

Abstract
C9ORF72 mutations are found in a significant fraction of patients suffering from amyotrophic lateral sclerosis and frontotemporal dementia, yet the function of the C9ORF72 gene product remains poorly understood. Here, we show that mice harboring loss-of-function mutations in the ortholog of C9ORF72 develop splenomegaly, neutrophilia, thrombocytopenia, increased expression of inflammatory cytokines, and severe autoimmunity, ultimately leading to a high mortality rate. Transplantation of mutant bone marrow into wildtype recipients was sufficient to recapitulate the phenotypes observed in the mutant animals, including autoimmunity and premature mortality. Reciprocally, transplantation of wildtype marrow into mutant mice improved their phenotype. We conclude that C9ORF72 serves an important function within the hematopoietic system to restrict inflammation and the development of autoimmunity.
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of motor neurons, resulting in paralysis and death (1). Genetic findings suggest ALS can result from mutations in genes acting in several cellular processes (2). To date, the most common mutation found in ALS patients, as well as in frontotemporal dementia (FTD), is a hexanucleotide repeat expansion in the first intron of C9ORF72 (3). This mutation is present in 4-8% of patients with sporadic ALS (4). The repeat expansion is also found in 40% of familial ALS cases from Europe and the USA and is even more prevalent in Scandinavia(3,4). Identification of unaffected elderly carriers suggests the mutation's effects are incompletely penetrant (5).
The expanded GGGGCC repeat in C9ORF72 has been proposed to mediate its effects through one or a combination of three mechanisms: the creation of long repetitive RNAs(3,6), translation of these RNAs into toxic dipeptides(7), or silencing of the mutant allele (3,8,9). Histological and gene expression studies indicate that all three effects of the repeat expansion manifest themselves in individuals harboring it. However, which effect, or effects of the mutation contribute to the degenerative phenotypes seen in patients remains unresolved. The normal protein product of C9ORF72 contains a DENN domain (10) and may act as a guanine nucleotide exchange factor (11). Previous studies in fish and worms suggested that C9ORF72 might function in the nervous system(8,12). We found that while the murine ortholog of C9ORF72 (3110043O21Rik hereafter referred to as C9orf72) is widely expressed, it was enriched in motor neurons known to degenerate in ALS (13). However, a recent study demonstrated that conditional excision of C9orf72 in cells of the nervous system had no overt effects raising the question of whether it plays any role in the nervous system or overall health (14).
Resolving the function of the endogenous C9ORF72 gene product remains important as it could provide insight into whether the reduction in gene product found in patients with the repeat expansion contributes to their disease (3,8,9). Furthermore, efforts are proceeding to develop therapeutic approaches for knocking down the gain of function products of the repeat expansion (9). As knockdown strategies could inadvertently depress transcription of the normal gene product, it will be critical to understand the phenotypic ramifications of long-term depression of gene expression from this locus.
Here, we report an initial study of mice harboring loss-of-function mutations in C9orf72. We found that homozygous mutant animals produced through homologous recombination as well as CRISPR/Cas9 targeting developed several classical features of autoimmunity. Transplantation studies demonstrated that mutant bone marrow cells were sufficient to cause autoimmunity and premature mortality when placed into otherwise wildtype recipients. Thus, we conclude that the gene product encoded by C9orf72 likely acts within the hematopoietic system to play an important role in the promotion of immunological tolerance.
Results
Characterization of C9orf72 loss of function mutants
We recently identified the mouse ortholog of C9ORF72, and generated heterozygous mice harboring a LacZ insertion replacing exons 2-6 of the gene on an inbred C57BL/6 background(13) (Fig 1A). Because this targeting event eliminated highly conserved 5′ exons of C9orf72 we reasoned that it might result in a loss of gene function, allowing the importance of this gene to be investigated. After intercrossing heterozygous (+/-) animals, we found that mice of the three expected genotypes were recovered at expected Mendelian frequencies (Fig 1B). We designated this strain “KOMP” as the gene targeting to create this allele was originally performed by the Knock Out Mouse Project (KOMP) Consortium.
Fig. 1. A loss of function mutation in the C9orf72 ortholog.
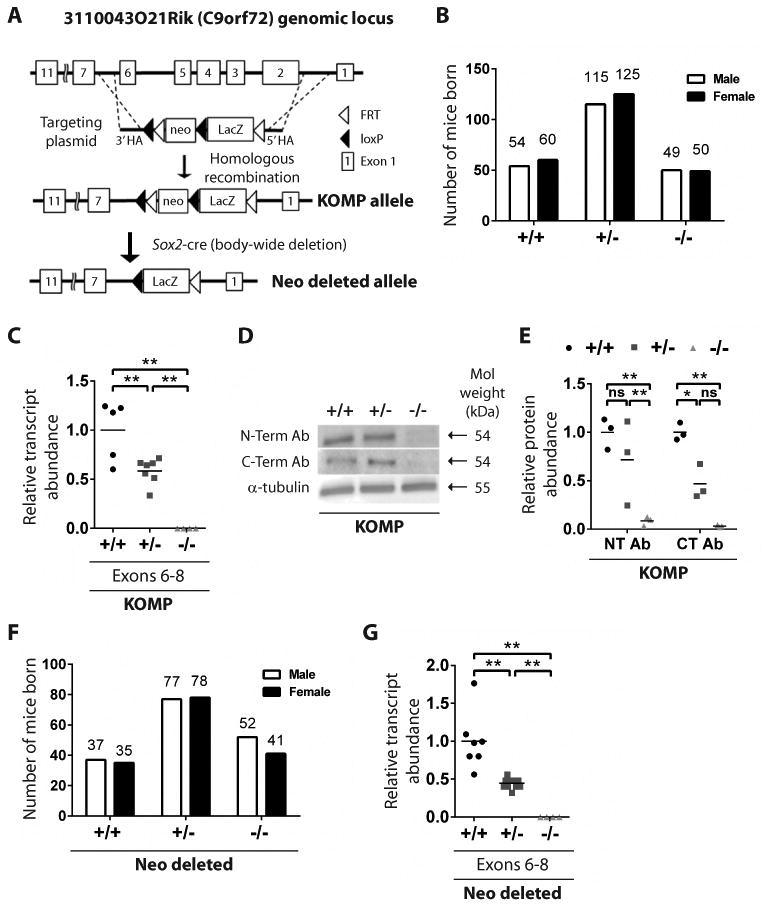
(A) The schematic illustrates replacement of exons 2-6 to generate the KOMP allele and crosses with Sox2-cre mice to generate the Neo-deleted allele. (B) Frequency of genotypes born from KOMP +/- crosses. (C) C9orf72 expression in KOMP whole blood by quantitative RT-PCR. **p<0.01 Tukey's multiple comparisons. ns not significant. (D) Western blotting of cortical tissue from the 3 KOMP genotypes using anti-C9ORF72 antibodies. (E) Quantification of Western blot bands. *p<0.05 **p<0.01 Tukey's multiple comparisons. (F) Frequency of genotypes born from Neo deleted +/- crosses. (G) C9orf72 expression in Neo deleted whole blood by quantitative RT-PCR. **p<0.01 Tukey's multiple comparisons.
We next sought to use animals from the +/- intercrosses to determine whether the targeted mutation reduced expression of C9orf72. Using exon-spanning primers we detected a significant, dose dependent reduction in the abundance of transcript sequences from exons 6 through 8 in whole blood of +/- and -/- animals relative to that found in wild type controls (Fig 1C; **p<0.01). To assess whether the mutation also resulted in decreased protein levels, we employed two anti-C9orf72 antibodies, one raised against C-terminal regions of the protein and the other against its N-terminus. In each case, a band with the predicted mass of C9orf72 was significantly depleted in mutant animals (Fig 1D-E; *p<0.05**p<0.01).
There are historical examples of the transcriptional units encoding drug resistance used in gene targeting leading to off-target phenotypes(15). We therefore removed the selection cassette used for gene targeting by outcrossing C9orf72 targeted KOMP mice with outbred animals that expressed the Cre-recombinase under control of the Sox2 promoter (16) (hereafter referred to as “Neo deleted”) (Fig 1A; Fig S1A). Intercrosses of +/- Neo deleted animals resulted in recovery of +/+, +/- and -/- animals at the expected Mendelian frequencies (Fig 1F). Importantly, Neo deleted +/- and -/- mice exhibited a significant and dose dependent reduction in C9orf72 transcript abundance (Fig 1G; **p<0.01). As proper interpretation of mouse models relies upon the fidelity of the genetic modifications introduced, we performed whole genome sequencing of representative +/+, +/-, and -/- KOMP animals as well as -/- Neo deleted mice. These studies revealed proper gene targeting in each strain and the absence of off-target insertions (Fig S1B). We were also able to confirm the C57BL/6 inbred nature of KOMP animals and outbred nature of Neo deleted animals (Fig S1C-F).
To determine the effects of C9orf72 loss of function, we intercrossed +/- KOMP animals and monitored littermates for 400 days (Fig 2A; n=163; +/+ n=50; +/- n=84; -/- n=29). At 70 days of age, we noted no obvious phenotypes (Fig 2A). However, as the cohort continued to age, the risk of death in both -/- and +/- animals grew to a high level of significance (Fig 2A; *p<0.05). We also examined Neo deleted mice generated by +/- intercrosses (Fig 2B; n=50, +/+ n=15, +/- n=21, -/- n=14). At later ages we similarly observed significantly increased mortality in +/- and -/- mice (Fig 2B; *p<0.05). As C9orf72 has been genetically implicated in ALS, we asked whether increased mortality in mutant animals was associated with neural degeneration. However, we did not find obvious changes in the number of spinal motor neurons (Fig S2). Nor did we observe differences in the gross histology of the mutant motor cortex (Fig S3) or other brain regions of mutant animals (Fig S4). We did however observe evidence for increased neural inflammation in the cortex of mutant animals as evidenced by focal staining with anti-GFAP antibodies (Fig S5).
Fig. 2. C9orf72 mutations lead to premature death of mice.
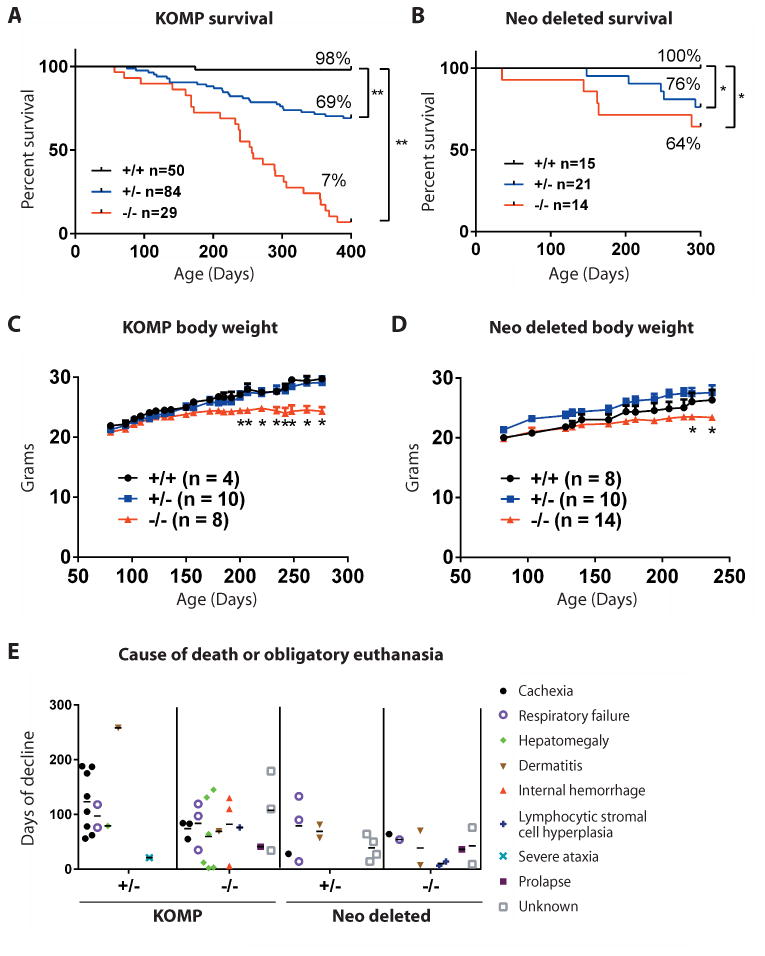
Mice harboring loss of function mutations in C9orf72, generated by homologous recombination using a targeting vector in embryonic stem cells on C57BL/6 background (KOMP) and those outcrossed with Sox2-cre expressing mice to remove the neomycin cassette (Neo deleted), were aged for survival studies. Kaplan Meier survival curves for (A) KOMP and (B) Neo deleted mice. *p<0.05, **p<0.01 Generalized Wilcoxon test. Body weight of female (C) KOMP mice and (D) Neo deleted animals over time. *p<0.05 Dunnett's multiple comparisons. (E) Cause of death or obligatory euthanasia in KOMP and Neo deleted mice. Days of decline indicates time between maximum body weight (onset) and death or obligatory euthanasia.
We next sought to establish the onset of decline in these mutant animals, to quantify the duration of their decline and to better understand the causes of their premature death. We weighed mice weekly and performed necropsy on animals found dead or that we were obliged to euthanize due to pre-established animal welfare criteria. We noted that while animals of all genotypes initially exhibited similar weights, as they aged the weight of both KOMP and Neo deleted -/- animals plateaued, and became significantly lower than wild type controls (Fig 2C-D; *p<0.05). By defining phenotypic onset for each animal as the age at which it reached its maximum body weight, we were then able to quantify the number of days that a given animal was burdened by decline prior to its death. Heterozygous animals were burdened by decline for on average 118±67 days (KOMP; n=13) and 56±38 days (Neo deleted; n=10), while -/- animals were burdened by decline for on average 76±51 days (KOMP; n=21) and 37±29 days (Neo deleted; n=9)(Fig 2E). During these studies we determined that the causes of demise included cachexia, respiratory failure, hepatomegaly, dermatitis, internal hemorrhage, lymphocytic stromal cell hyperplasia, severe ataxia and prolapse (Fig 2E; Fig S6).
Upon necropsy of end stage animals, we observed enlarged spleens in KOMP -/- (n=10/11) and Neo deleted -/- (n=6/6) mice (Fig 3A-B). Splenomegaly can result from any one of several underlying pathologies including infection, myeloproliferative disease, chronic lymphocytic leukemia, lymphoma and autoimmunity. To investigate whether any of these was the cause of splenomegaly in mutant animals we visually inspected day 300 spleens from Neo deleted animals (+/+ n=6, +/- n=3, -/- n=7) and then subjected them to histological analysis. We observed a clear demarcation between red and white pulp regions in +/+ and +/- spleens, while -/- spleens displayed a disruption of red and white pulp boundaries (Fig 3B). We next asked when splenomegaly first appeared in these animals. We found that while day 25 Neo deleted -/- animals exhibited normal spleen size, by day 50 -/- animals had developed significantly enlarged spleens relative to their littermates (Fig 3D; **p<0.01). This significant increase in spleen weight was maintained in day 100, day 200 and day 300 -/- animals (Fig 3D; **p<0.01). We reasoned that splenic vein thrombosis and spontaneous microbial infection were less likely causes of splenomegaly in -/- animals due to the synchronicity and penetrance at which this phenotype developed.
Fig. 3. Identity of cells within the enlarged mutant spleens of Neo deleted mice.
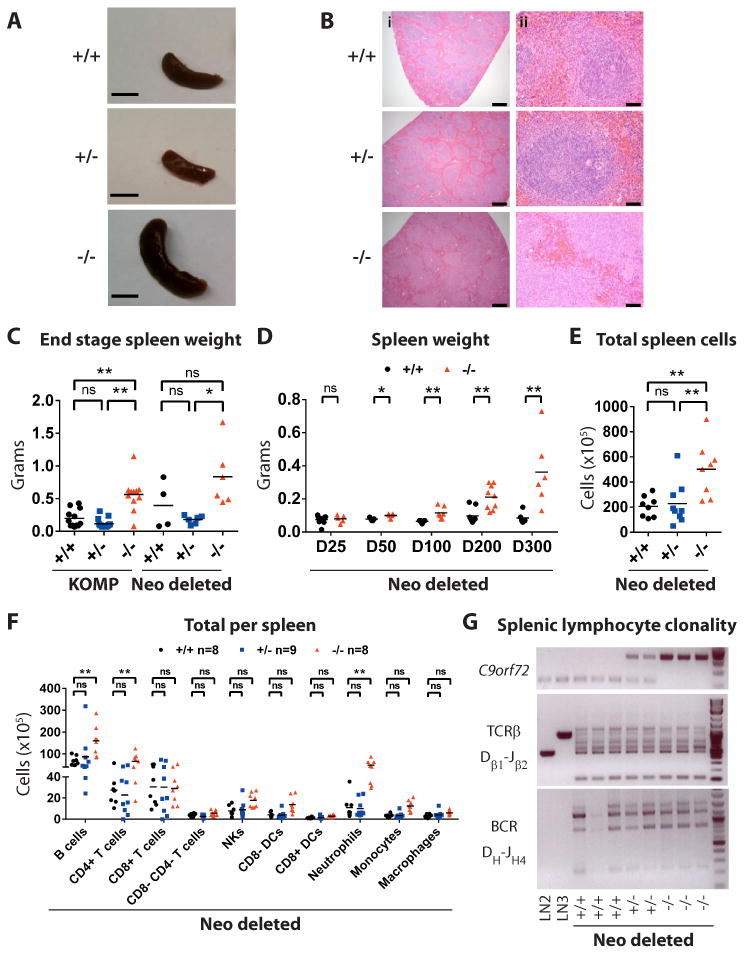
(A) Spleens from day 300 Neo deleted animals. Scale bar, 1 cm. (B) Hematoxylin & eosin staining of spleens from Neo deleted mice at day 300. Scale bar (i) 500 micron and (ii) 50 micron. (C, D) Quantification of spleen weight in (C) end stage KOMP and Neo deleted animals and (D) aged Neo deleted animals. C,E,F *p<0.05, **p<0.01 Tukey's multiple comparisons. (D) *p<0.05, **p<0.01 Student t-test. ns not significant. (E) Splenocyte counts from day 200 Neo deleted mice. (F) Quantification of splenocyte subsets in day 200 Neo deleted mice. (G) PCR analysis of T cell and B cell clonality in the spleens of day 400 Neo deleted mice. LN2 and LN3 represent ESCs generated by nuclear transfer from lymph node-derived T cells that harbor monoclonal TCRβ rearrangements.
We next considered the possibility that splenomegaly was the result of lymphoma. In lymphoma, individual clonal populations of either B or T cells can dominate the splenic compartment. However, mutant animals displayed an increase in splenocytes derived from several lineages including CD19+ B220+ B cells, CD3+ CD4+ T cells and CD11b+ Ly6G+ neutrophils (Fig 3E-F; Fig S7; *p<0.05**p<0.01). Furthermore, analysis of V(D)J recombination in spleens of mutant animals (17) revealed polyclonal populations of B and T cells, arguing against the type of clonal expansion observed in lymphoma (Fig 3G). We also did not observe histological changes in the cellularity of -/- bone marrow (Fig S8), which can be a site of lymphocytic transformation (18).
In addition to the nearly ubiquitous splenomegaly observed in -/- animals, we also noted that a subset of end stage KOMP -/- (n=4/21) and Neo deleted -/- (n=3/9) animals developed grossly enlarged cervical lymph nodes (Fig S9). Enlarged lymph nodes can result from inflammation, lymphoma, autoimmunity or an inability of lymphocytes to escape this compartment (19,20). Although we noted modest changes in the relative proportion of B and T cells, both of these populations were greatly expanded within enlarged cervical lymph nodes of mutant animals (fig. S9), again arguing against clonal transformation as an explanation. Instead we observed an increase in T cell activation state, with a significant reduction in CD62Lhi CD44lo cells and a significant increase in CD62Lhi CD44hi and CD62Llo CD44hi cells when compared to controls (Fig S9; *p<0.05**p<0.01).
Finally, we noted that a small but significant subset of both KOMP and Neo deleted mutant animals developed hepatomegaly (Fig 2E; Fig S10). Histological analysis suggested that this liver overgrowth was likely the result of immune cell infiltration (Fig S10). Such immune infiltrates in liver can be seen in the context of both leukemia and autoimmunity(21). In this case, infiltrating hematopoietic cells appeared morphologically diverse, rather than clonal in nature, which again seemed less parsimonious with the diagnosis of leukemia and more consistent with chronic inflammation (Fig S10).
Changes in peripheral blood were consistent with autoimmunity not leukemia
Assessing the composition of peripheral blood can yield insight into pathways that disrupt the hematopoietic system. We therefore performed whole blood cell counts on mutant mice greater than 300 days of age (Fig 4A-H). Numbers of white blood cells, platelets and red blood cells in KOMP +/+ and Neo deleted +/+ animals were similar to historic ranges in inbred mouse strains(22). In contrast, total white blood cell counts were modestly but significantly elevated in KOMP -/- and Neo deleted -/- mice (Fig 4A; **p<0.01), which was due to a significant increase in the number of circulating neutrophils (Fig 4B; **p<0.01). We observed no significant changes in the relative proportion of peripheral lymphocytes (Fig 5C) or monocytes in mice of differing genotypes (Fig 4D).
Fig. 4. Mice with C9orf72 mutations develop hematological phenotypes.
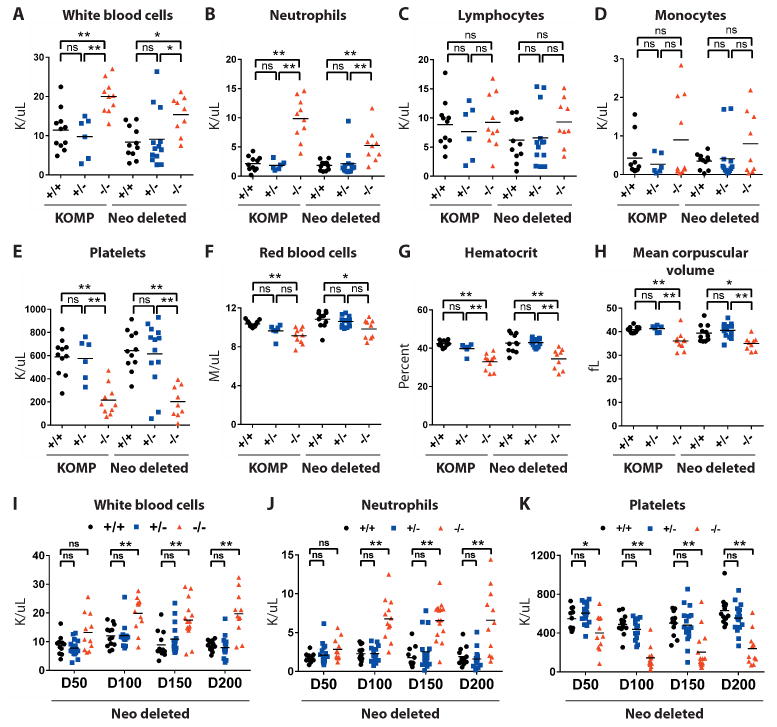
(A-H) Peripheral blood counts assessed for day 300+ KOMP and Neo deleted animals. *p<0.05, **p<0.01 Tukey's multiple comparisons. ns not significant. (A) White blood cells (B) Neutrophils (C) Lymphocytes (D) Monocytes (E) Platelets (F) Red blood cells (G) Hematocrit (H) Mean corpuscular volume. (I-K) Peripheral blood counts assessed for aged Neo deleted animals. (I) White blood cells (J) Neutrophils (K) Platelets.
Fig. 5. Mice with C9orf72 mutations show increased cytokines, chemokines and auto-antibodies.
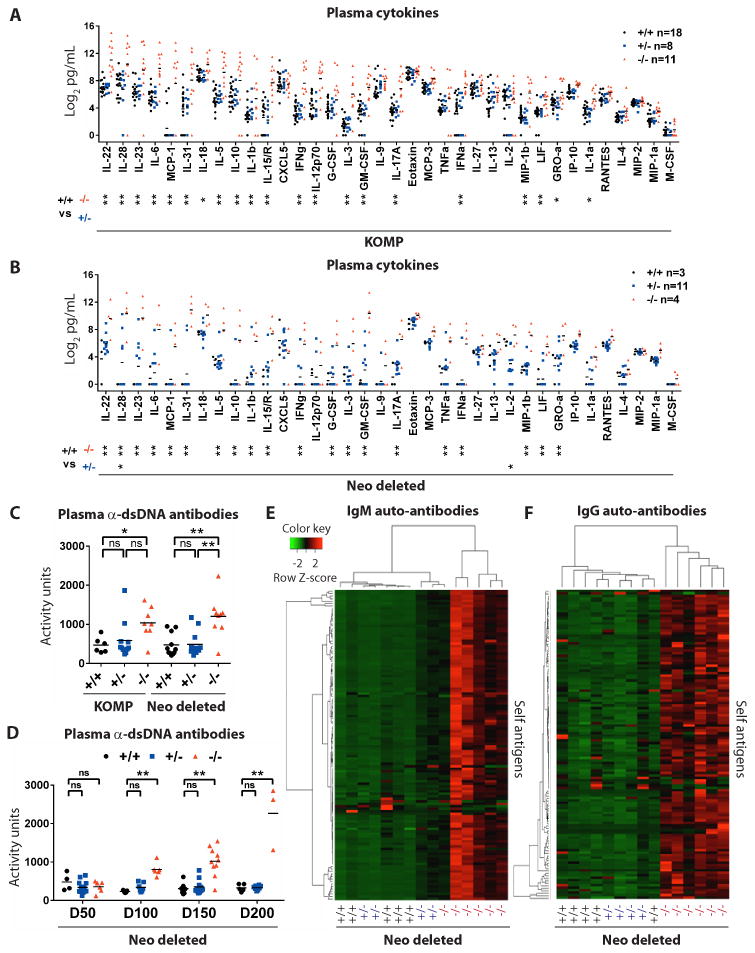
(A, B) Analysis of 36 plasma cytokines and chemokines in day 300+ (A) KOMP and (B) Neo deleted animals. *p<0.05, **p<0.01 Tukey's multiple comparisons. ns not significant. (C, D) Anti-double stranded (ds)DNA antibody reactivity in plasma of (C) day 300+ KOMP and Neo deleted animals and (D) aged Neo deleted animals. (E, F) Plasma from day 300+ Neo deleted mice assessed for (E) IgM and (F) IgG reactivity against 124 self-antigens (26). Unsupervised hierarchical clustering grouped individual animals (x-axis) and self-antigens (y-axis).
We also noted that platelet count was significantly reduced in KOMP -/- as well as Neo deleted -/- animals (Fig 4E; **p<0.01) and that there was a modest but significant reduction of red blood cells in KOMP -/- and Neo deleted -/- (Fig 4F; **p<0.01) animals that was accompanied by a modest but significant reduction in hematocrit (Fig 4G; **p<0.01). Mean corpuscular volume was also reduced in KOMP -/- and Neo deleted -/- animals (Fig 4H; **p<0.01), consistent with a modest microcytic anemia. Although there can be many causes for changes in red cell and platelet counts, similar phenotypes are seen in autoimmune hemolytic anemia and thrombocytopenic purpura, in which red blood cells or platelets, respectively, are targeted by auto-antibodies leading to their destruction (23).
To understand how changes in the peripheral blood developed, we bled Neo deleted animals (n=164) ranging from 50-200 days of age (Fig 4I-K; +/+ n=49, +/- n=65, -/- n=49). We noted that platelet count was already reduced by day 50 in -/- mice and was persistently depressed in older animals (Fig 4K; *p<0.05). Although young animals displayed normal neutrophil and white cell numbers, at 100 days of age -/- mice began to exhibit a persistent and significant increase in neutrophil numbers (Fig 4J; **p<0.01), which was the primary contributor to a significant increase in total white cell counts (Fig 4I; *p<0.05). In contrast, numbers of lymphocytes in the blood remained unchanged relative to wild type controls. Thus, neutrophilia, thrombocytopenia and splenomegaly were highly penetrant phenotypes, which all developed between day 50 and day 100 in -/- animals, suggesting an interconnected pathological relationship. In contrast, enlarged lymph nodes and immune cell infiltrates in the liver were observed later and at lower frequencies in mutant animals suggesting that they could be secondary effects of changes to the hematopoietic compartment.
Loss of C9orf72 causes early and chronic cytokine induction
If the splenic and blood phenotypes we observed in -/- animals developed in response to a chronic inflammatory process, we reasoned their emergence might be associated with an increased abundance of inflammatory chemokines and cytokines. To test this idea, we measured the levels of 36 chemokines and cytokines in plasma from day 300 KOMP (Fig 5A; +/+ n=18, +/- n=8, -/- n=11) and Neo deleted animals (Fig 5B; +/+ n=3, +/- n=11, -/- n=4). Eighteen of 36 cytokines and chemokines were significantly elevated in both KOMP -/- and Neo deleted -/- animals relative to +/+ controls, including IL-22, IL-28, IL-23, IL-6, MCP-1, IL-31, IL-5, IL-10, IL-1β, IL-15/IL-15R, IFNγ, IL-3, GM-CSF, IL-17A, IFNα, MIP-1B, LIF and GROα (Fig 5A-B; *p<0.05**p<0.01). No cytokines or chemokines were significantly changed in KOMP +/- mice relative to controls (Fig 5A) although we did note that IL-28 was significantly elevated and IL-2 was significantly reduced in +/- Neo deleted animals relative to controls (Fig 5B; *p<0.05). We next analyzed cytokine levels in cohorts of mice at earlier times points. We found that already at 50 days of age, IL-31, IL-15/IL-15R and GM-CSF were significantly elevated in Neo deleted -/- mice relative to wild type mice (Fig S11; *p<0.05). By 100 days of age Neo deleted -/- animals displayed significantly increased levels of 19/36 cytokines and chemokines tested, fully recapitulating the pattern observed in older animals at both day 200 and day 300 (Fig S11; *p<0.05**p<0.01). Thus the development of an inflammatory cytokine profile was an early event in mutant animals that either preceded or was coincident with other blood phenotypes and a decline in health.
Mutant animals rapidly develop persistent autoimmunity
We next considered the possibility that the changes in cytokine levels we found were due to autoimmunity. Auto-antibodies targeting double-stranded (ds)DNA are a common diagnostic marker of autoimmune syndromes including systemic lupus erythematosus (24). We found significant accumulation of anti-dsDNA antibody activity between 50 and 100 days of age in Neo deleted -/- animals (Fig 5D; **p<0.01), which was sustained in plasma from KOMP -/- and Neo deleted -/- animals greater than 300 days of age (Fig 5C; *p<0.05**p<0.01).
While the accumulation of anti-dsDNA antibodies occurs under conditions of autoimmunity, it can also be indicative of rapid but transient cell death (25). To distinguish between these two possibilities we used established auto-antigen microarrays to test the plasma of day 300 Neo deleted littermates for the presence and abundance of IgM and IgG antibodies targeting 124 auto-antigens from disparate cell types (26) (Fig 5E, F). In -/- plasma, IgM auto-antibodies targeting 117/124 antigens were significantly elevated relative to controls. Similarly, IgG auto-antibodies targeting 113/124 antigens were significantly elevated in -/- animals (Table S1; *p<0.05). In +/- plasma, IgM auto-antibodies targeting 1/124 antigens (Liver kidney microsomal type 1; LKM1) were significantly elevated and IgG auto-antibodies targeting 2/124 antigens (Collagen II and Mi-2) were significantly elevated relative to controls (Table S1; *p<0.05). Hierarchical clustering revealed that the pattern of IgM auto-antibody activity in 5/6 -/- animals was significantly distinct from +/+ controls and 2/4 +/- animals (Fig 5E). Intriguingly, the other 2/4 +/- animals displayed an intermediate pattern of IgM auto-antibody reactivity, clustering with the remaining -/- mice (Fig 5E). Clustering based on IgG-recognized auto-antigens placed all mutants in one distinct group, with no obvious distinction between +/- and +/+ found in this case (Fig 5F). Thus, we conclude that loss of C9orf72 results in the accumulation of a wide array of self-reactive antibodies, which is indicative of an autoimmune phenotype.
As T regulatory cells restrict inflammation, we measured the percentage of CD25+ CD4+ cells, which are enriched for this T cell subtype, in spleens from 400 day old Neo deleted animals and found that CD25+ CD4+ cells were significantly elevated in -/- spleens relative to +/+ animals (Fig S12; **p<0.01).
C9orf72 promotes tolerance in the immune system
The Immgen database(27) indicates that C9ORF72 and its murine ortholog are expressed in blood cells. We therefore asked whether C9orf72 acts within the hematopoietic compartment. To address this, we performed bone marrow transplantation (BMT) between animals of distinct genotypes. Because of their inbred nature, we performed reciprocal BMT experiments using KOMP mice. To serve as phenotypic negative and positive controls, we transplanted -/- bone marrow into -/- recipients (-/-→-/-)(n=6) and +/+ marrow into +/+ recipients (WT→WT)(Fig 6A). To test whether a -/- genotype in bone marrow-derived cells was sufficient to cause autoimmunity, we transplanted -/- bone marrow into +/+ recipients (-/-→WT)(n=12). Reciprocally, we asked whether loss of C9orf72 in the blood was necessary for the development of autoimmunity by attempting to ameliorate the phenotype of -/- animals with transplantation of +/+ marrow (WT→-/-)(n=7). B6.SJL-PtprcaPepcb/BoyJ mice were used as wildtype bone marrow donors and recipients to facilitate tracking of reconstitution efficiency (Fig 6A). No animals perished immediately following BMT. By 17 weeks, 29/30 transplanted animals exhibited >90% reconstitution of CD45+ cells including CD11b+ Ly6G- monocytes, CD11b+ Ly6G+ neutrophils, and B220+ B cells (Fig 6B-C; Fig S13), indicating engraftment of donor hematopoietic cells was efficient and sustained. However, we did note that CD3+ T cell reconstitution was incomplete, ranging from 40 to 80% donor-derived cells (Fig 6B-C; Fig S13), which can be observed due to the relative radio-resistance and long-lived nature of this cell type(28).
Fig. 6. C9orf72 acts in bone marrow derived cells to prevent autoimmunity.
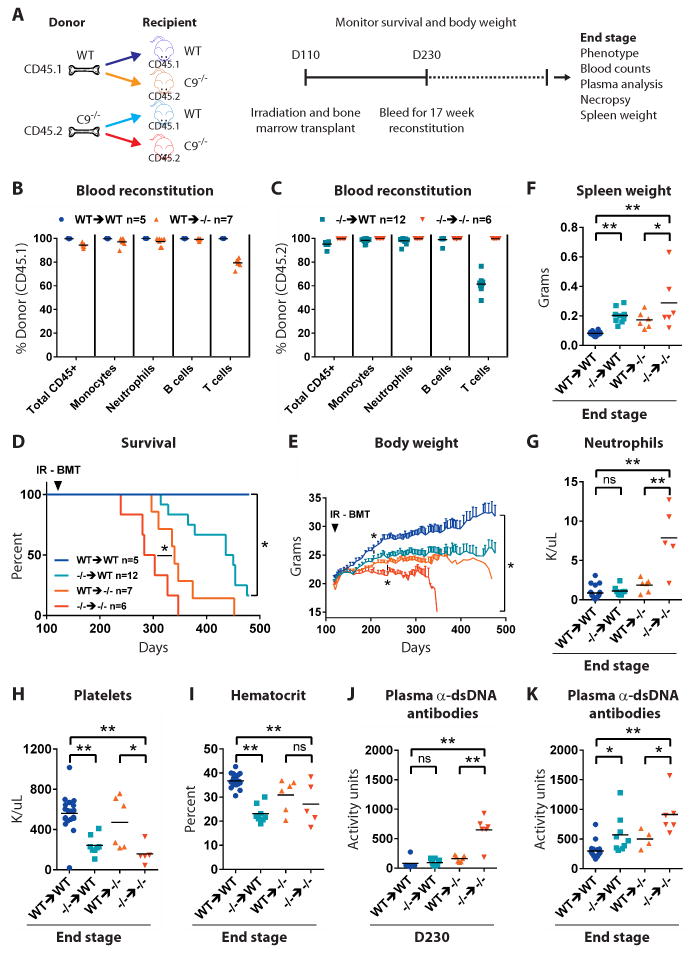
(A) Wild type or C9orf72 deficient animals were lethally irradiated at day 110 and reconstituted with wild type or mutant bone marrow. Recipient mice were regularly weighed, monitored for survival and bled for whole blood counts and plasma analyses and necropsied at end stage. (B, C) Quantification of flow cytometry-based assessment of 17 week post-transplant peripheral blood reconstitution. (B,C, F-K) Each dot represents one mouse. (D) Survival curves for transplanted mice. *p<0.05 Generalized Wilcoxon test. (E) Average body weight ± SEM. *p<0.05 Dunnett's multiple comparisons. (F) End-stage spleen weight. (F-K) *p<0.05, **p<0.01 Tukey's multiple comparisons. ns not significant. (G) End-stage peripheral blood neutrophil counts. (H) End-stage peripheral blood platelet counts. (I) End-stage hematocrit. (J-K) Plasma anti-dsDNA antibody activity in animals at (J) day 230 and (K) end-stage.
All WT→WT animals survived beyond 460 days of age (Fig 6D) and progressively gained weight (Fig 6E). In line with our previous findings (Fig 2), we noted that mice lacking C9orf72 (-/-→-/-) failed to gain weight as fast as their wild type (WT→WT) counterparts, a difference that became significant by 174 days of age (Fig 7E; *p<0.05). Moreover, all -/-→-/- animals (n=6/6) died by 350 days, with a median survival of 293 days (Fig 6D; *p<0.05), and the causes of death or obligatory euthanasia in -/-→-/- animals were consistent with those observed previously (Fig 2E; Fig S13; Video 1). At end stage, -/-→-/- mice had developed splenomegaly (Fig 6F; **p<0.01), neutrophilia (Fig 6G; **p<0.01), thrombocytopenia (Fig 6H; **p<0.01) and anemia (Fig 6I; **p<0.01). Anti-dsDNA antibody activity was significantly elevated in -/-→-/- at an age that preceded mortality (day 230) (Fig 6J; **p<0.01) as well as at end stage (Fig 6K; **p<0.01). Thus, lethal irradiation and bone marrow transplantation did not affect the development of autoimmunity in -/- animals.
Fig. 7. CRISPR/Cas9 induced mutations in C9orf72 lead to autoimmunity.
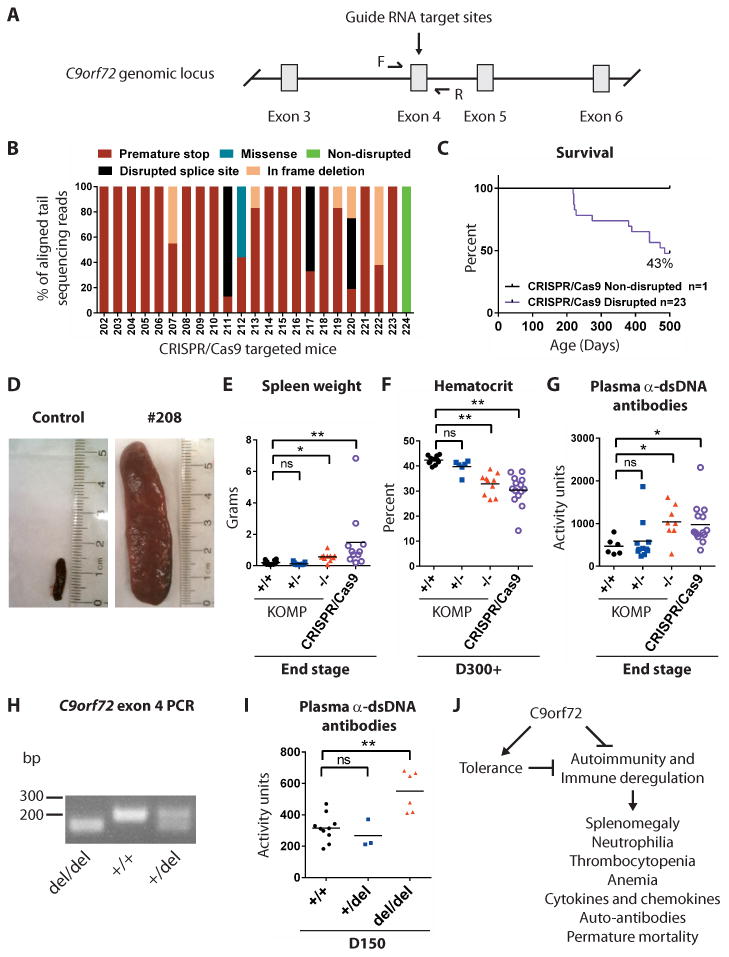
(A) Schematic showing the CRISPR/Cas9 targeting strategy to cause DNA double strand breaks in exon 4 of C9orf72, resulting in several distinct mutations. (B) DNA sequences from CRISPR/Cas9-targeted mice indicated that 23/24 animals harbored a stop codon in exon 4 of the C9orf72 gene. (C) Survival of CRISPR/Cas9 targeted mice. (D) Spleens from a CRISPR/Cas9-targeted mouse at end-stage and age-matched C57BL/6 control. (E) Spleen weights for CRISPR/Cas9 mutant and KOMP mice. (E-G) Each dot represents one mouse. *p<0.05 **p<0.05 Dunn's multiple comparisons. (F) Hematocrit for day 300+ CRISPR/Cas9-targeted animals. (G) Plasma anti-dsDNA antibody activity in day 300+ CRISPR/Cas9 targeted animals. (H) Mice harboring a 38-nucleotide deletion in exon 4 of C9orf72 were bred to heterozygosity (+/del) and homozygosity (del/del), as visualized by PCR of tail DNA using primers flanking the deletion site. (I) Plasma anti-dsDNA antibody reactivity in day 150 +/+, +/del and del/del animals. **p<0.01 Tukey's multiple comparisons. ns not significant. (J) Proposed model for how C9orf72 may act in bone marrow-derived cells to limit fatal immune deregulation.
We asked which of the phenotypes found in -/- animals could be observed in +/+ animals receiving -/- bone marrow. We found -/-→WT animals were significantly smaller than WT→WT animals at 206 days of age, after which their body weight plateaued (Fig 6E; *p<0.05). Strikingly, 9/12 -/-→WT mice died by 460 days of age with a median survival of 443 days (Fig 6D; *p<0.05 relative to WT→WT ; Video 2). At end stage, all -/-→WT (n=9/9) had larger spleens (Fig 6F; **p<0.01), while a subset of -/-→WT mice (3/12) had developed enlarged cervical lymph nodes (Fig S13). Relative to WT→WT mice, -/-→WT animals had similar numbers of neutrophils (Fig 6G), but displayed a significantly lower platelet count (Fig 6H; **p<0.01), and had significantly reduced hematocrit (Fig 6I; **p<0.01). IL-22, IL-31 and MIP-1B were significantly elevated in -/-→WT animals relative to WT→WT at end stage (Fig S13; *p<0.05). Anti-dsDNA antibody activity was similar in -/-→WT and WT→WT animals at day 230 (Fig 6J), yet by end stage -/-→WT displayed significantly elevated anti-dsDNA antibody activity (Fig 6K; *p<0.05).
Next we asked whether transplantation of WT bone marrow into -/- animals could improve their phenotype. By 230 days of age, WT→-/- mice had gained more weight than their -/-→-/- counterparts (Fig 6E; *p<0.05). Moreover, WT→-/- animals lived significantly longer than -/-→-/- animals, with the longest lived WT→-/- animals surviving to 452 days of age and with a median survival of 340 days, representing a 43 day extension of lifespan (Fig 6D; *p<0.05 relative to -/-→-/-; Video 3). Splenomegaly was apparent in all WT→-/- animals at end stage, but on average their spleens were smaller than -/-→-/- mice (Fig 6F; **p<0.01). Relative to -/-→-/- animals, WT→-/- mice had significantly fewer neutrophils (Fig 6G; **p<0.01) and significantly elevated platelet counts (Fig 6H; **p<0.01), but hematocrit was not significantly improved (Fig 6I). IL-31, IL-6, MIP-1B, IL-10, IL-17A and IL-15/IL-15R were significantly reduced in WT→-/- relative to -/-→-/- at end stage (Fig S13; *p<0.05). Anti-dsDNA activity was significantly reduced in WT→-/- relative to -/-→-/- animals at day 230 (Fig 6J; **p<0.01) and at end stage (Fig 6K; *p<0.05).
Generation of C9orf72 mutant mice using CRISPR/Cas9
We noted that some non-coding regions within the deleted locus showed a degree of conservation that raised the possibility that their disruption could contribute to the phenotypes observed in KOMP and Neo deleted animals. To rule out this possibility, we utilized CRISPR/Cas9 technology to induce deletion mutations in exon 4 of C9orf72 (Fig 7A), an exon present in all transcribed isoforms of both human and murine orthologs. Next generation sequencing of PCR amplicons spanning the target site revealed disruption of C9orf72 exon 4 in 23/24 CRISPR/Cas9 targeted mice (Fig 7B, Fig S14; Table S2). In 11/24 CRISPR/Cas9 targeted animals we identified only premature stop codon mutations and no wild type sequences, suggesting that these mice were compound heterozygous loss of function mutants for C9orf72 (Fig 7B, Fig S14; Table S2).
All CRISPR/Cas9 targeted mice survived to adulthood, yet as they aged their propensity to die increased with only 10/24 animals surviving beyond 500 days of age (Fig 7C). Splenomegaly was apparent in all mice (n=7/7) necropsied (Fig 7D-E), while protrusions in the neck area indicative of enlarged cervical lymph nodes, were apparent in 3/24 animals (Fig S14). Analysis of blood from mutant mice at day 310 revealed a significant reduction in hematocrit (Fig 7F; **p<0.01) and a significant increase in anti-dsDNA antibody activity (Fig 7G; *p<0.05).
As CRISPR/Cas9 targeted animals were mosaic for mutations in C9orf72, we crossed F0 mice with C57BL/6 to isolate F1 progeny harboring one wildtype allele and one mutated allele (Fig S14). We then interbred F1 progeny containing a 38-nucleotide deletion in C9orf72 exon 4, predicted to generate a premature stop codon after 183 amino acids, to generate animals that were wild-type (+/+), heterozygous (+/del), and homozygous (del/del) for this particular mutation (Fig. 7H and fig. S14). We again found that C9orf72 del/del F2 mutants displayed significantly elevated anti-dsDNA antibody activity by day 150 (Fig. 7I; **P < 0.01).
Discussion
Here we report that eliminating function of mouse C9orf72 predisposes animals to fatal immune defects and autoimmunity. We observed that these classic features of autoimmunity (24,29) developed synchronously, well before the onset of premature mortality. Furthermore, heterozygous animals were more susceptible than control mice to the generation of a limited repertoire of auto-antibodies and were at increased risk of early mortality. Chronic inflammation is a known contributor to most, if not all of the causes of death we observed in +/- and -/- animals. Notably, the pattern of inflammatory cytokines chronically upregulated in -/- animals included several members of the IL-23/IL-17 immune axis, which plays a pathogenic role in rheumatoid arthritis, psoriasis, Crohn's disease and multiple sclerosis (30).
Our findings led us to posit that C9orf72 serves an important function in the hematopoietic system. This hypothesis was substantiated by transplant studies in which mutant bone marrow transferred autoimmune phenotypes to wildtype recipients and wildtype bone marrow suppressed immune deregulation and significantly extended lifespan in mutant animals. However, while the phenotype of mutant animals that received wildtype marrow was improved relative to mutant mice receiving mutant bone marrow, their phenotype was still substantially worse than wildtype animals receiving wildtype bone marrow. Failure of wildtype bone marrow to completely rescue mutant mice leaves open the possibility that C9orf72 also functions in a radiation-resistant cell population, such as microglia, which are peripheral antigen presenting cells the dysfunction of which could lead to improper priming of adaptive immune responses., C9orf72 might also function in the thymic epithelium, which is critical for the establishment of peripheral tolerance. Alternatively, T cells themselves are well known modulators of immunity and the incomplete replacement of this compartment after transplantation provides another potential explanation for the incomplete rescue observed. Our findings, and those that indicate T cell function becomes compromised in mice harboring mutations in additional ALS-implicated genes (19,31), suggest that investigating whether C9orf72 functions in this cell type is warranted. Although we observed an increase in the abundance of CD25+CD4+ cells, which are enriched for T regulatory cells, in the spleens of mutant animals, whether mutant T regulatory cells harbor functional deficits that contribute to the phenotypes we observed remains to be determined. Elevation of T regulatory cells could represent a compensatory reaction to reduced functionality of this cellular subset or a homeostatic response to persistent inflammation.
We note that our study does have certain limitations and leaves several questions unresolved. While our studies demonstrate that bone marrow cells play a role in phenotypes we observed and suggests that C9orf72 functions in hematopoietic derivatives of the bone marrow, we cannot rule out the possibility that C9orf72 acts through marrow resident mesenchymal cells. Additionally, we did not perform an exhaustive census of transplanted animals to monitor the extent of engraftment into solid tissues including the brain. Therefore, we cannot address whether incomplete imposition or rescue of mutant phenotypes after transplant might be due to variable infiltration of cells into a given solid organ(32), rather than due to the function of C9orf72 in non-hematopoietic cell types. In the future, it will be important to resolve the identity of the specific cell types in which C9orf72 functions to preserve a normal immune response. We previously showed that C9orf72 was highly expressed in neuronal cell types that are most sensitive to cell death in ALS and FTD, but is more modestly expressed in microglia. Despite the high level of C9orf72 expression in the neuronal lineage, the data we report here suggests this gene acts through bone-marrow derived cells in which it is expressed. It remains to be determined whether C9orf72 serves some non-essential function in neurons that express it.
Recently, haploinsufficiency for TBK1, a kinase functioning in interferon signaling and selective autophagy(33), has been shown to cause ALS and FTD (Reviewed in 34). Whereas Tbk1 -/- mice display an embryonic lethal phenotype, deletion of this gene on an outbred background leads to monocytosis, splenomegaly and infiltration of immune cells into the skin, lung, liver and kidneys (35). Interestingly, Cre-mediated ablation of Tbk1 in T cells resulted in their activation and over accumulation in the spleen and lymph nodes(19). These phenotypic commonalities observed upon loss of Tbk1 and C9orf72 are striking. Whether these gene products act in a common pathway requires further investigation, however it is notable that at least one report has suggested that C9ORF72 may also function in autophagy (11).
Whether malfunctions in the immune system directly contribute to degenerative phenotypes in ALS patients remains unresolved(36,37). Studies have identified infiltrating T cells and hyper-activation of resident immune cells in the post-mortem spinal cords of ALS patients(38,39). Patients with a rapid course of disease were also found to have fewer circulating T regulatory cells than did individuals with slower disease progression(40), while levels of the pro-inflammatory cytokines IL-17 and IL-23 were found to be elevated in plasma and cerebral spinal fluid from at least one ALS cohort(41). Recently, a large epidemiologic study found substantially higher rates of autoimmune history in patients eventually developing ALS(42). However, studies of auto-antibodies in ALS patients have been mixed, with some finding evidence of their presence, while others have not(36,37). The ever-improving understanding of the genetic contributors to ALS suggests that revisiting immune phenotypes in patients that have been stratified by C9ORF72, TBK1 and other genotypes, could now be warranted.
While our manuscript was under review, additional phenotypic studies of C9orf72-deficient mice were reported (43,44). These reports both provided direct confirmation of our finding that C9orf72 mutant animals develop splenomegaly and lymph adenopathy. Furthermore, Atanasio and colleagues found, as we report here, that mutant animals exhibited elevated levels of plasma cytokines and autoantibodies (43). However, there were a number of differences between our findings and these recent studies that warrant future resolution. Most notably, while both we as well as O'Rourke and colleagues generated C9orf72 mutant animals using the same targeting vector generated by the Knock Out Mouse Project Consortium, heterozygous and homozygous mutants in our colony displayed an increased risk of mortality while mutants in their colony did not. This discrepancy suggests that either environmental factors or differences in genetic background may influence the longevity of mice lacking C9orf72, as is the case with other models of autoimmunity(29). Additionally, both Atanasio and O'Rourke found an increase in macrophage number in mutant animals and suggested that inflammatory phenotypes found in these mice may be due to dysfunction in this or other phagocytic cell-types. Our conclusion that C9orf72 functions in a bone-marrow derived cells is consistent with this idea. However, we also observed a much broader proliferative disorder impacting both myeloid and lymphoid lineages, leaving open the question of whether this gene may act in additional hematopoietic cell-types. In the future, conditional ablation of C9orf72 in specific blood lineages should address where precisely this gene functions to suppress pathological levels of inflammation.
Overall, our findings have implications for planned therapeutic intervention in ALS patients harboring the C9ORF72 repeat expansion. Regardless of whether loss of function in C9ORF72 plays a central role in the development of ALS, our experiments do strongly suggest that therapeutic efforts to reduce expression of the repeat expansion, such as with anti-sense oligonucleotides (9), should be designed and carried out with caution. If not, chronic depression of the limited quantity of normal gene product that still remains in these ALS patients could occur, potentially resulting in autoimmunity.
Materials and Methods
Study design
The goal of this study was to characterize the consequences of C9orf72 loss of function mutations in mice. The experimental design involved long-term survival studies paired with age-matched and genotype-matched histologic, cellular and biofluidic analysis. Pre-defined endpoint criteria included greater than 15% loss of body weight, signs of distressed breathing, enlarged abdomen, severe dermatitis, lymph nodes greater than 15 mm that inhibited mobility, inability of the mouse to right itself, or severe prolapse. Exploratory experiments were performed in excess of five mice per genotype. Sample sizes for BMT studies were calculated using G-power analysis based on previously defined effect sizes. Donor and recipient mice were randomized into treatment groups and all outliers were included in data analysis. All experimental protocols and procedures were approved by the Animal Committee of Harvard University.
RNA extraction and quantitative PCR
Total RNA was isolated using Trizol(GIBCO). First-strand cDNA was synthesized using iScript(Biorad). C9orf72 transcripts abundance we analyzed using SYBR Green on CFX96 Real-time System(ABI). C9orf72 exon 6-8 primers: 5′-GCAGTGCAGAGAAAGTAAATAAGATAG-3′ and 5′-ACTGCCTGTTGCATCCTTTAG-3′.
Fixation, sectioning and tissue staining
Mice were anesthetized with Avertin, perfused with PBS then 4% PFA. Tissues were harvested and post-fixed in 4% PFA overnight at 4°C. Tissue was dehydrated, embedded in paraffin, sectioned at 6um and stained for hematoxylin and eosin. Sections examined in a blinded manner and imaged using Zeiss AX10.
Western blotting
Total protein was extracted from frozen brain with a reducing sample buffer (RIPA) containing complete inhibitor cocktail(Roche). 15ug of protein was separated on 4-20% SDS-polyacrylamide gel, transferred to pvdf membrane, blocked with 5% BSA, incubated with primary antibodies (anti-C9ORF72-N-terminal(ProteinTech), anti-C9ORF72-C-terminal(Abgent), anti-α-tubulin(Abcam)) and detected by enhanced chemiluminescence(GE Amersham).
Blood and cytokine measures
Peripheral blood was collected via mandible puncture into EDTA-coated tubes. Blood counts were assessed using a Hemavet(Abaxis), then centrifuged to pellet cells and plasma harvested from supernatant. Luminex-based multiplexed fluorescence assay was used to assess 36 cytokines and chemokines.
Auto-antibody profiling
Plasma was diluted 1:200 and assessed using mouse α-dsDNA total-Ig kit(Alpha Diagnostic International). Antibodies against 124 autoantigens were measured on autoantigen arrays(26).
Spleen and lymph node FACS
Spleens were mashed between two glass slides and passed repeatedly through a 25 5/8G needle to dissociate single cells. Following RBC lysis, cells were quantified using a Countess(Invitrogen). Lymph nodes were processed as above without RBC lysis. Cells stained using these antibodies(Biolegend): B220(RA3-6B2), CD19(6D5), Kappa(RMK-45), Lambda(RML-42), CD11c(N418), CD8α(53-6.7), CD3ε(145-2C11), CD4(GK1.5), CD62L(MEL-14), CD44(IM7), CD25(PC61), Pan-NK(DX5), CD11b(M1/70), Ly6G(1A8), CD115(AFS98), F4/80(BM8), and DAPI(Sigma).
Bone marrow transplantation
Female B6.SJL-PtprcaPepcb/BoyJ(Jackson Labs) mice were used as age matched WT bone marrow donors and recipients. Female KOMP C9orf72 -/- mice were used as age matched bone marrow donors and recipients. At D110, marrow from femur and tibias were harvested from two WT and two -/- donors, flushed, single cell dissociated, counted, pooled and diluted to 5E6 cells/100uL. Recipient animals received two rounds of 550rad separated by 3 hours. 5E6 bone marrow cells were injected into the tail vein. Recipients were maintained on pH 3.0 water for two weeks. Reconstitution efficiency in RBC-lysed peripheral blood at 17 weeks post-transplant was measured using these antibodies(Biolegend): CD45.2(104), CD3ε(145-2C11), B220(RA3-6B2), Ly6G(1A8), CD11b(M1/70), CD45.1(A20).
CRISPR/Cas9 sequencing
Tail DNA was PCR amplified with primers containing flanking adaptors followed by PCR barcoding with 24 unique barcodes. Samples were pooled, cleaned (Promega Wizard DNA-cleanup kit), sequenced via MiSeq and analyzed(45).
Statistics
All statistical calculations were performed using Graphpad Prism. Tests between two groups used two-tailed Student t-test. Tests between multiple groups used One-way ANOVA with Tukey's multiple comparisons. Tests between multiple groups over time used Two-way ANOVA with Dunnett's multiple comparisons. Survival curves were evaluated by Generalized Wilcoxon.
Supplementary Material
Supplementary Materials
Fig. S1 Validation of C9orf72 loss of function allele.
Fig. S2 Analysis of spinal motor neurons.
Fig. S3 Motor Cortex histology.
Fig. S4 Histology of Thalamus, CA1 and Cerebellum.
Fig. S5 GFAP expression in the CNS
Fig. S6 Individual mouse weight curves after disease onset.
Fig. S7 Splenocyte gating scheme.
Fig. S8 Bone marrow histology and cell counts.
Fig. S9 Analysis of B cells and T cells in cervical lymph node.
Fig. S10 Hematopoietic liver infiltrates occur in a subset of mutant animals.
Fig. S11 Analysis of cytokines and chemokines
Fig. S12 Analysis of CD4+ CD25+ cells.
Fig. S13 Cellular and organismal phenotypes following bone marrow transplant.
Fig. S14 CRISPR/Cas9-targeted mutations in exon 4 of C9orf72.
Table S1 Analysis of autoantibodies in Neo deleted mice.
Table S2 CRISPR/Cas9-induced mutations in C9orf72 exon 4.
Video 1 End stage -/- marrow donor to -/- recipient.
Video 2 End stage -/- marrow donor to WT recipient.
Video 3 End stage WT marrow donor to -/- recipient.
Editor's Summary.
C9orf72 acts in hematopoietic cells to suppress autoimmunity
Mutations in C9ORF72 are a common contributor to amyotrophic lateral sclerosis and frontotemporal dementia, yet the function of this gene is still poorly defined. In new work, Burberry et al. demonstrate that mutations disrupting the normal function of C9orf72 cause mice to develop features of autoimmunity. They further found that transplantation of normal bone marrow into mutant animals ameliorated their condition, whereas transplantation of mutant bone marrow into normal animals was sufficient to cause autoimmunity. The authors conclude that C9orf72 acts through hematopoietic cells to maintain normal immune function and suggest that investigations into whether disruptions in immunity contribute to disease in patients are warranted.
Acknowledgments
The authors thank Eggan lab, HSCRB Histology Core, M. Charlton, A. Wagers, D. Scadden and K. Hochedlinger for advice, technical support and manuscript review.
Funding: K.E. is supported by HHMI, p2ALS, Target ALS and NIH5R01NS089742. F.M. was supported by Wellcome Trust, MRC and NIH5K99NS083713.
Footnotes
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.
Competing interests: Authors declare no competing financial interests.
Data materials availability: Whole genome sequencing data for this study have been deposited in Sequence Read Archive SRP073407.
Author contributions: K.E. conceived project; K.E., A.B. and N.S. designed experimental plan. A.B., N.S., J.W., A.S, K.K. and S.S-U supported animal experiments. R.M. supported DNA sequence analysis. D.M. evaluated staining. S.G. and Q-Z.L. performed auto-antibody array. A.S. M.S., J.J.T., D.J.R., L.Z. and L.D.N. performed hematological analysis. A.B., F.M. performed CRISPR/Cas9 design. A.B., N.S., R.M. and K.E. wrote manuscript. K.E. supervised project.
References
- 1.Ludolph AC, Brettschneider J, Weishaupt JH. Amyotrophic lateral sclerosis. Curr Opin Neurol. 2012;25:530–535. doi: 10.1097/WCO.0b013e328356d328. [DOI] [PubMed] [Google Scholar]
- 2.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 3.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GYR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, McLaughlin RL, Iyer PM, O'Brien C, Phukan J, Wynne B, Bokde AL, Bradley DG, Pender N, Al-Chalabi A, Hardiman O. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11:232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, Nicholson GA. Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry. 2013;84:931–935. doi: 10.1136/jnnp-2012-304529. [DOI] [PubMed] [Google Scholar]
- 6.Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 8.Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74:180–187. doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 9.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinforma Oxf Engl. 2013;29:499–503. doi: 10.1093/bioinformatics/bts725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RAK, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–3595. doi: 10.1093/hmg/ddu068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PloS One. 2013;8:e83450. doi: 10.1371/journal.pone.0083450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki N, Maroof AM, Merkle FT, Koszka K, Intoh A, Armstrong I, Moccia R, Davis-Dusenbery BN, Eggan K. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci. 2013;16:1725–1727. doi: 10.1038/nn.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koppers M, Blokhuis AM, Westeneng H-J, Terpstra ML, Zundel CAC, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015 doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rijli FM, Dollé P, Fraulob V, LeMeur M, Chambon P. Insertion of a targeting construct in a Hoxd-10 allele can influence the control of Hoxd-9 expression. Dev Dyn Off Publ Am Assoc Anat. 1994;201:366–377. doi: 10.1002/aja.1002010408. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi S, Lewis P, Pevny L, McMahon AP. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev. 2002;119(Suppl1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 17.Whitehurst CE, Chattopadhyay S, Chen J. Control of V(D)J Recombinational Accessibility of the Dβ1 Gene Segment at the TCRβ Locus by a Germline Promoter. Immunity. 1999;10:313–322. doi: 10.1016/s1074-7613(00)80031-x. [DOI] [PubMed] [Google Scholar]
- 18.Rozman C, Montserrat E, Rodriguez-Fernandez JM, Ayats R, Vallespi T, Parody R, Rios A, Prados D, Morey M, Gomis F. Bone marrow histologic pattern--the best single prognostic parameter in chronic lymphocytic leukemia: a multivariate survival analysis of 329 cases. Blood. 1984;64:642–648. [PubMed] [Google Scholar]
- 19.Yu J, Zhou X, Chang M, Nakaya M, Chang JH, Xiao Y, Lindsey JW, Dorta-Estremera S, Cao W, Zal A, Zal T, Sun SC. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun. 2015;6:6074. doi: 10.1038/ncomms7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore SW, Schneider JW, Schaaf HS. Diagnostic aspects of cervical lymphadenopathy in children in the developing world: a study of 1,877 surgical specimens. Pediatr Surg Int. 2003;19:240–244. doi: 10.1007/s00383-002-0771-x. [DOI] [PubMed] [Google Scholar]
- 21.Murakami J, Shimizu Y, Murakami J, Shimizu Y. Hepatic Manifestations in Hematological Disorders, Hepatic Manifestations in Hematological Disorders. Int J Hepatol Int J Hepatol. 2013:e484903. doi: 10.1155/2013/484903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell ES, Neufeld EF, Higgins CT. Comparison of normal blood picture of young adults from 18 inbred strains of mice. Proc Soc Exp Biol Med Soc Exp Biol Med N Y N. 1951;78:761–766. doi: 10.3181/00379727-78-19210. [DOI] [PubMed] [Google Scholar]
- 23.Freedman J. Autoimmune Hemolysis: A Journey through Time. Transfus Med Hemotherapy. 2015;42:278–285. doi: 10.1159/000437195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giles BM, Boackle SA. Linking complement and anti-dsDNA antibodies in the pathogenesis of systemic lupus erythematosus. Immunol Res. 2013;55:10–21. doi: 10.1007/s12026-012-8345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su KY, Pisetsky DS. The Role of Extracellular DNA in Autoimmunity in SLE. Scand J Immunol. 2009;70:175–183. doi: 10.1111/j.1365-3083.2009.02300.x. [DOI] [PubMed] [Google Scholar]
- 26.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–3439. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heng TSP, Painter MW. Immunological Genome Project Consortium, The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 28.Bosco N, Swee LK, Bénard A, Ceredig R, Rolink A. Auto-reconstitution of the T-cell compartment by radioresistant hematopoietic cells following lethal irradiation and bone marrow transplantation. Exp Hematol. 2010;38:222–232.e2. doi: 10.1016/j.exphem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Crampton SP, Morawski PA, Bolland S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis Model Mech. 2014;7:1033–1046. doi: 10.1242/dmm.016451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A. 2008;105:17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwartz M, Baruch K. Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. J Autoimmun. 2014;54:8–14. doi: 10.1016/j.jaut.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Chau TL, Gioia R, Gatot JS, Patrascu F, Carpentier I, Chapelle JP, O'Neill L, Beyaert R, Piette J, Chariot A. Are the IKKs and IKK-related kinases TBK1 and IKK-ε similarly activated? Trends Biochem Sci. 2008;33:171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Bettencourt C, Houlden H. Exome sequencing uncovers hidden pathways in familial and sporadic ALS. Nat Neurosci. 2015;18:611–613. doi: 10.1038/nn.4012. [DOI] [PubMed] [Google Scholar]
- 35.Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, Goutagny N, Kuang W, Askew GR, Roberts V, Benoit S, Zhou T, Ling V, Pfeifer R, Stedman N, Fitzgerald KA, Lin LL, Hall JP. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol. 2010;88:1171–1180. doi: 10.1189/jlb.0210071. [DOI] [PubMed] [Google Scholar]
- 36.Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol. 2013;8:888–899. doi: 10.1007/s11481-013-9489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pagani MR, Gonzalez LE, Uchitel OD, Pagani MR, Gonzalez LE, Uchitel OD. Autoimmunity in Amyotrophic Lateral Sclerosis: Past and Present. Neurol Res Int. 2011:e497080. doi: 10.1155/2011/497080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Troost D, van den Oord JJ, de Jong JM, Swaab DF. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol. 1989;8:289–294. [PubMed] [Google Scholar]
- 39.Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 40.Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5:64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rentzos M, Rombos A, Nikolaou C, Zoga M, Zouvelou V, Dimitrakopoulos A, Alexakis T, Tsoutsou A, Samakovli A, Michalopoulou M, Evdokimidis J. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand. 2010;122:425–429. doi: 10.1111/j.1600-0404.2010.01333.x. [DOI] [PubMed] [Google Scholar]
- 42.Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology. 2013;81:1222–1225. doi: 10.1212/WNL.0b013e3182a6cc13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KMV. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. doi: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AKMG, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–1329. doi: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmid-Burgk JL, Schmidt T, Gaidt MM, Pelka K, Latz E, Ebert TS, Hornung V. OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 2014;24:1719–1723. doi: 10.1101/gr.176701.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials
Fig. S1 Validation of C9orf72 loss of function allele.
Fig. S2 Analysis of spinal motor neurons.
Fig. S3 Motor Cortex histology.
Fig. S4 Histology of Thalamus, CA1 and Cerebellum.
Fig. S5 GFAP expression in the CNS
Fig. S6 Individual mouse weight curves after disease onset.
Fig. S7 Splenocyte gating scheme.
Fig. S8 Bone marrow histology and cell counts.
Fig. S9 Analysis of B cells and T cells in cervical lymph node.
Fig. S10 Hematopoietic liver infiltrates occur in a subset of mutant animals.
Fig. S11 Analysis of cytokines and chemokines
Fig. S12 Analysis of CD4+ CD25+ cells.
Fig. S13 Cellular and organismal phenotypes following bone marrow transplant.
Fig. S14 CRISPR/Cas9-targeted mutations in exon 4 of C9orf72.
Table S1 Analysis of autoantibodies in Neo deleted mice.
Table S2 CRISPR/Cas9-induced mutations in C9orf72 exon 4.
Video 1 End stage -/- marrow donor to -/- recipient.
Video 2 End stage -/- marrow donor to WT recipient.
Video 3 End stage WT marrow donor to -/- recipient.