

Abstract
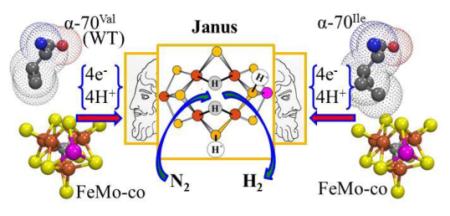
We have proposed a reductive elimination/oxidative addition (re/oa) mechanism for reduction of N2 to 2NH3 by nitrogenase, based on identification of a freeze-trapped intermediate of the α-70Val→Ile substituted MoFe protein as the Janus intermediate that stores four reducing equivalents on FeMo-co as two [Fe-H-Fe] bridging hydrides (denoted E4(4H)). The mechanism postulates that obligatory re of the hydrides as H2 drives reduction of N2 to a state (denoted E4(2N2H)) with a moiety at the diazene (HN=NH) reduction level bound to the catalytic FeMo-cofactor. In the present work, EPR/ENDOR and photophysical measurements show that a state freeze-trapped during N2 reduction by wild type (WT) MoFe protein is the same Janus intermediate, thereby establishing the α-70Val→Ile intermediate as a reliable guide to mechanism, and enabling new experimental tests of the re/oa mechanism with WT enzyme. These allow us to show that the re/oa mechanism accounts for the longstanding Key Constraints on mechanism. Monitoring the S = ½ FeMo-co EPR signal of Janus in WT MoFe during N2 reduction under mixed-isotope condition, H2O buffer/D2, and the converse, establishes that the bridging hydrides/deuterides do not exchange with solvent during enzymatic turnover, thereby explaining earlier observations and verifying the re/oa mechanism. Relaxation of E4(2N2H) to the WT resting-state is shown to occur via oa of H2 and release of N2 to form Janus, followed by sequential release of two H2, demonstrating the kinetic reversibility of the re/oa equilibrium. The relative populations of E4(2N2H) and E4(4H) freeze-trapped during WT turnover furthermore show that the rapidly reversible re/oa equilibrium between [E4(4H) + N2] and [E4(2N2H) + H2] is roughly thermoneutral (ΔreG0 ~ −2 kcal/mol), whereas hydrogenation of gas-phase N2 would be highly endergonic. These findings establish (i) that re/oa satisfies all key constraints on mechanism, (ii) that Janus is the key to N2 reduction by WT enzyme, which (iii) indeed occurs via the re/oa mechanism. Thus emerges a picture of the central mechanistic steps by which the nitrogenase MoFe protein carries out one of the most challenging chemical transformation in biology, the reduction of the N≡N triple bond.
Graphical abstract
Introduction
By catalyzing biological nitrogen fixation — the reduction of N2 to two NH3 molecules — nitrogenase generates the nitrogen-containing nutrients that support most of the biosphere, including over half the human population.1 But an understanding of the mechanism of N2 reduction by nitrogenase has been elusive. A ‘kinetic’ foundation for the mechanism of the Mo-dependent nitrogenase was developed through extensive studies in the 1970’s and 1980’s.2-4 The culmination of these measurements was the Lowe-Thorneley (LT) kinetic scheme for nitrogenase function, Fig 1,2-4 which describes the transformations among catalytic intermediates, denoted En where n is the number of steps of electrons/protons delivered from the nitrogenase Fe protein to the MoFe protein, which contains the active site iron-molybdenum cofactor ([7Fe-9S-Mo-C-R-homocitrate, denoted M]; FeMo-co) Fig 2. A defining feature of this scheme is the obligatory formation of one mole of H2 per mole of N2 reduced, which leads to a limiting stoichiometry for enzyme-catalyzed nitrogen fixation given by eq 1,
| (1) |
in agreement with stoichiometric measurements by Simpson and Burris.5 However, the obligatory requirement for H2 formation has not been universally accepted, and was even questioned in the culminating review of Burgess and Lowe: “Thus the data that support the obligatory evolution of one H2 for every N2 reduced are much less compelling than the data that require us to believe that some H2 will always be evolved during N2 reduction.”2
Figure 1.

Simplified Lowe-Thorneley (LT) kinetic scheme for nitrogen reduction2-4 that focus on the electron-accumulation and FeMo-co activation (boxed) stages. In the En notation, n = number of [e−/H+] added to FeMo-co; in parentheses, the stoichiometry of H/N bound to FeMo-co, except n = 7, 8, where structures are shown.
Figure 2.
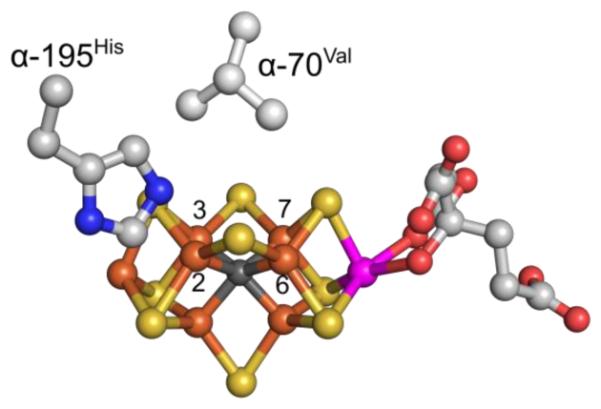
Crystal structure of FeMo-co. Fe is show in rust, Mo in magenta, S in yellow, carbide in dark-grey, carbon in gray, N in blue and O in red. The Fe atoms of catalytic 4Fe-4S face are labelled as 2, 3, 6, and 7. Two amino acids, α-70Val and α-195His, that approach the FeMo-co are also shown. The image was created using PDB coordinate 2AFI.
We recently proposed6,7 that obligatory H2 formation during nitrogenase N2 reduction is in fact required to explain the catalytic function of nitrogenase. This proposal originates with the characterization of an intermediate (FeMo-co spin S = ½) trapped during turnover of MoFe protein having the α-70Val→Ile substitution, which apparently inhibits access by all substrates to the active site, other than protons.8 A combination of 1,2H/95Mo ENDOR spectroscopy8,9 and cryoannealing ‘electron counting’10 showed this state to be the key E4(4H) ‘Janus’ intermediate, which has accumulated four of the eight reducing equivalents required by eq 1, storing them as two [Fe-H-Fe] bridging hydrides.8-10 E4(4H) sits at a transition in the N2 reduction pathway, Fig 1, poised to ‘fall back’ to the E0 resting state by successive release of two H2,10 but equally poised to eliminate H2 and proceed to the reduction of N2 to two NH3 through the accumulation of four more equivalents, hence the appellation, ‘Janus’.7
The discovery that the four reducing equivalents accumulated by E4(4H) are stored as bridging hydrides forged a connection between nitrogenase catalysis and the organometallic chemistry of metal hydrides11-13 that offered explanations of a multitude of features of nitrogenase mechanism that had defied explanation for decades.2 At the most basic level, this hydride formation helped to explain how a constant-potential electron donor, reduced Fe protein (Fered), could reduce FeMo-co by four equivalents. Moreover, bridging hydrides are less susceptible to protonation to form H2, or to solvent exchange, than terminal hydrides. As a consequence of the latter, bridging hydrides diminish the tendency of FeMo-co to ‘fall back’ to resting state through the formation of two H2 (Fig 1) However, the bridging mode also lowers hydride reactivity, relative to that of terminal hydrides. How the release of H2 contributes to the activation of this ‘deactivated’ intermediate for the hydrogenation of N2 to a moiety at the N2H2-reduction level is thus a central ‘mystery’ of N2 reduction by nitrogenase.
Importantly, reference to the inorganic chemistry of metal-dihydrides offered an explanation to this mystery, as well. Once it is recognized that E4(4H) contains two bridging hydrides, then the chemistry of metal-dihydride complexes11-13 identifies the LT E4(4H)↔E4(2N2H) mechanistically coupled equilibrium (Fig 1) as the reductive elimination (re) of H2 and its reverse, the oxidative addition (oa) of H2, Fig 3. We proposed that for nitrogenase, the re of H2 carries off two of the four reducing equivalents formally stored as the H−, driving the first, and most difficult, step of N2 cleavage; reduction of the N2 triple bond to a diazene-level (2N2H) moiety bound to FeMo-cofactor, Fig 3, by the remaining two reducing equivalents. This process fulfills one of the long-standing key constraints on mechanism: Chart 1, (i).2
Figure 3. Schematic of re/oa Equilibrium.
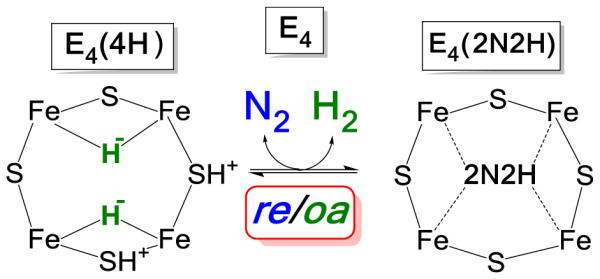
The cartoon represents the Fe 2,3,6,7 face of FeMo-co, and the ‘2N2H’ implies a species at the diazene reduction level of unknown structure and coordination geometry. In the indicated equilibrium the binding and activation of N2 is mechanistically coupled to the re of H2, as described in the text. Some of the potential complexities associated with this enzymatic process that underlie this cartoon are discussed in reference 18..
Chart 1.

Key Constraints on Nitrogenase Mechanism2
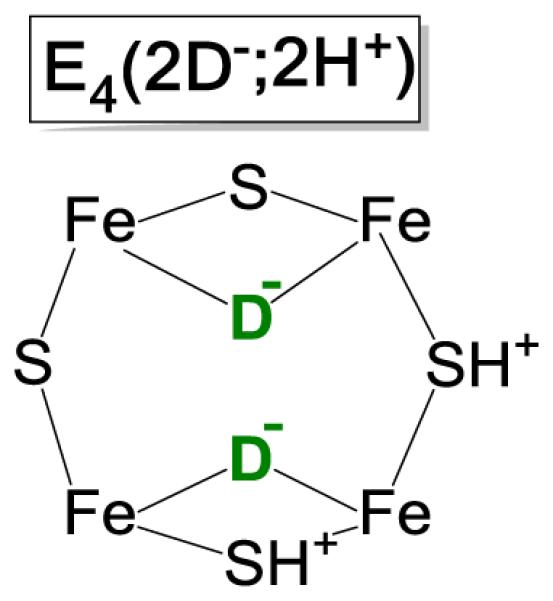
The proposal that N2 fixation requires the obligatory re of H2 was first supported by its ability to provide explanations of key constraints on nitrogenase mechanism summarized in Chart 1.2 These include not only the En state at which N2 is first reduced (Key Constraint (i)), but most especially previously baffling results from turnover in the presence of D2/T2. It was found that: Key Constraints (ii)), D2 only reacts with nitrogenase during turnover under an N2/D2 atmosphere; (iia) in this reaction D2 is stoichiometrically reduced to two HD (with H2O buffer); (iib), corresponding turnover with T2 does not lead to exchange of T+ into H2O solvent.2 These constraints are explained as arising through the reverse of the re process, Fig 3, namely the oxidative addition (oa) of H2 with loss of N2.6,7 According to the mechanism, during turnover under D2/N2, reaction of the E4(2N2H) intermediate with D2 generates dideutero-E4 with two [Fe-D-Fe] bridging deuterides which do not exchange with solvent.14 This E4(4H) isotopologue, which we denote E4(2D−;2H+) (Chart 2), would relax through E2(D−;H+) to E0 with successive loss of two HD 7,10.
Chart 2.
The proposed formation of the E4(2D−;2H+) and E2(D−;H+) states through the thermodynamically allowed reverse of re, the oa of D2 with accompanying release of N2, led to a successful test of their formation. During turnover under N2/D2, the non-physiological substrate acetylene (C2H2) intercepted these states to generate deuterated ethylenes (C2H3D and C2H2D2).14 More recently, we identified an S = ½ EPR signal that appears during N2 reduction by wild type (WT) MoFe protein as arising from FeMo-co of the E4(2N2H) state formed by re of H2 with N2 reduction, Fig 3, and in so doing confirmed the prediction that the (re/oa) activation equilibrium is not only thermodynamically, but also kinetically reversible.15 Characterization of the E4(4H) ‘Janus’ intermediate as carrying four reducing equivalents in the form of two [Fe-H-Fe] bridging hydrides thus laid the foundation for the re/oa mechanism, Fig 3, with its obligatory formation of one H2 per N2 reduced and resultant limiting stoichiometry of eq 1,15 while the success of predictions based on the mechanism provides powerful support for the mechanism.
We here report two major types of advance in our understanding of nitrogenase catalysis. The first was motivated by our recognition that, although the predictions of re/oa were tested in WT MoFe, the mechanism was founded on the presence and properties of the two [Fe-H-Fe] bridging hydrides in the Janus intermediate trapped and characterized in the α-70Val→Ile MoFe protein, which shows a much decreased ability to bind and reduce N2. This raised the question: Do the properties of the Janus intermediate in the MoFe variant accurately reflect those of the WT intermediate, and are mechanistic conclusions based on studies of the variant applicable to WT enzyme? This report answers these coupled questions: yes. We here establish that the same E4(4H) Janus intermediate, with its two [Fe-H-Fe] bridging hydrides, in fact does participate in catalysis by the WT enzyme through the re/oa mechanism for N2 reduction, thereby showing the α-70Val→Ile Janus intermediate to be a reliable guide to mechanism.
The second type involves observations founded on the ability to monitor both the E4(4H) and E4(2N2H) partners of the re/oa equilibrium in WT enzyme (Fig 3). This ability enables us to experimentally demonstrate that the re/oa mechanism indeed satisfies the Key Constraints of Chart 1, and beyond that to measure the energetics and kinetics of the equilibrium interconversion. Overall, this report yields a picture of the key mechanistic steps by which nitrogenase carries out one of the most challenging chemical transformation in biology, the reduction of the N≡N triple bond.16
Materials and Methods
Materials and Protein purifications
All the reagents were obtained from SigmaAldrich (St. Louis, MO) or Fisher Scientific (Fair Lawn, NJ) and were used without further purification. Argon and N2 gases were purchased from Air Liquide America Specialty Gases LLC (Plumsteadville, PA). Experiments were carried out with WT Azotobacter vinelandii MoFe protein expressed and purified as described elsewhere.17 The handling of all buffers and proteins were done anaerobically unless stated otherwise.
EPR and ENDOR samples
Samples were prepared as described under turnover conditions specified in figure legends; X-band samples contain 50 μM MoFe protein, 75 μM Fe protein, and the concentration of intermediates trapped during turnover is typically ~ 20 μM.15,18 For Q band samples, 100 μM MoFe and 150 μM Fe was used. All the samples were allowed to turnover for 20-25s, transferred to a X or Q band tube, and frozen with liquid nitrogen..15,18 It was found that during turnover of WT enzyme under low partial pressures of N2 a g1 = 2.15 species, shown below to be the E4(4H) intermediate in WT enzyme could be trapped in populations adequate for study, typically along with its ‘equilibrium partner’ the E4(2N2H) intermediate. The properties of g1 = 2.15, E4(4H) are compared with those of E4(4H) trapped in the α-70Val→Ile and α-70Val→Ile/α-195His→Gln MoFe proteins as described.17,18
EPR and ENDOR Measurements
X-band CW EPR spectra and Q-band CW EPR and 1,2H ENDOR spectra, including those during in situ photolysis by constant 450nm diode laser illumination, were collected as described.18 Data points for kinetics of photolysis and annealing were obtained as amplitudes of EPR signals at the well-resolved g1 features of the E4 states linked by re/oa. Tests of this protocol by EPR simulations show that the error of this quantitation approach should not exceed 15% when both E4(4H) and E4(4D) intermediates are present. We recall a time constant for photolysis is inversely proportional to the intensity of illumination and photolysis quantum yield.18 As a result the kinetic isotope effect on photolysis corresponds to the effect on quantum yield.
Results and Discussion
Enhancing the population of the WT g1 = 2.15 intermediate
Figure 4 presents EPR spectra of WT MoFe protein freeze-quenched under turnover at high and low partial pressure of N2, along with a spectrum of the E4(4H) intermediate trapped during turnover of α-70Val→Ile MoFe protein. The spectrum for the α-70Val→Ile enzyme shows the signal from the E4(4H) Janus intermediate, g = [2.15, 2.007, 1.965]. The WT enzyme turned over under high partial pressure of N2 (P(N2)) shows the EPR signal from the E4(2N2H) intermediate, in which FeMo-co binds a diazene-level product of N2 binding/reduction, g = [2.09, 1.99, 1.97]. However, WT enzyme freeze-trapped under low P(N2) turnover with high solution concentration of H2 (high effective P(H2), see figure legend and ref 15) shows only traces of the signal from E4(2N2H), and instead shows an EPR signal of an intermediate with g1 = 2.15, which is indistinguishable from that of E4(4H) in the MoFe α-70Val→Ile variant.8,10 Closer inspection of the high-P(N2) signal then shows a trace of the g1 = 2.15 feature, associated with a very low population of this intermediate, confirming its presence under all turnover conditions.
Figure 4.

X-band EPR spectra of WT nitrogenase turnover samples trapped under 1 atm of N2 (with stirring to facilitate transfer of H2 formed during turnover into the headspace15), and under low P(N2) in H2O buffer (without stirring) shown in comparison with spectrum of E4(4H) state trapped during turnover of α-70Val→Ile MoFe protein of the same concentration (see M&M). EPR conditions: temperature, 12 K; microwave frequency, 9.36 GHz; microwave power, 10 mW; modulation amplitude, 9 G; time constant, 160 ms; field sweep speed, 20 G/s.
Such a correlation of relative intensities with P(N2) and effective P(H2) is as expected if this g1 = 2.15 signal is indeed from WT E4(4H). In this case its concentration relative to that of E4(2N2H) is governed by the equilibrium of Fig 3, when, as expected, the forward and reverse steps of the re/oa equilibrium are rapid compared to the slow delivery of the next electron from Fered (Fig 1),
| (2) |
Here Kre is the equilibrium constant for the re/oa equilibrium, and a simple proportionality to P(N2) follows from our observation that at these high enzyme concentrations, turnover produces a roughly constant (saturating) concentration of H2 regardless of P(N2).15 Not only does this P(N2) dependence supports the idea that the g1 = 2.15 state is indeed the WT E4(4H), but of central importance to this study, the ability to prepare samples whose dominant EPR signal is that of the g1 = 2.15 state enables its characterization and identification, as now described.
1,2H Hyperfine Coupling to Bridging [Fe-H/D-Fe] in WT MoFe
ENDOR spectroscopy revealed the presence in E4(4H) of α-70Val→Ile and α-70Val→Ile/α-195His→Gln MoFe protein of two [Fe-H-Fe] bridging hydrides with anisotropic hyperfine tensors whose principal values are virtually identical, A1 = [11, 25, 37] MHz, A2 = [10, 24, 33] MHz, but whose orientations differ.8 To begin this investigation, we examined how the hyperfine coupling to the two hydrides influence the EPR spectrum of E4(4H) of α-70Val→Ile MoFe protein. Figure 5A presents CW X-band EPR spectra of this intermediate prepared in H2O and D2O buffers, along with expansions of the g1 and g3 regions of the spectra plus simulations that incorporate the ENDOR-derived hyperfine couplings to the hydrides/deuterides (the accompanying protons have only small couplings and do not significantly contribute). The difference in breadth of the g1 feature in H2O and D2O buffers is well-captured by the simulated 1H hyperfine broadening from the two hydrides, as calculated using the ENDOR-derived couplings. In particular, at g3, the EPR spectrum of the intermediate in H2O buffer even shows the 1-2-1 pattern arising from comparable couplings to the two 1H hydrides, and this too is reproduced by the simulation. The loss of this pattern in D2O buffer definitively confirms its identification with coupling to the two hydrides.
Figure 5.

(A) EPR spectra of WT and α-70Val→Ile turnover samples trapped in H2O (red) and D2O (blue) buffers. Spectra are shown after normalization to the same g1 feature amplitude and their extended g1 and g3 fragments are compared with EPR simulations obtained with hydrides/deuterides hyperfine interaction parameters known from previous ENDOR study.8 Red arrows indicate resolved features associated with hydrides hyperfine interaction. EPR conditions: temperature, 12 K; microwave frequency, 9.36 GHz; microwave power, 10 mW; modulation amplitude, 2.3 G; time constant, 160 ms; field sweep speed,10 G/s; 4 scans. (B) Mims 2H ENDOR spectra of WT low P(N2) turnover in D2O (middle) shown in comparison with previously obtained stochastic CW ENDOR of hydrides of E4(4H) trapped in α-70Val→Ile/α-195His→Gln protein during turnover in H2O (top).18 Spectra of 2H and 1H are scaled to the same Larmor frequency. In D2O spectra, down triangles indicate 'blind spots' of Mims ENDOR spectra with suppressed hyperfine couplings of A = n/τ , n = 1, 2, …; *-labeled signal in the H2O background Mims τ = 400 ns spectrum is associated with 5th harmonics of the matrix 1H response. Mims ENDOR conditions: microwave frequency, 34.743 GHz; π/2 = 50 ns; RF 20 μs; repetition time 50 ms; ~ 400-800 scans; temperature, 2 K.
The EPR spectrum of the WT g1 = 2.15 species in H2O buffer has the same breadth at g1 = 2.15 as that of E4(4H) of α-70Val→Ile, and shows the same narrowing in D2O. At g3 the signals from residual Fered and E4(2N2H) largely obscure the g1 = 2.15 intermediate signal for the WT enzyme, but the spectrum in H2O buffer nonetheless shows clear evidence for the same 1-2-1 hyperfine-coupling pattern from two [Fe-H-Fe] hydrides as seen for the α-70Val→Ile variant, a feature that is lost in D2O buffer as with the α-70Val→Ile intermediate. Thus, the WT and α-70Val→Ile enzymes have equivalent hyperfine-coupled hydrides.
Comparison of ENDOR responses from E4(4H) in α-70Val→Ile MoFe protein and the g1 = 2.15 intermediate of WT enzyme confirms the presence of the two bridging hydrides in the WT intermediate. Figure 5B shows a 2 K Q-band stochastic CW 1H ENDOR spectrum collected at g2 = 2.01 for E4(4H) α-70Val→Ile/α-195His→Gln.18 This spectrum is one component of the 2D field-frequency pattern of spectra collected across the EPR envelope used to carry out the ENDOR analysis. In this spectrum, taken at the field of maximum EPR intensity,8 the strongly-coupled 1H signals from the two hydrides are completely overlapped, yielding a single composite (structured) doublet whose feature of maximum intensity corresponds to a hyperfine splitting, A(1H) ~ 24 MHz.
The population of the g1 = 2.15 intermediate trapped during turnover of WT enzyme in H2O buffer is low, even when enhanced by low P(N2), which prevented collection of a satisfactory ENDOR response for strongly-coupled [Fe-1H-Fe] protons. However, during turnover of the WT enzyme in D2O buffer the intermediate is trapped with a more than two-fold higher population. As a result, we could obtain 2H ENDOR signals from strongly-coupled [Fe-2H-Fe] deuterons. The Q-band 2H Mims pulsed-ENDOR spectrum at g = 2.01 shows a narrow ‘distant deuteron’ signal centered at the 2H Larmor frequency (Fig 5B), but in addition shows a pair of peaks offset from the 2H Larmor frequency by ~ ±1.85 MHz. The identification of this pair as a hyperfine-split doublet with A(2H) ~ 3.7 MHz is confirmed by its suppression when the interval, τ, in the Mims microwave pulse sequence is chosen appropriately for the hyperfine coupling, in this case A(2H)τ ~ 2, and by the absence of the signal in a sample prepared in H2O buffer, Fig 5B.19 When scaled by the nuclear g-factors, the coupling for this feature corresponds to a proton coupling of A(1H) ~ 24 MHz, the value seen in the 1H spectrum from E4(4H) α-70Val→Ile/α-195His→Gln.
Together, the EPR and ENDOR measurements demonstrate that the WT g1 = 2.15 intermediate not only has the same g-values as α-70Val→Ile E4(4H) (vide supra), but also exhibits hyperfine couplings that correspond to the pair of [Fe-H-Fe] bridging hydrides of α-70Val→Ile E4(4H), thereby establishing that the intermediate in WT MoFe protein corresponds to that in the α-70Val→Ile variant: it is the E4(4H) Janus intermediate of N2 reduction by the WT enzyme.
Photoinduced re of WT and α-70Val→Ile E4(4H) Hydrides; oa of H2 by cryoannealing E4(2H)*
The assignment of the WT g1 = 2.15 intermediate to E4(4H), with its two [Fe-H-Fe] bridging hydrides, is confirmed by the behavior of this intermediate under photolysis. We were inspired to take this approach by considering that the photolysis of transition metal dihydride complexes (with mutually cis hydride ligands) commonly results in the release of H2,20-28 which represents “a typical example of reductive elimination”, while the thermal reverse reaction “is the prototype example of an oxidative addition reaction.”20 Thus, we tested whether the two bridging hydrides of E4(4H) would behave in this fashion. ENDOR and EPR measurements showed that photolysis of E4(4H) α-70Val→Ile at 4 K and above generates a new FeMo-co state, denoted E4(2H)*, through the photoinduced re of the two bridging hydrides as H2. The E4(2H)* thus trapped relaxes to the initial, equilibrium, E4(4H) form during cryoannealing of the frozen solid at temperatures above 175 K, where the oxidative addition (oa) of the eliminated H2 by E4(2H)* becomes kinetically allowed. The photolysis quantum yield is temperature invariant at liquid helium temperatures and shows a large kinetic isotope effect, KIE ≈ 10. These observations imply the photoinduced release of H2 involves a barrier to the combination of the two nascent H atoms and further suggest that H2 formation involves nuclear tunneling through that barrier.
Figure 6 compares EPR signals collected during the in situ irradiation by 450 nm light of E4(4H) α-70Val→Ile, WT E4(4H), and E4(2N2H) held at 12 K in an EPR cavity. Figure 7A plots the time course for the intensity of the EPR signals of these intermediates during the in situ photolysis. Irradiation converts E4(4H) α-70Val→Ile (g = [2.15, 2.007, 1.965) to E4 (2H)* (g = [2.098, 2.0, 1.956]), Fig 6. The figure shows that irradiation of WT E4(4H) likewise converts this state to an E4(2H)* state with g-values identical to those in the α-70Val→Ile MoFe variant. In contrast, the EPR spectra of Fig 6 and progress curves of Fig 7A, show that the E4(2N2H) signal is unaffected by photolysis. That a bound nitrogenous moiety, the N2-derived 2N2H moiety of E4(2N2H), is not photodissociable supports the idea that the photosensitivity is associated with the presence of bound hydrides. The photolyis results thus confirm the presence of the two bridging hydrides in the WT g = 2.15 intermediate, and its identification as E4(4H).
Figure 6.

Photoinduced changes in EPR spectra of WT and α-70Val→Ile freeze-trapped during enzymatic turnover in H2O during 24.5 minutes of 450 nm diode laser irradiation at 12 K. EPR conditions: the same as in Figure 4.
Figure 7.

(A) Time courses of E4(4H) and E4(4D) states in α-70Val→Ile and WT and E4(2N2H) in WT 1 atm N2 turnover in H2O during irradiation with 450 nm diode laser at 12 K. Data points in this and following kinetics plots were obtained as described in Materials and Methods; decays of E4(4H) and E4(4D) are fitted as stretched exponential with following parameters: τ = 9.2 min, m = 0.51 for H2O and τ = 83.3 min, m = 0.45 for D2O. EPR conditions: for this and the following kinetics figures are the same as in Figure 4. (B) Time courses of E4(4H)/ E4(4D) recovery during 193 K annealing of irradiated WT and α-70Val→Ile turnovers in H2O/D2O. Time constants obtained from exponential fit: 8.4 min (H2O) and 41.5 min (D2O), KIE ~ 5.
The progress curves for in situ irradiation of E4(4H) in WT and α-70Val→Ile MoFe protein (Fig 7A) are the same, within error, showing that the quantum yield for re of H2 is independent of environment - WT enzyme vs α-70Val→Ile variant. As a result, the re progress curves for WT and α-70Val→Ile in H2O buffer have been jointly fit to the stretched exponential behavior (exp(−(t/τ)m))29,30 that is a consequence of photolysis in a non-glassy sample,18 and likewise for those in D2O buffer. As reported for photolysis of E4(4H) α-70Val→Ile, the joint progress curves show a large KIE, defined as the ratio of the median decay times for D2O and H2O buffers, KIE ~ 9, which implies that photoinduced re of the two hydrides and release of H2 involves a barrier to the combination of the two nascent H atoms. Likewise, as reported for the α-70Val→Ile intermediate, the decay time for the WT intermediate is temperature invariant, within error, for T = 4-12 K, which suggests that the formation of H2 involves nuclear tunneling through that barrier.31
Figure 7B plots the timecourse for the oa of H2 by the E4(2H)* and E4(2D)* to regenerate the corresponding E4(4H)/E4(4D) states during 193 K cryoannealing of WT and α-70Val→Ile MoFe samples.18 In these experiments the sample is annealed at 193 K multiple times, with cooling for collection of EPR spectra at 12 K between annealing periods. For both H2O and D2O buffers the relaxation of E4(2H)* oa of H2 regenerates the E4(4H) state formed during turnover, and the progress curves are well-fitted as a single-exponential processes. As with photoinduced re, the progress curves during cryoannealing oa are the same for the two MoFe variants, and their joint fit yields KIE ~ 5, rather larger than seen for closed-shell monometallic complexes.32,33 Combined with a strong temperature dependence of the time-constant (not shown), this KIE implies that oa of H2 involves traversal of an energy barrier.
Overall, the measurements of photoinduced, re loss of H2 from E4(4H) and thermal regeneration of the E4(4H) state via oa of H2 by the resulting E4(2H)* confirm the identification of the WT and α-70Val→Ile intermediates as the same Janus, in showing that both the excited and ground state energy surfaces associated with these processes are essentially the same in the WT and variant MoFe proteins.
Accumulation of E4(4H) Isotopologues During Mixed-Isotope Turnover; Bridging Hydrides are Exchange-Inert
The kinetic reversibility of the re/oa mechanism in WT enzyme, as well as the stability of the bridging hydrides to solvent exchange, as required by Key Constraint (ii, b), Chart 1, are here established by the use of EPR and photolysis to measure the isotopic composition of the bridging hydrides in WT Janus trapped during N2 turnover under isotopically mixed conditions: H2O buffer under an atmosphere that include D2 and conversely, Fig 8. These measurements show that the re/oa mechanism satisfies Key Constraints, Chart 1, on catalysis by WT nitrogenase
Figure 8.

(A) The g1 features of Janus state EPR recorded for WT protein turnovers trapped under mixtures of 0.1 atm N2 with 0.9 atm of H2 or D2 in H2O and D2O buffers with stirring. EPR conditions: temperature, 12 K; microwave frequency, ~9.36 GHz; microwave power, 10 mW; modulation amplitude, 4.5 G; time constant, 160 ms; field sweep speed, 5 G/s; 4-8 scans. (B) Photolysis of WT Janus intermediate formed through isotopically-mixed turnover; irradiation with 450 nm light at 12 K. Progress curves for N2/H2, H2O and N2/D2, D2O are fitted as stretched exponential decays with parameters shown in the figure. Kinetics of other two samples can be well fitted with following parameters: τ = 52.6 min, m = 0.47 for N2/D2 turnover in H2O and τ = 38.3 min, m = 0.46 for N2/H2 turnover in D2O. Dotted lines present alternative fits as sums of two decays corresponding to photolysis of E4(4H) in H2O and E4(4D) in D2O turnover samples with ratios shown in the figure.
According to the re/oa mechanism, N2 turnover in H2O buffer under D2 would generate E4(4H) through turnover accumulation of reducing equivalents and protons derived from solvent (Fig 1), but in addition, oa of D2 from the gas phase by E4(2N2H) would generate E4(2D−;2H+), with two [Fe-D-Fe] bridges and two bound protons, and conversely for turnover in D2O buffer under H2 (Fig 3). As a result, during N2 turnover under both mixed-isotope conditions, two isotopologues of Janus are expected to accumulate, one in which the two bridges have the solvent H/D isotope, the other with the two bridges generated through oa of the diatomic D2/H2 in the gas phase.14
Figure 8A shows that the g1 = 2.15 feature from the EPR spectrum of the WT E4(4H) intermediate, as trapped in H2O buffer during turnover under N2/D2, is distinctly narrower than for turnover under N2/H2, thus demonstrating the accumulation of an intermediate in which the reverse of the re/oa equilibrium has generated [Fe-D-Fe] bridging deuterides with loss of the 1H hyperfine broadening (Fig 5A, above). Conversely the g1 = 2.15 feature for the WT Janus intermediate in D2O buffer is correspondingly broader for turnover under N2/H2 than for turnover under N2/D2, demonstrating the accumulation of an intermediate with bridging [Fe-H-Fe], which contribute 1H hyperfine broadening. Simulations that sum roughly equal contributions of the limiting spectra for E4(4H) and E4(4D) in fact reproduce the mixed-isotope spectrum quite well (not shown). Note especially, that the demonstration that deuterides/hydrides acquired by oa of the gas-phase diatomic accumulate in the catalytic intermediate, rather than exchanging with a solvent of opposite isotopic composition, confirms our proposal6,7 that these bridging hydrides are exchange-inert, and that their formation during oa of a gas-phase diatomic explains why turnover under N2/T2 does not lead to the exchange of T+ into the solvent.2
The accumulation of mixtures of E4(4H) isotopologues during turnover by oa of H2/D2 under isotopically-mixed conditions is actually seen most dramatically when comparing measurements of the KIE for the photolysis of WT Janus trapped during turnover under N2 in isotopically homogeneous conditions – H2O buffer with addition of 0.9 atm of H2, or D2O buffer with added D2 – with those for isotopically-mixed turnover conditions – H2O buffer with added D2 or D2O buffer with added H2. As expected, the photolysis traces from isotopically homogeneous intermediates, formed in H2O buffer with N2/H2 and in D2O buffer with N2/D2, Fig 8B, show a large KIE ~ 11, within error the same as seen for re of the two hydrides/deuterides formed in H2O/D2O buffers without the diatomics in the atmosphere (Fig 7A).
However, according to the re/oa mechanism, oa of D2 by E4(2N2H) during turnover in H2O buffer under D2 generates E4(2D−;2H+), with two [Fe-D-Fe] bridges and two bound protons, and conversely for turnover in D2O buffer under H2. To a good approximation each of these E4(4H) mixed isotopologues should undergo photoinduced re with quantum yield associated with the isotopic composition of the bridges, as determined by the complementary diatomic (see above), independent of the isotopic character of the protons/deuterons on sulfur, as determined by the solvent. Thus, if the H/D bridges introduced from the diatomic do not exchange with solvent, the intermediates formed in mixed isotope turnover should have apparent rates of photolysis roughly midway between those of E4(4H) and E4(4D). Indeed, Fig 8B shows that the Janus intermediate formed during turnover under N2/D2 in H2O buffer photolyzes more slowly than with N2/H2 in H2O, while the intermediate formed under N2/H2 in D2O buffer photolyzes more rapidly than with N2/D2 in D2O. The photolysis of these mixed-isotope samples each can be described by time constant roughly midway between those of the two isotopically homogeneous samples (see figure legend). Alternatively, as shown in Fig 8B, each isotopically mixed trace can be fit as the sum of a roughly f ~ 50% contribution from the progress curve for photolysis of E4(4H), with two bridging hydrides, plus a contribution of (1-f) from the curve for E4(4D) with two bridging deuterides.34
These EPR and photochemical observations thus confirm that turnover of WT MoFe protein under N2 involves a rapidly reversible re/oa equilibrium between the E4 Janus intermediate and the E4 state with diazene-level dinitrogen reduction product, Fig 3. Furthermore, the buildup of E4(2D−;2H+) with bridging deuterides during N2/D2 turnover in H2O buffer, and the converse, confirm that the bridging hydrides/deuterides of the Janus intermediate do not exchange with solvent during turnover. Thus, these measurements show that the re/oa mechanism indeed satisfies Key Constraints (i), (ii)b,c, Chart 1, as proposed.6,7
Kinetic reversibility of the re/oa equilibrium during N2 reduction by WT MoFe
The E4(4H) α-70Val→Ile intermediate was shown to have accumulated four reducing equivalents by a quench-cryoannealing relaxation protocol corresponding to that described above for the regeneration of E4(4H) by oa of H2 to the photogenerated E4(2H)*. Keeping the sample frozen prevents any additional accumulation of reducing equivalents because binding of reduced Fe protein to and release of oxidized protein from the MoFe protein both are abolished in a frozen solid. As recently confirmed,35 the frozen intermediate can relax towards the resting state only through steps that release a stable species from FeMo-co, with the En states formed prior to N2 binding losing two equivalents per relaxation step in the release of H2. By this approach, E4(4H) was identified by its relaxation to the resting state E0 through the release of a total of four reducing equivalents in a two-step process, each step involving hydride protonation with release of H2 (two equivalents per step), with formation of E2(2H) in the first step (Fig 1).10
As an extension of this procedure, the FeMo-co S = ½ E4(2N2H) intermediate trapped during catalytic turnover of WT enzyme was identified by the finding that the decay of this state in a frozen reaction mixture is accelerated by increasing [H2] and slowed by increasing [N2], which directly demonstrated that the intermediate is the product of the kinetically, as well as thermodynamically reversible (re/oa) activation equilibrium, Fig 3. However, in that study the E4(4H) precursor to re was accumulated in such low levels that its kinetic progress curve during cryoannealing could not be monitored directly, and we were forced to analyze the decay of E4(2N2H) to resting state, Chart 3, through a steady-state approximation for the concentration of E4(4H).
Chart 3.
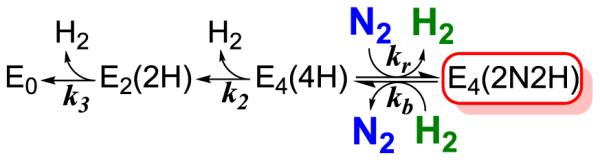
The preparation of freeze-quenched WT samples that contain significant populations of both E4(4H) and E4(2N2H), obtained through control of P(N2) during turnover, has now enabled us to directly monitor the progress curves in WT enzyme of all LT species on the cryoannealing relaxation pathway of E4(2N2H) to resting state (En states with n ≤ 4, even), Chart 3. In so doing we directly track the kinetics of the species linked through the oa reverse, of the re/oa equilibrium, Figs 1, 3. These progress curves for the relaxation of E4(2N2H) and kinetically linked intermediates (Chart 3) during cryoannealing of WT enzyme at −50 °C are presented in Fig 9. These are well described by the curves calculated by fitting them to the sequential kinetic scheme of Fig 9, which corresponds to the reverse of the LT scheme starting at E4(2N2H), Fig 1 and Chart 3. The fitting procedure allowed for each step to exhibit distributed kinetics, and the time-constants and ‘stretch’ parameters are collected in the legend to Fig 9. The amplitudes of the kinetic phases correspond to the steady-state turnover populations of the En states, and of course vary widely depending on P(N2) and the electron flux (see Fig 4). In the sample represented by Fig 9 these correspond to: E0 ~ 8%; E2 ~ 16%; E4(4H) ~8%; E4(2N2H) ~ 12 %. Thus, in this sample the two states linked by the re/oa equilibrium correspond to fully ~ 20% of the MoFe protein. The remainder of the MoFe protein is in EPR-silent En, n = odd states, ~ 56%; these cannot relax to E0 during cryoannealing.35
Figure 9.
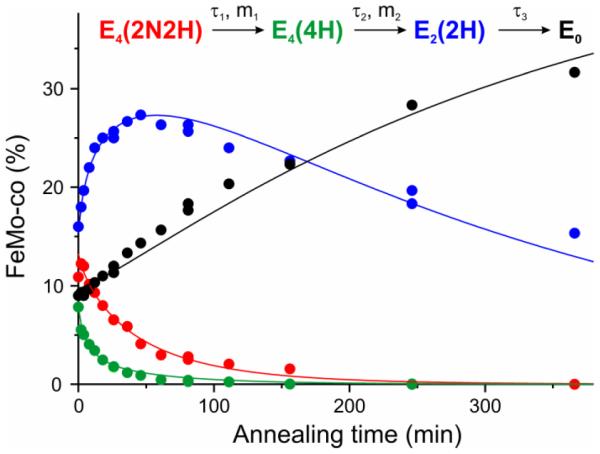
Time courses of four EPR detected states during −50 °C cryoannealing of WT low P(N2) ~ 0.05 atm turnover in H2O. The data colors correspond to those in the kinetic scheme (Top) and the lines correspond to fits to that scheme as previously described.10 Stretched exponential parameters of the first two fast steps are τ1 = 43 min, m1 = 0.79 and τ2 = 6 min, m2 = 0.8, the third slow step fitted as exponential with τ3 = 330 min. Intensities of E2 and E0 states obtained and quantitated with previously described procedures;15 intensities of E4 states converted to concentration units by scaling three-step kinetic scheme for corresponding decays. EPR conditions: for E4(4H) spectra the same as in Fig 4; for E4(2N2H), E2 and E0 the same as used in previous studies.15
The kinetic coupling of the loss of E4(2N2H) through oa of H2, with the formation of E4(4H) directly establishes the operation of the re/oa mechanism and its kinetic reversibility. The above demonstration that oa of D2 by E4(2N2H) in H2O yields E4(2D−;2H+) with exchange-inert deuteride bridges then shows that in this case each of the two steps by which this WT mixed-isotope intermediate relaxes to E0 would involve protonation of the deuteride bridges by protons from solvent, generating 2HD, confirming that the re/oa mechanism explains the final Key Constraint (ii a), Chart 1, as proposed.6,7
Thermodynamic reversibility of the re/oa equilibrium during N2 reduction by WT MoFe
The mere observation of both E4(4H) and E4(2N2H) in samples freeze-quenched during N2 fixation (Fig 4) establishes that the equilibrium constant for re/oa, Kre = kr/kb (Fig 3; Chart 3), is small. The cryoannealing experiments only give kb, not kr, but as a rough quantitative estimate of Kre, we assign the relative concentrations of E4(2N2H) and E4(4H) during turnover under low N2 partial pressure to the zero-time values determined by the fit to the annealing kinetics (Fig 9), E4(2N2H)/E4(4H) ~3/2, in keeping with the conclusion (vide supra) that the forward and reverse steps of the re/oa equilibrium are rapid compared to other steps in the catalytic cycle (Fig 1). In this case we can rewrite eq 2 to approximate Kre as,
| (3) |
The N2 partial pressure is fixed by the experimental conditions; earlier observations suggest that saturating concentrations of H2 are formed during turnover under all P(N2), which suggests an effective P(H2) ~ 1atm.15 As a result, one obtains:
| (4) |
| (5) |
The LT kinetic measurements likewise yielded values for the re process: Kre ~ 0.7 and ΔreG0 ~ +0.2 kcal/mol.2,4 Given the difference in methodologies – direct observation of species in equilibrium in the present study, analysis of turnover kinetics in the former – and the differences in origin of the MoFe proteins – Azotobacter vinelandii in the present study and Klebsiella pneumoniae in the former - we consider the measurements to be in excellent agreement: nitrogenase catalysis, driven by the re of H2, turns the highly endothermic first step in the reduction of the N2 triple bond, (to the diazene level) into the essentially thermoneutral re/oa equilibrium conversion of Fig 3.
Conclusions/Summary
The re/oa mechanism for N2 reduction by nitrogenase postulates that the reduction of the N2 triple bond to a 2N2H (diazene) level is driven by the obligatory formation of one H2 for each N2 reduced. This proposal was based on identification of a FeMo-co S = ½ state trapped during turnover of the α-70Val→Ile MoFe protein as the Janus intermediate: the E4(4H) state, which stores four reducing equivalents as two [Fe-H-Fe] bridging hydrides. Once this identification is made, then the LT E4(4H)↔E4(2N2H) equilibrium (Fig 1) ceases to be a ‘mystery’: the connection with the organometallic chemistry of metal-dihydride complexes identifies this process as the mechanistically coupled reductive elimination (re) of H2 (Fig 3). The H2 formed during re carries away two of the four reducing equivalents stored in E4(4H) and drives the reaction, while the metal-ion core of FeMo-co becomes activated to reduce N2 through the simultaneous acquisition of two reducing equivalents. The mechanism was first supported by its proposed explanation of the Key Constraints of Chart 1, and then two new predictions regarding turnover by WT MoFe were promptly verified,14,15 adding direct experimental support.
As we now summarize, the present report establishes the presence of Janus in WT enzyme, its participation in N2 reduction, and the operation of the re/oa equilibrium during N2 reduction. Experiments on WT enzyme further show that this mechanism satisfies all the Key Constraints imposed on N2 reduction by decades of careful study by others summarized in Chart 12 and that the re/oa equilibrium in WT MoFe is indeed kinetically and thermodynamically reversible, thereby establishing the role of the re/oa equilibrium in N2 reduction (Fig 3).6,7
(i) EPR/ENDOR, and photophysical measurements establish that the intermediate with g1 = 2.15 trapped during nitrogen fixation by WT MoFe in fact is the E4(4H) Janus intermediate of N2 reduction, which has accumulated four reducing equivalents stored as [Fe-H-Fe] bridging hydrides, whose properties are identical to those of the Janus intermediate first trapped in the α-70Val→Ile MoFe variant. This observation thereby establishes the freeze-trapped α-70Val→Ile Janus intermediate as a reliable guide to mechanism, but most importantly, enables direct observation of the participation of the Janus intermediate in N2 reduction and the re/oa process during catalysis by the WT enzyme. The new findings and conclusions that build on this foundation are summarized next.
(ii) Examination of the isotopic composition of WT Janus during turnover in H2O buffer under D2 (or D2O buffer under H2) establishes that oa of D2 from the gas phase by E4(2N2H) accumulates E4(2D−;2H+), with two [Fe-D-Fe] bridges and two bound protons from solvent (or E4(2H−;2D+)) (Fig 3), and that the bridging hydrides/deuterides do not exchange with solvent during turnover. This demonstrates experimentally that the re/oa mechanism accounts for the longstanding Key Constraints on mechanism, Chart 1, (i),(ii)b,c.
(iii) The observation and successful modeling of the entire relaxation pathway of WT enzyme by which E4(2N2H) relaxes to the resting-state, E0, including the oa of H2, to form Janus with release of N2, and two subsequent steps of hydride protonation each with release of H2, Fig 1, eq 2, demonstrates the kinetic reversibility of the re/oa equilibrium, Fig 3, Chart 3, and shows that re/oa satisfies the last of the Key Constraints, (ii)a, in addition to confirming that it satisfies (ii)c.
(iv) An estimate of the free energy for the re of H2 by FeMo-co that has accumulated four reducing equivalents, with reduction of N2 to generate a 2N2H-level species (Fig 3) in WT enzyme, quantifies the thermodynamic reversibility of the first step in the reduction of N2 by nitrogenase: this reaction is essentially thermoneutral, ΔreG0 ~ −2 kCal/M, whereas direct hydrogenation of gas-phase N2 is highly endergonic.16
In summary, in this report we have described the central mechanistic steps by which the WT nitrogenase carries out one of the most challenging chemical transformation in biology, the reduction of the N≡N triple bond.
Acknowledgements
This work was funded by the NIH (GM 111097;BMH), NSF (MCB 1515981 to BMH) and U.S. Department of Energy, Office of Science, Basic Energy Sciences (BES) (DE-SC0010687 and DE-SC0010834; LCS and DRD).
References
- (1).Smil V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. MIT Press; Cambridge, MA: 2001. [Google Scholar]
- (2).Burgess BK, Lowe DJ. Chem. Rev. 1996;96:2983. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- (3).Thorneley RNF, Lowe DJ. Met. Ions Biol. 1985;7:221. [Google Scholar]
- (4).Wilson PE, Nyborg AC, Watt GD. Biophys. Chem. 2001;91:281. doi: 10.1016/s0301-4622(01)00182-x. [DOI] [PubMed] [Google Scholar]
- (5).Simpson FB, Burris RH. Science. 1984;224:1095. doi: 10.1126/science.6585956. [DOI] [PubMed] [Google Scholar]
- (6).Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Chem. Rev. 2014;114:4041. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Acc. Chem. Res. 2013;46:587. doi: 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Igarashi RY, Laryukhin M, Dos Santos PC, Lee HI, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2005;127:6231. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- (9).Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2010;132:2526. doi: 10.1021/ja910613m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. PNAS. 2007;104:1451. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hartwig J. Organotransition metal chemistry: from bonding to catalysis. University Science Books; Sausalito, CA: 2010. [Google Scholar]
- (12).Crabtree RH. The organometallic chemistry of the transition metals. 5th Wiley; Hoboken, N.J.: 2009. [Google Scholar]
- (13).Peruzzini M, Poli R, editors. Recent Advances in Hydride Chemistry. Elsevier Science B.V.; Amsterdam, Netherlands: 2001. [Google Scholar]
- (14).Yang Z-Y, Khadka N, Lukoyanov D, Hoffman Brian M, Dean Dennis R, Seefeldt Lance C. PNAS. 2013;110:16327. doi: 10.1073/pnas.1315852110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lukoyanov D, Yang ZY, Khadka N, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2015;137:3610. doi: 10.1021/jacs.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). ΔrG0[H2(g) + XY(g) → H2XY(g)]; N2, ~ +50 kCal/mol; CO, ~ +4kCal/mol.
- (17).Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR. Biochemistry. 1998;37:12611. doi: 10.1021/bi981165b. [DOI] [PubMed] [Google Scholar]
- (18).Lukoyanov D, Khadka N, Yang ZY, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2016;138:1320. doi: 10.1021/jacs.5b11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Schweiger A, Jeschke G. Principles of Pulse Electron Paramagnetic Resonance. Oxford University Press; Oxford, UK: 2001. [Google Scholar]
- (20).Perutz RN. Pure Appl. Chem. 1998;70:2211. [Google Scholar]
- (21).Colombo M, George MW, Moore JN, Pattison DI, Perutz RN, Virrels IG, Ye TQ. J. Chem. Soc., Dalton Trans. 1997:2857. [Google Scholar]
- (22).Whittlesey MK, Mawby RJ, Osman R, Perutz RN, Field LD, Wilkinson MP, George MW. J. Am. Chem. Soc. 1993;115:8627. [Google Scholar]
- (23).Ballmann J, Munha RF, Fryzuk MD. Chem. Commun. 2010;46:1013. doi: 10.1039/b922853e. [DOI] [PubMed] [Google Scholar]
- (24).Ozin GA, Mccaffrey JG. J. Phys. Chem. 1984;88:645. [Google Scholar]
- (25).Dugan TR, Holland PL. J. Organomet. Chem. 2009;694:2825. doi: 10.1016/j.jorganchem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Inorg. Chem. 2006;45:5742. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]
- (27).Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J. Am. Chem. Soc. 2006;128:756. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- (28).Yu Y, Sadique AR, Smith JM, Dugan TR, Cowley RE, Brennessel WW, Flaschenriem CJ, Bill E, Cundari TR, Holland PL. J. Am. Chem. Soc. 2008;130:6624. doi: 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Phillips JC. Rep. Prog. Phys. 1996;59:1133. [Google Scholar]
- (30).Berberan-Santos MN, Bodunov EN, Valeur B. Chem. Phys. 2005;315:171. [Google Scholar]
- (31). Work in progress suggests the process involves initial formation of an H2 complex.
- (32).Abu-Hasanayn F, Goldman AS, Krogh-Jespersen K. J. Phys. Chem. 1993;97:5890. [Google Scholar]
- (33).Campian MV, Perutz RN, Procacci B, Thatcher RJ, Torres O, Whitwood AC. J. Am. Chem. Soc. 2012;134:3480. doi: 10.1021/ja210568t. [DOI] [PubMed] [Google Scholar]
- (34). We are aware that this partitioning is simplified in that it does not consider other isotopologues that may be formed during steady-state turnover.
- (35).Lukoyanov D, Yang ZY, Duval S, Danyal K, Dean DR, Seefeldt LC, Hoffman BM. Inorg. Chem. 2014;53:3688. doi: 10.1021/ic500013c. [DOI] [PMC free article] [PubMed] [Google Scholar]