors receiving adoptive transfer of MRC1−/Lo or MRC1+/Hi TAMs. C, LLC1 regrowth after the last cyclophosphamide injection (day 0). D and E, vascularization (D) and proliferation (E) in LLC1 tumors receiving MRC1Hi or MRC1Lo (n = 5/group). Costaining of cyclophosphamide-treated LLC1 tumor sections with antibodies against the pan-leukocyte marker (CD45) and BrdUrd. Fewer than 1% of proliferating cells were leukocytes in either group. *, P < 0.05. Bars, 50 μm.
MRC1+ TAMs are proangiogenic in untreated tumors and express the important proangiogenic mediator VEGFA (9, 26). Moreover, myeloid-specific deletion of VEGFA is known to have profound effects on the vascularization and progression of untreated mouse tumors (27). We, therefore, investigated the role of VEGFA derived from MRC1+ TAMs in tumor-relapse after chemotherapy. As reported previously in mouse tumors (25), VEGFA was expressed by both TAMs and other (F4/80−) cells in control LLC1s (Fig. 3A and Supplementary Fig. S3A). However, within 48 hours of the last dose of cyclophosphamide, VEGFA was expressed almost exclusively by MRC1+ TAMs (Supplementary Fig. S3B). As the expression of VEGFA was closely associated with MRC1+ TAMs, and because the latter were more abundant in cyclophosphamide-treated LLCs (Fig. 3B and C), we postulated that the macrophage population as a whole found within cyclophosphamide-treated LLCs might be capable of producing higher levels of VEGFA. Indeed, the release of VEGFA was found to be significantly greater for TAMs sampled from cyclophosphamide-treated LLCs (Fig. 3D).
Figure 3.
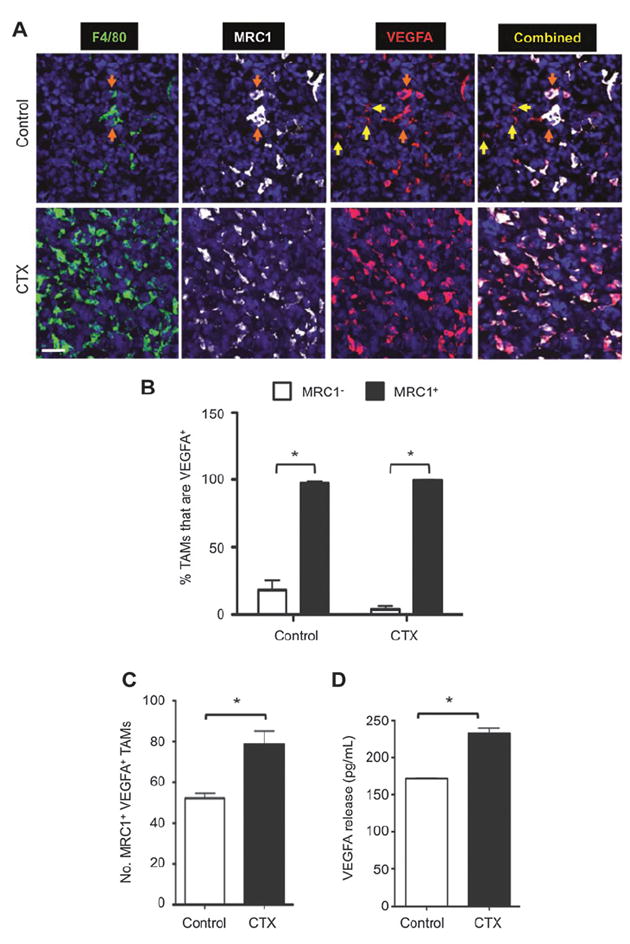
Effect of cyclophosphamide (CTX) on the number of MRC1+ VEGFA+ TAMs in LLC1 tumors and VEGFA release by TAMs in vitro. A, representative immunostaining for F4/80+, MRC1+, and VEGFA+. VEGF-expressing F4/80− cells (yellow arrows) and F4/80+ TAMs (orange arrows). B, VEGFA colocalization with MRC1 in TAMs. Bar, 50 μm. C, number of MRC1+ VEGFA+ TAMs. D, VEGFA release by CD45+CD11b+Ly6G−F4/80+ TAMs (VEGFA release in medium over 16 hours, standardized by live TAM numbers). *, P < 0.05.
MDSCs (CD11b+Gr1+) in tumor-bearing mice are also reported to express MRC1 raising the possibility that some VEGFA-expressing MRC1+ cells in cyclophosphamide-treated tumors might have been MDSCs (28). However, we found that both granulocytic (CD45+CD11b+Ly6G+) and monocytic (CD45+CD11b+Ly6G−Ly6C+) MDSC subsets were depleted in cyclophosphamide-treated tumors, and that the expression of MRC1 on monocytic MDSCs was significantly (P < 0.05) lower than that on TAMs (Supplementary Fig. S3C). Taken together, these data suggest that MDSCs are unlikely to represent a significant proportion of the MRC1Hi cells in our LLC1 tumors.
We previously showed that a subset of TAMs accumulates in hypoxic areas of untreated mouse and human tumors (29), where they respond to hypoxia by upregulating hypoxia-inducible factors (HIF) 1 and 2 and a wide array of HIF-dependent M2 genes (30). As we detected some MRC1+ TAMs in the hypoxic regions of cyclophosphamide-treated LLC1 tumors (Supplementary Fig. S4A), albeit at much lower numbers than in the well-vascularized, normoxic (PIMO−) regions (Fig. 4A), we investigated the possibility that hypoxia-regulated transcriptional programming (via HIFs 1 and 2) might contribute to the relapse-promoting functions of TAMs. LLC1s were implanted into LysMCre/+HIF1fl/fl or LysMcre/+HIF2fl/fl myeloid-specific knockout mice and treated with vehicle or cyclophosphamide. HIF knockout was seen to have no effect on the number of MRC1+ TAMs in either the hypoxic or PIMO− areas in cyclophosphamide-treated LLC1s, or on tumor relapse (Supplementary Fig. S4B and S4C). IHC staining demonstrated a reduction of >90% in TAMs expressing HIF1α or 2α in control LLC1s, confirming high efficiency of LysCre-targeted HIF allele ablation in our mice (Supplementary Fig. S4D). These data suggest that hypoxia and HIF1/2-regulated transcriptional programming do not regulate the relapse-promoting functions of MRC1+ TAMs.
Figure 4.
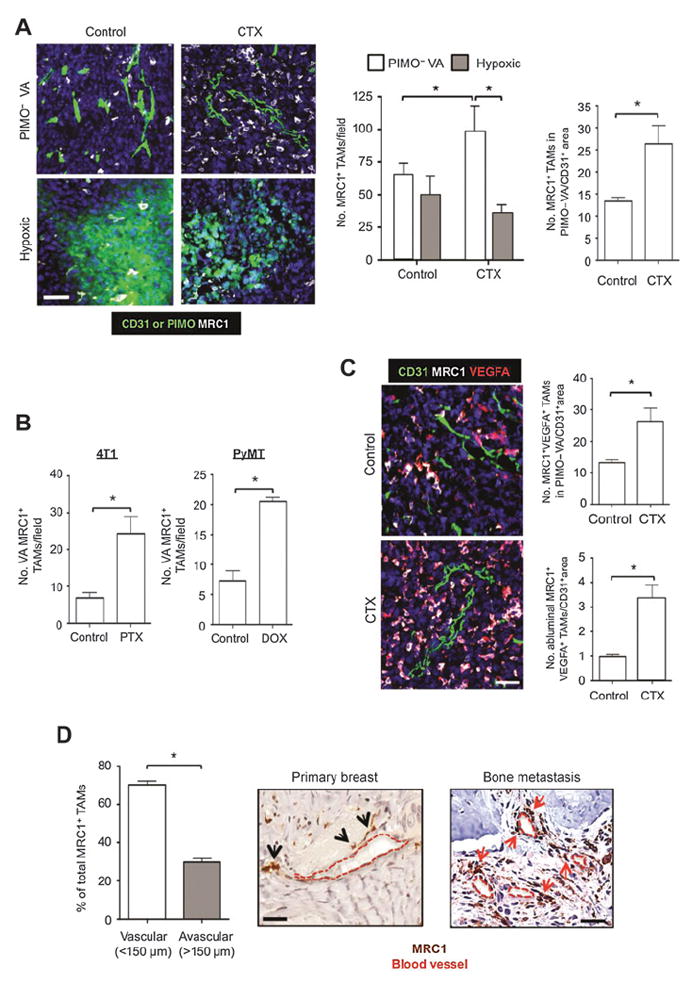
Effect of cytotoxic drugs on the distribution of MRC1+ TAMs in mouse and human tumors: colocalization with VEGFA. A, immunostaining of MRC1+ TAMs in PIMO−VA and hypoxic (PIMO+) areas of LLC1 tumors 48 hours after last injection of cyclophosphamide (CTX; left and middle), and their abundance in PIMO− VA areas normalized by CD31+ area (right; n = 4/group). B, MRC1+ TAM accumulation in vessel-associated (VA) areas in 4T1 (n = 8–9/group) and MMTV-PyMT tumor implants after treatment with paclitaxel (PTX) or doxorubicin (DOX) respectively (n = 7/group). C, overall number of MRC1+VEGFA+ TAMs in PIMO− VA (top) and the number in direct contact with CD31+ vessels (abluminal; bottom) in control and cyclophosphamide-treated LLCs 48 hours after the final dose of cyclophosphamide (both normalized by total CD31+ area). D, MRC1+ TAMs in vascular or avascular areas of human primary carcinomas 3 weeks after three cycles of paclitaxel treatment (black arrows; n = 4 biopsies). MRC1+ macrophages near vessels in bone metastases from patients with advanced breast cancer after treatment (red arrows; n = 4 biopsies). Bars, 50 μm. *, P < 0.05.
MRC1+ TAMs accumulate in well-vascularized areas of tumors after chemotherapy in both mouse and human tumors and promote tumor relapse: role of VEGFA
Previous studies have shown that MRC1+ TAMs can reside close to blood vessels in untreated mouse tumors (10). Furthermore, the aforementioned HIF knockout study demonstrated an increase in MRC1+ TAMs in PIMO− vascularized areas (PIMO− VA) in cyclophosphamide-treated tumors (Supplementary Fig. S4B). We, therefore, conducted a more detailed immunofluorescence staining analysis of the distribution of these cells in control and cyclophosphamide-treated LLC1s. This showed that, although MRC1+ TAMs were evenly distributed between PIMO− VA and PIMO+ hypoxic areas in size-matched control tumors, there was a significant (P < 0.05) increase in the number of F4/80+MRC1+ TAMs in the former in cyclophosphamide-treated LLC1s (Fig. 4A). As cyclophosphamide treatment also resulted in reduced tumor vascularity (Supplementary Fig. S1A), we normalized MRC1+ TAM numbers to CD31+ area in PIMO− VA of control and cyclophosphamide-treated LLC1s. Interestingly, this showed that the increase in MRC1+ TAMs in PIMO− VA is independent of changes in the vasculature (Fig. 4A, far right). Further immunofluorescent analysis of 4T1 and PyMT-MMTV tumors demonstrated a similar increase in the number of vessel-associated MRC1+ TAMs after treatment with paclitaxel and doxorubicin, respectively (Fig. 4B). Furthermore, because the majority of MRC1+ TAMs coexpressed VEGFA in both control and cyclophosphamide-treated LLCs, this meant that there was also a significant increase in VEGFA+ TAMs in perivascular areas (Fig. 4C, top). Interestingly, there was also a significant (P < 0.05) increase in the number of MRC1+VEGFA+ TAMs in direct contact with the abluminal surface of the tumor vasculature in cyclophosphamide-treated tumors (Fig. 4C, bottom). However, the preferential accumulation of MRC1+VEGFA+ TAMs in the PIMO–VA regions of chemotherapy-treated tumors was no longer present after relapse, suggesting that the accumulation of MRC1+ TAMs was an acute, transient response to therapy (Supplementary Fig. S5A). MRC1+ TAMs were also seen to preferentially localize in perivascular areas (i.e., within 150 μm of blood vessels) in human breast carcinomas 21 to 28 days after three cycles of neoadjuvant paclitaxel (Fig. 4D, left and middle), and in breast cancer metastases in the bone after chemotherapy (with or without bisphosphonates; Fig. 4D, right).
Immunofluorescence staining of VEGFA in orthotopic MMTV-PyMT tumors excised at day 6 indicated that only MRC1+, not MRC1−, TAMs expressed detectable levels of VEGFA in vehicle and doxorubicin-injected Cre− tumors. Indeed, >98% of vessel-associated MRC1+ TAMs were VEGFA+ in both groups of tumors (Fig. 5A and B). So, we investigated the role of VEGFA expression by these MRC1+ TAMs in tumor relapse using the Csf1r-Mer-iCre-Mer inducible cre-recombinase/estrogen receptor fusion protein model for the tamoxifen-induced ablation of VEGFA selectively in monocytes/macrophages (23).
Figure 5.
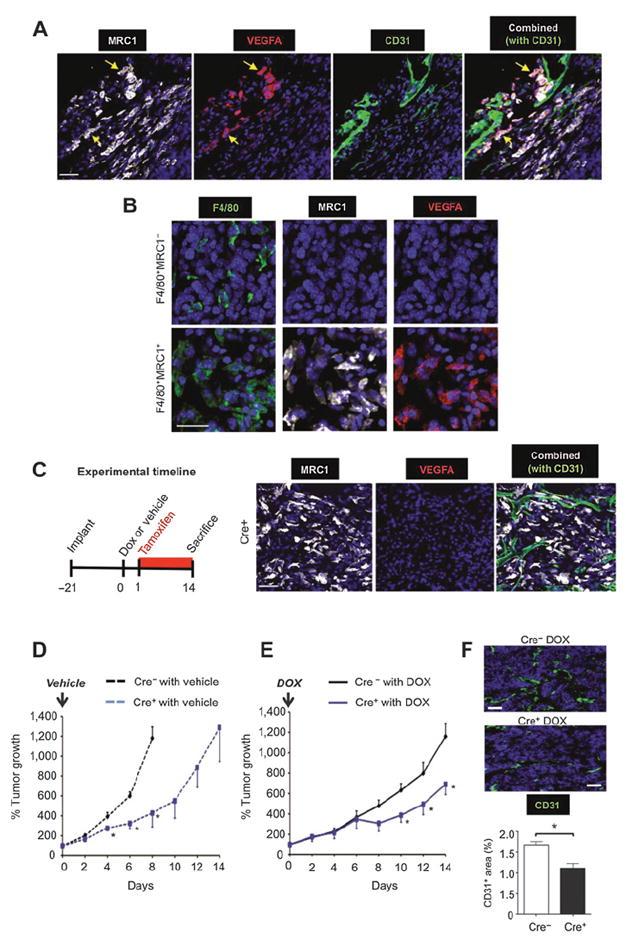
Effect of TAM-derived VEGFA on relapse of orthotopic MMTV-PyMT tumors after doxorubicin (DOX) treatment. A, representative immunostaining for VEGFA in MRC1+ TAMs in vascularized (CD31+) areas of doxorubicin-treated, Cre−, MMTV-PyMT tumors. This was not seen for MRC1− TAMs anywhere in the same tumors (B). C, female Tg(Csf1r-Mer-iCre-Mer)1jwp; Vegfafl/fl) mice bearing MMTV-PyMT tumors were administered tamoxifen for 24 hours to delete VEGFA expression in TAMs (right panels) after a single injection of either vehicle alone or doxorubicin. D and E, growth of tumors treated with vehicle alone in Cre+ and Cre− mice (n = 3–6/group; D) and regrowth of tumors in Cre+ and Cre− mice after treatment with doxorubicin (n = 3–4/group; E). F, CD31 staining of vessels in tumors in Cre− or Cre+ mice given doxorubicin. Bars, 50 μm. *, P < 0.05 with respect to tumors at the same time point in the respective Cre− group.
Female Csf1r-Mer-iCre-Mer FVB/n mice were orthotopically implanted with syngeneic MMTV-PyMT tumors and administered tamoxifen 24 hours after a single injection of either vehicle or doxorubicin. In addition, tamoxifen was given continuously thereafter to ensure VEGFA knockout until mice were sacrificed at the end of the experiment (day 14), with the same treatment given to control Cre-recombinase negative mice (Fig. 5C). As the implanted PyMT cells in these tumors did not carry the Csf1r-driven Cre recombinase and expression of Csf1r is largely confined to TAMs in such tumors, the knockdown of VEGFA was restricted to the macrophage lineage, a significant source of VEGFA in the PyMT model (as in cyclophosphamide-treated LLC1s; ref. 31). Tamoxifen-induced ablation of this TAM-derived VEGFA (Fig. 5C) caused a significant (P < 0.05) delay in the growth of vehicle-treated MMTV-PyMT tumors (Fig. 5D). Furthermore, relapse of doxorubicin-treated tumors lacking VEGFA+ TAMs (Cre+) was significantly (P < 0.05) slower than that of doxorubicin-treated tumors in VEGFA-expressing (Cre−) mice (Fig. 5E). This was accompanied by a significant (P < 0.05) decrease in vessel area in these tumors (Fig. 5F).
Pharmacologic blockade of CXCR4 prevents perivascular accumulation of MRC1+VEGFA+TAMs after chemotherapy and delays tumor relapse
The majority of F4/80+MRC1+ TAMs expressed the CXCL12 receptor, CXCR4 in both control and cyclophosphamide-treated LLC1s (Fig. 6A) and 98% of all CXCR4-expressing cells were TAMs in cyclophosphamide-treated LLC1 tumors, compared with 78% in control LLCs (where other cell types were also CXCR4+; Fig. 6B). As reported previously (32, 33), LLC1 cells do not express CXCR4 (Fig. 6A and B). Moreover, less than 1% of CD31+ blood vessels expressed CXCR4 in cyclophosphamide-treated tumors (Fig. 6C). MRC1Hi TAMs in both control and cyclophosphamide-treated tumors expressed elevated levels of CXCR4 compared with the MRC1Lo TAMs (Fig. 6D).
Figure 6.
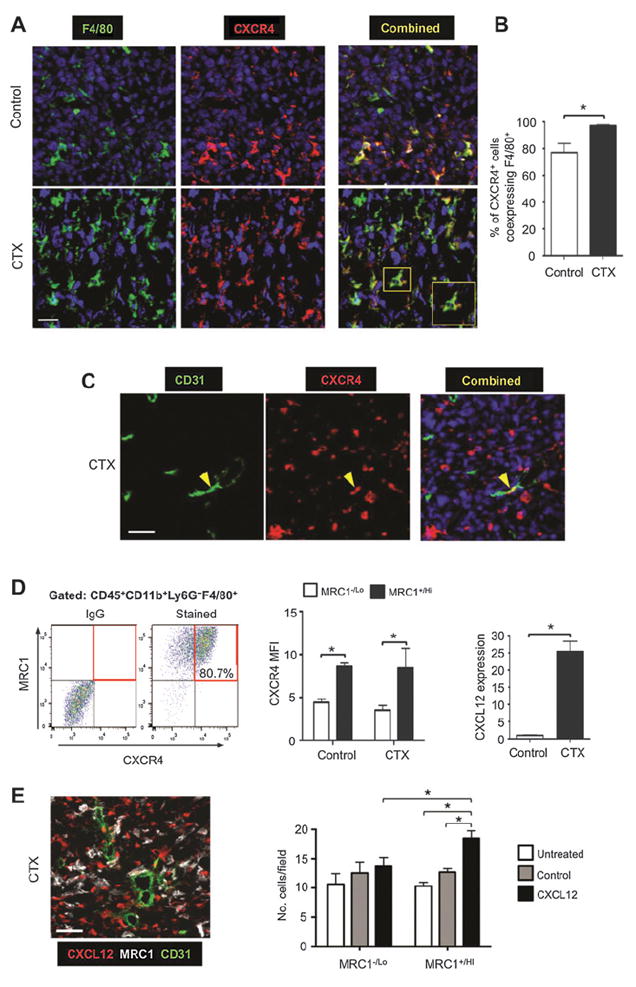
Expression of CXCR4 by MRC1+ TAMs and upregulation of CXCLl2 in LLC1 tumors after cyclophosphamide (CTX) treatment. A and B, immunostaining of F4/80 and CXCR4 (A), and the percentage of CXCR4+ cells coexpressing F4/80 in control and cyclophosphamide-treated LLCs (n = 4/group; B). C, immunostaining of CXCR4 and CD31 in cyclophosphamide-treated LLCs. D, flow-cytometric analysis of TAM expression of CXCR4 and MRC1 in dispersed cyclophosphamide-treated LLCs (left), CXCR4 MFI on MRC1+ vs. MRC1− TAMs (middle; n = 4–5/group), and tumor levels of immunodetectable CXCL12 protein in control and cyclophosphamide-treated tumors (n = 4/group; right). E, CXCL12+ cells were perivascular in cyclophosphamide-treated LLC1s (left) and exogenous recombinant human CXCL12 was chemotactic for MRC1+/Hi (but not MRC−/Lo) TAMs isolated from LLC1 tumors. Bars, 50 μm. *, P < 0.05.
These changes in CXCR4+ cells after cyclophosphamide were accompanied by a marked increase in tumor levels of its ligand, CXCL12, on day 6 (Fig. 6D, far right). These CXCL12+ cells were CD31− (Fig. 6E, left) and most likely tumor cells and/or fibroblasts (34). As a hypoxia-inducible gene (35), it was not surprising to find CXCL12 more highly expressed in hypoxic than normoxic areas of control LLC1 tumors. However, after cyclophosphamide, tumor levels of HIF1 and -2 were markedly reduced in all areas of tumors (Supplementary Fig. S6A, i and ii), but CXCL12 was abundant in both hypoxic and normoxic, vascularized areas (Supplementary Fig. S6B). This suggested CXCL12 upregulation by factors other than hypoxia in such tumors.
Cyclophosphamide is known to induce oxidative stress (36), a cellular response known to regulate the expression of CXCL12 (37), so we investigated the expression of a well-defined marker for oxidative stress, heme oxygenase-1 (HMOX-1) in control versus cyclophosphamide-treated tumors. Interestingly, this was found to be upregulated in perivascular, PIMO−, CXCL12-rich areas of tumors after cyclophosphamide treatment but not in control tumors (Supplementary Fig. S6C, i and ii). Both MRC1+ TAMs and MRC1− cells expressed HMOX-1 in these vascularized areas (Supplementary Fig. S6C, iii).
We then investigated whether CXCL12 might recruit and/or retain CXCR4+MRC1+ TAMs in LLC1 tumors. First, we showed in an in vitro chemotaxis assay that CXCL12 is selectively chemotactic for MRC1+/Hi TAMs isolated from LLC1 tumors (Fig. 6E, right). Then we administered cyclophosphamide to LLC1-bearing mice alone or in combination with the CXCR4 antagonist, plerixafor (AMD3100; Fig 7A). This was feasible as TAMs were the predominant cell type expressing CXCR4 after cyclophosphamide (Fig. 6B). CXCR4 blockade significantly inhibited cyclophosphamide-induced accumulation of F4/80+ MRC1+ TAMs in the PIMO− VA (while the numbers in PIMO+ areas of LLC1 tumors were unchanged, as was F4/80+ MRC1− TAMs in either of these areas) and delayed tumor relapse (Fig. 7A and B; Supplementary S5B). At day 10 (when tumors had relapsed in the cyclophosphamide alone group), a similar distribution of MRC1+ TAMs was seen compared with day 6 (the start of the relapse period; Fig. 7B and C). In addition, tumors administered cyclophosphamide + AMD3100 contained significantly (P < 0.05) shorter CD31+ blood vessels, higher levels of hypoxia, and fewer BrdUrd+ (proliferating) cells than tumors exposed to cyclophosphamide with the vehicle for AMD3100 (PBS; Fig. 7D; Supplementary Fig. S5C).
Figure 7.
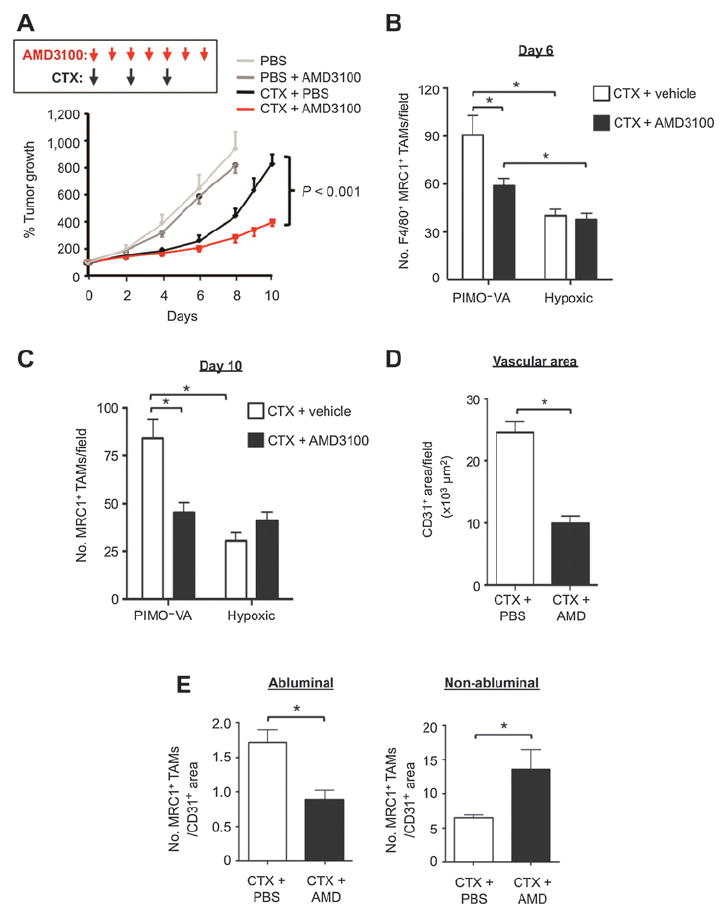
Effect of the CXCR4 inhibitor AMD3100 on LLC1 relapse after cyclophosphamide (CTX): role of perivascular MRC1+CXCR4Hi TAMs. A, regrowth of tumors in mice treated with PBS, PBS + AMD3100, cyclophosphamide + PBS, or cyclophosphamide + AMD3100 (n = 5–9/group). B and C, F4/80+MRC1+ TAMs in the PIMO− VA and hypoxic (PIMO+) areas of cyclophosphamide + PBS or cyclophosphamide + AMD3100 treated tumors (n = 4–6/group) at day 6 (B) and day 10 (n = 7–9/group; C). D, total CD31+ vessel area in tumors treated with cyclophosphamide + PBS and cyclophosphamide + AMD3100 (at day 6 in A). E, MRC1+ TAMs in direct contact (abluminal) or not in contact with (non-abluminal) with CD31+ endothelial cells in PIMO− VA areas of either cyclophosphamide + PBS or cyclophosphamide + AMD3100-treated LLCs (normalized to total CD31+ area in each field). *, P < 0.05.
We then examined the effect of CXCR4 blockade on the distribution of MRC1+ TAMs across PIMO− VAs in cyclophosphamide-treated tumors. Interestingly, this was found to significantly (P < 0.05) reduce the number of MRC1+ TAMs in direct contact with the abluminal surface of CD31+ blood vessels (standardized by CD31+ vessel area as this differed between cyclophosphamide + PBS and cyclophosphamide + AMD3100 groups), and increase them elsewhere in the PIMO− VAs (Fig. 7E). These data suggest that CXCR4 regulates the direct association of alternatively activated TAMs with blood vessels in cyclophosphamide-treated LLC1s.
A recent study demonstrated a marked rebound in the growth of pulmonary metastases after treatment with an antibody to CCL2 (38). This was the result of increased mobilization of monocytes from the bone marrow after CCL2 inhibition, and increased blood vessel formation and cancer cell proliferation in the lungs. We, therefore, investigated the possibility of a similar rebound effect after CXCR4 inhibition using AMD3100 as this could disrupt the bone marrow niche. When we extended the length of the AMD3100 experiment, primary LLC1 tumors relapsed eventually, but not at an accelerated rate, and mice receiving cyclophosphamide + AMD3100 showed increased survival compared with those in the cyclophosphamide alone group (or controls). Importantly, there was also no rebound in pulmonary metastases (Supplementary Fig. S8).
Discussion
Our studies show that M2-skewed TAMs (MRC1+TIE2+CXCR4HiVEGFA+) selectively accumulate in vascularized, well-oxygenated areas of LLC1 tumors after treatment with cyclophosphamide. A similar increase in such vessel-associated, M2-skewed TAMs was also seen in orthotopic 4T1 and MMTV-PyMT implants after treatment with paclitaxel and doxorubicin, respectively, and in human breast carcinomas after neoadjuvant treatment with paclitaxel. This perivascular accumulation was found to be CXCR4-dependent, especially the increased, direct contact of this TAM subset with the abluminal surface of blood vessels in chemotherapy-treated tumors. When this was disrupted using a CXCR4 inhibitor, tumor revascularization and regrowth after chemotherapy were markedly impaired. We also show, using an inducible, macrophage-specific, gene knockdown model that VEGFA expressed by such MRC1+ TAMs mediates, in part, their ability to promote tumor relapse after therapy. Consistent with this, genetic deletion of HIF signaling in TAMs in hypoxic tumor areas had no effect on this rescue.
The mobilization and accumulation in tumors, of such BMDCs as myelomonocytic cells, MDSCs, endothelial progenitor cells (EPC) and macrophages after various forms of anti-cancer therapy are now well established (13-19, 39, 40). However, our data show that M2-skewed TAMs represent a significant proportion of such BMDCs in tumors after chemotherapy, and is not accompanied by TAM proliferation or increased numbers of stem/progenitor cells. These data suggest that increased recruitment of circulating monocytes precedes the accumulation of such M2 TAMs in tumors after chemotherapy. It remains to be seen whether monocytes are already M2-skewed upon arrival in treated tumors and/or activated by factors produced in the perivascular niche. Of note, the recruitment, activation, and perivascular retention of these cells does not appear to be just an acute response to chemotherapy-induced tumor damage, as increased numbers of perivascular M2 TAMs persisted throughout the relapse phase.
Our observations suggest that the accumulation of MRC1+VEGFA+ TAMs on and around the tumor vasculature plays an important part in tumor revascularization and relapse after chemotherapy. Furthermore, this was accompanied by a marked change in the pattern of CXCL12 expression in tumors. Although CXCL12 was mainly confined to hypoxic areas of control tumors, it was upregulated in vascularized, well-oxygenated areas after chemotherapy. This correlated with an increase in the expression of the enzyme, HMOX-1, a marker of oxidative stress, in such perivascular areas. Cytotoxic agents such as cyclophosphamide induce oxidative stress in tumors (41), which activates cellular expression of HMOX-1. This, in turn, generates carbon monoxide from the breakdown of heme, a gas known to upregulate both CXCL12 and VEGFA in neighboring cells (37). Interestingly, HMOX-1 also regulates expression of MRC1 by macrophages (42). So, chemotherapy induction of this stress pathway in perivascular areas could both retain TAMs via local CXCL12 induction, and upregulate their expression of VEGFA and MRC1. Interestingly, the CXCL12-induced retention of myeloid cells around the vasculature is also essential to the process of neovascularization in nonmalignant tissues (43).
Other factors may also contribute to the retention of MRC1+ TAMs. Our observation that MRC1+ TAMs expressed elevated TIE2 suggests that ANGPT2 expressed by the tumor endothelium (44) might also be involved. Indeed, ANGPT2 has an established role in retaining vascular modulatory myeloid cells in proximity to the vasculature in progressing mouse tumors (26).
In untreated mouse tumors, perivascular M2-skewed TAMs have an established vascular modulatory role and facilitate tumor growth and progression (26). Our observation that chemotherapy induces the accumulation of MRC1+VEGFA+ TAMs around tumor blood vessels strongly infers a proangiogenic role for these cells. This is supported by our finding that pharmacologic inhibition of their chemotherapy-induced accumulation resulted in reduced subsequent tumor revascularization. Furthermore, in doxorubicin-treated, MMTV-PyMT tumor implants VEGFA was found to be expressed predominantly by MRC1+ TAMs, consistent with our previous finding that TAMs are a major source of VEGFA in MMTV-PyMT tumors (31). The selective ablation of VEGFA in MRC1+ TAMs resulted in delayed tumor relapse, post-therapy. However, PyMT tumors still relapsed, albeit at a reduced rate, in the absence of TAM-derived VEGFA, indicating that other TAM-derived factors may also contribute to tumor relapse and/or the possible induction of resistance mechanisms in tumors to VEGFA knockout. The presence of the latter has been demonstrated in tumors after anti-VEGFA therapy and shown to include increased expression of such alternative proangiogenic mediators as FGF2 (45), ANGPT2 (46), or PlGF (47) and the recruitment of tumor-promoting, CD11b+ Gr1+ myeloid cells (48).
Recently, the Condeelis group has used intravital imaging to characterize a distinct subset of perivascular TAMs in untreated mouse tumors that make contact with endothelial cells and tumor cells expressing high levels of Mena (a protein that enhances their motility), and directly stimulate tumor cell intravasation (49). These cell trios have been termed “tumor microenvironments of metastasis” (TMEM). It is possible that some of the perivascular MRC1+ M2-skewed TAMs accumulating around tumor blood vessels after chemotherapy form TMEMs and promote metastasis as well as relapse, a dangerous combination in patients with inoperable tumors.
The effect of CXCR4 blockade on tumor relapse after chemotherapy in our study suggests that CXCR4 inhibitors might be successfully combined with chemotherapy. This combination could extend relapse-free survival in patients with inoperable tumors, although our data suggests that multiple rounds of the inhibitor would have to be administered to maintain a suppressive effect on relapse. Such sustained use of a CXCR4 antagonist after chemotherapy could conceivably disrupt the marrow niche. However, this is unlikely to lead to clinical problems as a recent, first-in-human clinical trial has shown that repeated daily injections of the CXCR4 antagonist, LY2510924 over a number of consecutive, 28-day cycles was well tolerated in advanced cancer patients (50).
Taken together, our data suggest that the selective targeting of relapse-promoting, perivascular TAMs could delay the relapse of both primary and metastatic tumors in patients after chemotherapy, thereby extending their relapse-free survival.
Acknowledgments
Grant Support
This work was supported mainly by a Cancer Research UK grant (C.E. Lewis), the grant support of the Swiss Cancer League (M. De Palma), Breast Cancer Research Foundation (O.C. Olson and J.A Joyce), a Chancellor’s Fellowship from the University of Edinburgh (B.-Z. Qian), and The Wellcome Trust (Senior Investigator Award; J.W. Pollard).
Footnotes
Authors’ Contributions
Conception and design: R. Hughes, B.-Z. Qian, M. Muthana, J.W. Pollard, C.E. Lewis
Development of methodology: R. Hughes, B.-Z. Qian, C. Addison, J.W. Pollard, C.E. Lewis
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): R. Hughes, B.-Z. Qian, C. Rowan, I. Keklikoglou, O.C. Olson, S. Tazzyman, C. Addison, M. Clemons, A.M. Gonzalez-Angulo, J.A Joyce, M. De Palma, J.W. Pollard, C.E. Lewis
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): R. Hughes, B.-Z. Qian, C. Rowan, S. Tazzyman, S. Danson, J.W. Pollard, C.E. Lewis
Writing, review, and/or revision of the manuscript: R. Hughes, B.-Z. Qian, S. Danson, C. Addison, A.M. Gonzalez-Angulo, J.A Joyce, M. De Palma, J.W. Pollard, C.E. Lewis
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): R. Hughes, M. Muthana
Study supervision: R. Hughes, M. Muthana, J.W. Pollard, C.E. Lewis
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Savir G, Huber KE, Saif MW. Locally advanced pancreatic cancer. Looking beyond traditional chemotherapy and radiation. JOP. 2013:14, 337–9. doi: 10.6092/1590-8577/1677. [DOI] [PubMed] [Google Scholar]
- 2.Strom HH, Bremnes RM, Sundstrom SH, Helbekkmo N, Aasebo U. Poor prognosis patients with inoperable locally advanced NSCLC and large tumors benefit from palliative chemoradiotherapy: a subset analysis from a randomized clinical phase III trial. J Thorac Oncol. 2014;9:825–33. doi: 10.1097/JTO.0000000000000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffelt SB, Lewis CE, Naldini L, Brown JM, Ferrara N, De Palma M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am J Pathol. 2010;176:1564–76. doi: 10.2353/ajpath.2010.090786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 7.Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochimica et biophysica acta. 2009;1796:11–8. doi: 10.1016/j.bbcan.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008;180:2011–7. doi: 10.4049/jimmunol.180.4.2011. [DOI] [PubMed] [Google Scholar]
- 9.Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood. 2009;114:901–14. doi: 10.1182/blood-2009-01-200931. [DOI] [PubMed] [Google Scholar]
- 10.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–26. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Daenen LG, Houthuijzen JM, Cirkel GA, Roodhart JM, Shaked Y, Voest EE. Treatment-induced host-mediated mechanisms reducing the efficacy of antitumor therapies. Oncogene. 2014;33:1341–7. doi: 10.1038/onc.2013.94. [DOI] [PubMed] [Google Scholar]
- 12.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–86. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 13.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–79. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70:5679–85. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest. 2011;121:1969–73. doi: 10.1172/JCI44562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–85. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 21.Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–86. [PubMed] [Google Scholar]
- 22.Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014;25:809–21. doi: 10.1016/j.ccr.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 25.Hilton JF, Amir E, Hopkins S, Nabavi M, DiPrimio G, Sheikh A, et al. Acquisition of metastatic tissue from patients with bone metastases from breast cancer. Breast Cancer Res Treat. 2011;129:761–5. doi: 10.1007/s10549-010-1264-6. [DOI] [PubMed] [Google Scholar]
- 26.Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19:512–26. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–8. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16:4583–94. doi: 10.1158/1078-0432.CCR-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–34. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 30.Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114:844–59. doi: 10.1182/blood-2008-12-195941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 32.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104:15735–40. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss ID, Jacobson O, Kiesewetter DO, Jacobus JP, Szajek LP, Chen X, et al. Positron emission tomography imaging of tumors expressing the human chemokine receptor CXCR4 in mice with the use of 64Cu-AMD3100. Mol Imaging Biol. 2012;14:106–14. doi: 10.1007/s11307-010-0466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46:2587–97. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Jiang L, Geng C, Cao J, Zhong L. The role of oxidative stress in acrolein-induced DNA damage in HepG2 cells. Free Radic Res. 2008;42:354–61. doi: 10.1080/10715760802008114. [DOI] [PubMed] [Google Scholar]
- 37.Lin HH, Chen YH, Chang PF, Lee YT, Yet SF, Chau LY. Heme oxygenase-1 promotes neovascularization in ischemic heart by coinduction of VEGF and SDF-1. J Mol Cell Cardiol. 2008;45:44–55. doi: 10.1016/j.yjmcc.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 38.Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–3. doi: 10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- 39.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14:263–73. doi: 10.1016/j.ccr.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Jungsuwadee P, Vore M, Butterfield DA, StClair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Mol Interv. 2007;7:147–56. doi: 10.1124/mi.7.3.6. [DOI] [PubMed] [Google Scholar]
- 42.Tu TH, Joe Y, Choi HS, Chung HT, Yu R. Induction of heme oxygenase-1 with hemin reduces obesity-induced adipose tissue inflammation via adipose macrophage phenotype switching. Mediators Inflamm. 2014;2014:290708. doi: 10.1155/2014/290708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–89. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 44.Stratmann A, Risau W, Plate KH. Cell type-specific expression of angio-poietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol. 1998;153:1459–66. doi: 10.1016/S0002-9440(10)65733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 46.Rigamonti N, Kadioglu E, Keklikoglou I, Rmili CW, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signalling blockade. Cell Rep. 2014;8:696–706. doi: 10.1016/j.celrep.2014.06.059. [DOI] [PubMed] [Google Scholar]
- 47.Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–75. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 48.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 49.Roussos ET, Goswami S, Balsamo M, Wang Y, Stobezki R, Adler E, et al. Mena invasive (Mena(INV)) and Mena11a isoforms play distinct roles in breast cancer cell cohesion and association with TMEM. Clin Exp Metastasis. 2011;28:515–27. doi: 10.1007/s10585-011-9388-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galsky MD, Vogelzang NJ, Conkling P, Raddad E, Polzer J, Roberson S, et al. A Phase I Trial of LY2510924, a CXCR4 peptide antagonist, in patients with advanced cancer. Clin Cancer Res. 2014;20:3581–8. doi: 10.1158/1078-0432.CCR-13-2686. [DOI] [PubMed] [Google Scholar]