

Abstract
The photochemical release of chemical reagents and bioactive molecules provides a useful tool for spatio-temporal control of biological processes. However, achieving this goal requires the development of highly efficient one- and two-photon sensitive photo-cleavable protecting groups. Thiol-containing compounds play critical roles in biological systems and bioengineering applications. While potentially useful for sulfhydryl protection, the 6-bromo-7-hydroxy coumarin-4-ylmethyl (Bhc) group can undergo an undesired photoisomerization reaction upon irradiation that limits its uncaging efficiency. To address this issue, here we describe the development of 6-bromo-7-hydroxy-3-methylcoumarin-4-ylmethyl (mBhc) as an improved group for thiol-protection. One- and two-photon photolysis reactions demonstrate that a peptide containing a mBhc-caged thiol undergoes clean and efficient photo-cleavage upon irradiation without detectable photoisomer production. To test its utility for biological studies, a K-Ras-derived peptide containing an mBhc–protected thiol was prepared by solid phase peptide synthesis using Fmoc-Cys(mBhc)-OH for the introduction of the caged thiol. Irradiation of that peptide using either UV or near IR light in presence of protein farnesyltransferase (PFTase), resulted in generation of the free peptide which was then recognized by the enzyme and became farnesylated. To show the utility of this caging group in biomaterial applications, we covalently modified hydrogels with mBhc-protected cysteamine. Using multi-photon confocal microscopy, highly defined volumes of free thiols were generated inside the hydrogels and visualized via reaction with a sulfhydryl-reactive fluorophore. The simple synthesis of mBhc and its efficient removal by one- and two-photon processes make it an attractive protecting group for thiol caging in a variety of applications.
Graphical abstract
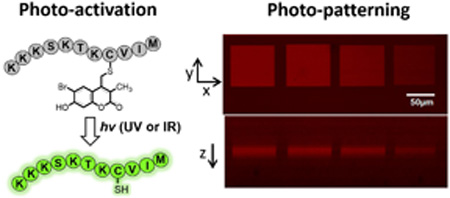
Introduction
Photo-removable protecting groups (also known as caging groups) allow spatio-temporally controlled release or activation of a variety of biomolecules, including peptides and inhibitors inside living systems.1,2 These protecting groups can be used to mask specific functionalities present in bioactive agents (generating caged inactive molecules) such that they can be cleaved on-demand upon irradiation and release the bioactive species.3,4 Recent advances in the development of two-photon cleavable protecting groups allow uncaging using near IR irradiation instead of UV light, with remarkably improved spatial resolution and increased penetration while causing significantly lower photo-toxicity.5,6 This has broadened the application of the caging strategy for photo-triggered release of biomolecules inside tissues or organisms useful for a variety of biological studies.7 Additionally, two-photon uncaging approaches have proved to be extremely useful for creating novel biomaterials; in that strategy, laser irradiation is used to unmask a specific caged functionality pre-incorporated into a hydrogel or matrix, such that it can be used to immobilize peptides, proteins or cells in a three dimensionally controlled fashion.8–10 Such highly tuned matrices allow artificial extracellular environments to be created that can be used to study cell migration, differentiation and cell-cell interactions.11
Differences in the chemical reactivity of various functional groups means that there is no single protecting group that can be universally employed for caging applications.1 Sulfydryl-containing compounds play critical roles in various aspects of cellular function.12,13 Hence, significant effort has gone into development of photo-activatable thiol-containing peptides or small molecule substrates as tools to elucidate or dissect cellular pathways;14,15 under many conditions, thiols are the most reactive nucleophiles present in biological systems. Importantly, they are prone to oxidation and are also relatively poor leaving groups compared with phosphates and carboxylates.16 Those features render the design of photoremovable thiol protecting groups challenging.
Ortho-nitrobenzyl (ONB) compounds are the most commonly used caging groups for sulfhydryl-protection.17 ONB groups provide free thiols in high yield upon photolysis, however, they are poor chromophores and they generally lack two-photon sensitivity. To address these limitations, coumarin-based protecting groups have been utilized due to their high one- and two-photon sensitivity. The fluorogenic character of coumarins can also be used as a tool to track the caged probes inside cells, tissue or in a polymeric matrix.18 Despite several reports that showed successful application of brominated hydroxy-coumarin (Bhc, 1) for thiol-protection,9,19,20 a recent study showed photolysis of Bhc-protected thiols often leads to the generation of unwanted photo-isomeric by-products.21 The two-step mechanism of this photo-rearrangement process was studied in detail by Distefano and coworkers, which set the stage for further modification of the Bhc structure to engineer reduced photoisomerization.
In this report, guided by mechanistic studies of the photo-triggered isomerization of Bhc-thiols, we developed 6-bromo-7-hydroxy-3-methylcoumarin-4-ylmethyl (mBhc, 3) as an alternative coumarin-based caging group that can afford efficient thiol release upon one- and two-photon irradiation. To test the efficiency of mBhc for thiol-protection in peptides, we have synthesized an mBhc-protected form of cysteine (Fmoc-Cys(mBhc)-OH) suitable for incorporation via solid phase peptide synthesis and subsequently used it to prepare a K-Ras-derived peptide. One- and two-photon photolysis of the caged peptide resulted in clean conversion to the free compound with no photo-isomerization. Irradiation of the caged peptide using a near-IR laser in the presence of an enzyme (protein farnesyltransferase, PFTase) resulted in the generation of a free thiol-containing peptide which was then enzymatically farnesylated. To further evaluate the utility of this novel caging group for biomaterial applications, an mBhc-protected thiol was covalently incorporated into a hydrogel. Using a 740 nm two-photon laser from a confocal microscope, patterns of free thiols were generated inside the matrix and visualized by reaction with maleimide functionalized fluorophores. Such 3D patterns could be useful for a variety of applications in tissue engineering.10,11
Results and Discussion
Design and synthesis of a coumarin-based caging group for efficient thiol protection
In previous work, we demonstrated that the major product of photolysis of Bhc-protected thiols (4) is not the free thiol, but rather an isomeric product (6) that is formed via the two step process illustrated in Scheme 2. We proposed that the first step of that mechanism involves a photo-induced 1,3 shift of the thiol from the exocyclic position to the endocyclic 3 position yielding intermediate 5 that undergoes tautomerization to produce the final photo-rearranged product 6. Those results illustrate why Bhc is not an efficient caging group for thiol protection. To address this limitation, we have reported nitrodibenzofuran (NDBF) as an alternative caging group for thiol protection, which showed remarkably high uncaging efficiency in both one- and two-photon processes.21
Scheme 2.

Photo-rearrangement mechanism of Bhc protected cysteine.
Despite the efficiency of NDBF for thiol caging, using coumarin-based caging groups for thiol protection is still advantageous due to their comparatively straightforward synthesis, higher water solubility (relative to NDBF) and the fluorogenic character of coumarin which is useful to track probes inside cells. Hence, we elected to investigate the development of an alternative coumarin-based caging group for thiol protection in small molecules and peptides.
We initially hypothesized that changing the substituents on the phenolic ring of Bhc could be used to decrease the extent of photo-rearrangement over uncaging. Therefore, we decided to study the photolysis of a thiol protected by hydroxycoumarin (Hc, 2, Scheme 1) lacking the bromine on the phenolic ring. Hence, Hc-protected Boc-cysteamine (7, Figure 1A) was synthesized following a previously reported procedure22 and studied as a model caged thiol for photolysis experiments. Solutions of compound 7 in buffered aqueous solution were irradiated using 365 nm light for varying times. Analysis of the photolysis reactions via LC-MS using extracted ion current data (EIC) clearly indicates the formation of the undesired photo-isomer upon photolysis as evidenced by the appearance of a new peak (tR = 7.71 min) with an m/z ratio identical to that of the starting material (Figure 1B, C); no Hc-OH (9, m/z = 231) generation was observed in the corresponding EIC chromatogram suggesting minimal uncaging occurred upon irradiation.
Scheme 1.

Coumarin-based caging groups discussed in this work.
Figure 1.

Photolysis reaction of Hc-protected Boc-cysteamine. (A) Structures used in this study. (B) EIC chromatogram (m/z = 334.09, calcd for [M(7)-tBu+K]+ = 334.01) of a 50 µM solution of 7 in photolysis buffer (50 mM phosphate buffer, pH 7.4 containing 1 mM DTT) before irradiation, (C) EIC chromatogram (m/z = 334.09, calcd for [M(7)-tBu+K]+ = 334.01) of a 50 µM solution of 7 in photolysis buffer after 6 min irradiation at 365 nm, this data clearly indicates generation of the photo-isomer, (D) EIC chromatogram (m/z = 231.01, calcd for [M(9) +K]+ of 7 after 6 min irradiation showing no evidence of generation of 9, this indicates photolysis leads predominantly to photo-isomerization rather than uncaging.
In addition to these data, it should be noted that Kotzur et al. previously reported photo-rearrangement occurring upon photolysis of 7-amino and 7,8-bis(carboxymethoxy) coumarin protected thio-carbamates.19 These results and observations indicate that photoisomerization is widespread in coumarin photochemistry and that simple alteration of the substituents on the phenolic ring would not be sufficient to shut down the photoisomerization reaction manifold. Given those results, we next elected to modify the endocyclic 3 position which is directly involved in the photo-rearrangement mechanism. We hypothesized that replacing the hydrogen atom at that position with an alkyl moiety should attenuate photo-isomerization due to several factors. First, the presence of an alkyl group on C-3 would block photoisomer formation since intermediate 12 cannot re-aromatize due to the absence of a hydrogen at the 3 position. Second, the syn pentane type interaction between the sulfur and the C-3 methyl group shown in Scheme 3 should make it sterically more difficult for the sulfur atom to migrate to the C-3 position. Computational analysis of the model compounds mBhc-SCH3 and Bhc-SCH3 shows that the lowest energy conformers for both molecules position the thiomethyl group 90° out of the coumarin plane (see Figure S1). However, the steric hindrance noted above is highly destabilizing for mBhc-SCH3 as evidenced by the large increase in conformational energy that occurs when the thiomethyl group is moved towards the coumarin plane; such a movement would be required in the thiol migration step.
Scheme 3.

Illustration of potential effects of C-3 substitution on photoisomerization process.
Based on this hypothesis, we decided to synthesize 6-bromo-7-hydroxy-3-methylcoumarin-4-ylmethyl bromide (mBhc-Br, 3a) and examine its utility for thiol protection, particularly for S-protection of cysteine containing peptides. The synthesis of mBhc-Br is depicted in Scheme 4. Dropwise addition of Br2 to an ice cold solution of ethyl-2-methylacetoacetate in CHCl3 followed by overnight stirring at room temperature gave 4-bromo-2-methylacetoacetate (14, 70% yield).23 That compound was subsequently treated with 4-bromoresorcinol in CH3SO3H overnight at room temperature to afford the desired bromide (3a) in 35% yield. After successful synthesis of mBhc-Br, we next sought to utilize this caging group for thiol protection in cysteine containing peptides to evaluate its uncaging efficiency in the context of biologically useful molecules.
Scheme 4.

Synthesis of Fmoc-Cys(mBhc)-OH and its incorporation into a K-Ras-derived peptide via SPPS.
Our strategy for creating caged peptides was to prepare mBhc-protected Fmoc-Cys-OH and incorporate that into a peptide of interest through solid phase peptide synthesis (SPPS). The synthesis of the desired mBhc S-protected cysteine suitable for SPPS is illustrated in Scheme 4. The phenolic hydroxyl group of mBhc was protected as a MOM ether via treatment with MOM-Cl and TEA to give compound 15 in 95% yield. That species was then used to alkylate Fmoc-Cys-OCH3 under mild acidic conditions using Zn(OAc)2 as a catalyst to produce 16 in 90 % yield. The resulting methyl ester was hydrolyzed via treatment with (CH3)3SnOH24 in refluxing CH2Cl2 to generate a caged form of Fmoc-cysteine (Fmoc-Cys(mBhc)-OH 17) in 85 % yield.
Synthesis of the desired caged peptide was performed via SPPS, in which Fmoc-protected amino acids were added sequentially to the growing chain anchored on Wang resin. Standard coupling conditions were used throughout the synthesis except for the incorporation of Fmoc-Cys(mBhc)-OH where the coupling time was increased to 6 hours to ensure quantitative incorporation. Final acidic treatment of the resin-bound peptide with Reagent K ensured removal of all side-chain protecting groups including the MOM group present on the mBhc moiety, and cleavage of the peptide from the resin to produce the desired caged molecule. The above procedure was successfully used to generate a caged form of a K-Ras-derived peptide (18) that was subsequently used to study the uncaging reaction and the utility of this new protecting group.
After synthesis and purification of the mBhc S-protected peptide, photolysis experiments were carried out to probe for the formation of free peptide and any possible photo-isomer. Thus, a solution of 18 was irradiated using 365 nm light and subsequently analyzed by LC-MS. As confirmed by the extracted ion-current (EIC) data shown in Figure 2, photolysis resulted in the generation of the desired uncaged peptide as evidenced by the appearance of the corresponding peak with the expected mass (tR = 0.75 min, m/z = 647). However, apart from the remaining caged peptide peak (18, m/z = 520), there was no evidence of any new peak bearing the same mass suggesting that no photo-isomer was generated upon photolysis. Photolysis experiments were carried out in presence of 1mM DTT to block possible disulfide formation, thus simplifying analysis of the crude reaction mixture. (see Figure S2). Similar photolysis experiments, previously reported by our group using the analogous Bhc-protected (lacking the methyl group at C-3) peptide, resulted in the photo-isomer being the predominant product with only low amounts of the desired uncaged peptide formed. Overall, the data presented here indicates that an mBhc caged thiol, unlike its Bhc-protected counterpart, undergoes clean conversion to the uncaged peptide with no significant formation of undesired photo-rearranged byproducts.
Figure 2.

Uncaging studies using a peptide with a mBhc-protected thiol. (A) Photo-triggered uncaging of mBhc-protected K-Ras peptide (18). (B) EIC chromatogram (m/z = 520.57, calcd for [M + 3H]3+= 520.58) of a 100 µM solution of 18 before irradiation, (C) EIC chromatogram (m/z = 520.57, calcd for [M + 3H]3+= 520.58) of a 100 µM solution of 18 after 60 s irradiation at 365 nm suggesting that no photo-isomer is generated and only the remaining starting peptide peak is present, (D) EIC chromatogram (m/z = 647.37, calcd for [M + 2H]2+= 647.37) of a 100 µM of 18 after 60 s irradiation at 365 nm which clearly indicates formation of free peptide 19.
Photo-physical properties of an mBhc protected thiol
The observations noted above suggest that mBhc should be useful as a caging group for thiol protection in peptides and other biomolecules. Accordingly, the spectral and photochemical properties mBhc were studied in detail in order to compare them with Bhc and other established caging groups. Perusal of the data summarized in Table 1 shows that λmax(ex) and λmax(em) of mBhc at pH 7.2, are minimally (~ 5 nm) red-shifted relative to those of Bhc due to the electronic effect of the methyl substituent. The molar absorptivity of mBhc was measured to be 14,500 M−1cm−1 which is comparable to that of Bhc. The one- and two-photon uncaging efficiencies of an mBhc - protected thiol were also quantified by irradiating solutions of 18 followed by analysis via RP-HPLC (Figure 3). For one-photon measurement, solutions of 18 were irradiated using 365 nm light in a Rayonet reactor for varying amounts of time ranging from 5 to 60 sec and analyzed by RP-HPLC to monitor the disappearance of 18 over time. The one-photon quantum yield for thiol uncaging of 18 was measured to be 0.01 by following the disappearance of caged peptide over different irradiation times and using 6-bromo-7-hydroxycoumarin-4-ylmethyl acetate (Bhc-OAc) as a reference, which was photolyzed under the same conditions (Fig 3B). In order to fully evaluate the photo-conversion yield of mBhc protected thiols to the free thiols, a fluorophore-labeled homolog of the caged peptide 18 (Figure S4) and also an mBhc-protected form of cysteamine (Figure S3) were prepared. Having the fluorophore group remain associated with the thiol moiety after photolysis allowed us to fully monitor the release of the free thiol or any other possible byproducts using analytical RP-HPLC via fluorescence detection That HPLC data shows essentially clean conversion of the caged compounds to the corresponding free thiols with no photo-isomer or byproduct formation. Further experiments were carried out to evaluate the two-photon uncaging efficiency of mBhc. For those measurements, solutions of 18 were irradiated at 800 nm using a pulsed Ti:Saphire laser and the photolysis products were again analyzed by RP-HPLC and confirmed by LC-MS (Figure 3C). The two-photon action cross-section of mBhc was measured to be 0.16 GM at 800 nm again using Bhc-OAc as a reference. The two-photon action cross-section and quantum yield for mBhc are comparatively high considering that thiols are poorer leaving groups relative to carboxylates and phosphates.
Table 1.
Photophysical properties of mBhc-thiol versus Bhc-OAc.
| λmax (ex) (nm) |
λmax (em) (nm) |
ε (λmax) (M−1cm−1) |
Qu (365 nm) | δu (800 nm) | |
|---|---|---|---|---|---|
| mBhc-thiol (18) | 374 | 480 | 14,500 | 0.01 | 0.16 |
| Bhc-OAc | 370 | 474 | 15,000 | 0.04 | 0.42 |
λmax (ex) and λmax (em): absorption and emission maximum in nm, respectively, ε: extinction coefficient in M−1cm−1 at wavelength indicated, Qu: quantum yield of one-photon uncaging at 365 nm, δu two-photon action cross-section in 10−50cm4s/photon (GM) for uncaging at 800 nm.
Figure 3.

Photophysical properties of mBhc. (A) Absorption and emission spectra of mBhc (3) in 50 mM PB, pH 7.4. (B) Time course of photolysis of 18 and Bhc-OAc as a reference at 365 nm and (C) Time course of photolysis of 18 and Bhc-OAc as a reference at 800 nm (pulsed Ti:Saphire laser, 210 mw, 80 fs pulse width) quantified by RP-HPLC. Photolysis reactions were performed in 100 µM (for UV), and 300 µM (for TP) solutions of 18 containing 1 mM DTT in 50 mM PB, pH 7.4.
One- and two-photon activation of protein prenylation
Since an mBhc protected thiol demonstrated good uncaging efficiency toward one- and two-photon excitation, we next sought to study its utility for photo-triggered activation of a peptide in a more biologically relevant context. Protein prenylation is a ubiquitous post-translational modification that plays critical roles in a variety of cellular functions including the regulation of cell growth, differentiation and cytoskeletal integrity. Prenylation involves the enzymatic attachment of a prenyl group through a thioether linkage to a conserved cysteine residue near the C-terminus of various proteins.25 This process is catalyzed by protein prenyltransferases including protein farnesyltransferase which transfers a farnesyl (C15) group. Among the proteins that undergo prenylation is Ras, which upon farnesylation migrates to plasma membrane where it participates in key cell signaling pathways including cell division. Mutations in the Ras protein have been linked to numerous types of cancers.26
To investigate the utility of mBhc, the K-Ras derived peptide described above was studied in in vitro prenylation reactions. It should be noted peptide 18 incorporates an mBhc-protected cysteine residue at the natural site of prenylation and hence should not be a substrate for protein farnesyltransferase in its caged state. However, upon irradiation, photo-cleavage of the protecting group should generate a peptide manifesting a free thiol suitable for prenylation by PFTase (see Figure 3A). To test this, in vitro farnesylation reactions using the caged K-Ras derived peptide 18 were performed under several different conditions. As predicted, incubation of the caged peptide with the enzyme and FPP did not result in the generation of any farnesylated peptide. LC-MS analysis of the mixture indicates only the presence of the caged peptide (m/z = 520.65, Figure 3B). Photolysis of 18 for 60 seconds at 365 nm in the absence of the enzyme, produced the free-thiol containing peptide (19) as evidenced by the appearance of a new peak with a lower retention time exhibiting the expected mass (m/z = 647.45). Importantly, photolysis of 18 in the presence of PFTase resulted in generation of a different peak corresponding to the expected farnesylated peptide (21). This was confirmed by the detection of a new peak eluting at 5.36 min with an m/z ratio of 500.06 which is in good agreement with the calculated value (m/z for [M+3H]3+ = 499.99).
Since, photo-triggered activation of the mBhc-protected peptide was successful using a one-photon (UV) process, we next sought to further evaluate its ability to be uncaged via two-photon excitation where IR light is used as the trigger in lieu of UV irradiation. This would open the door for employing such caged peptides in studies performed inside tissue or whole organisms where UV light has low penetration and causes phototoxicity. Accordingly, in vitro farnesylation experiments, similar to those described above for UV irradiation, were carried out using an 800 nm laser light source. Irradiation of 18 using 800 nm laser light for 5 min in the absence of PFTase resulted in the generation of free peptide (Figure S5). However, two-photon irradiation of caged peptide in the presence of FPP and PFTase generated the farnesylated peptide as confirmed by LC-MS analysis (Figure S5). This data clearly illustrates that mBhc-protected K-Ras peptide can be being activated and undergo farnesylation upon near IR irradiation, setting the stage for future studies in whole cells and tissue samples.
Two-photon patterning using a mBhc-caged thiol
In addition to their use for triggering biological activity as noted above, caged thiols are also useful for creating patterns of thiols that can be further functionalized for various material science applications. In particular, since the above experiments demonstrated that mBhc could be efficiently removed by two-photon excitation with 800 nm light, we reasoned that it should be possible to use an mBhc-protected thiol to create 3D patterns within a hydrogel matrix. This has been previously accomplished using a Bhc-protected thiol.9,27 However, the improved efficiency of thiol-uncaging obtained with mBhc relative to Bhc due to elimination of the photoisomerization pathway should increase the utility of this approach; in theory, higher levels of thiol uncaging should be obtained with mBhc for a given amount of irradiation. Accordingly, we sought to compare the thiol patterning obtained using mBhc versus Bhc. Hyaluronic acid (HA) hydrogels were modified with mBhc- or Bhc-protected thiols by coupling mBhc/Bhc-protected cysteamine with the carboxylate groups of HA, and furan functional groups, which are crosslinked with poly(ethylene glycol) (PEG)-bismaleimide (Scheme 5, Figure 5A). Unreacted furan groups are quenched with N-hydroxyethyl maleimide and then the functionalized, crosslinked hydrogels are extensively washed. The resulting material was then infused with sulfhydryl-reactive AlexaFluor546-maleimide to allow visualization of any uncaged thiols formed followed by two-photon irradiation at 740 nm using a confocal microscope. Square tile patterns were created by scanning a square region of interest 5–20 times in the x-y plane at a fixed z-dimension. The overall dimensions for each square tile were 80×80 µm, with the plane of the patterned tile positioned 150 µm from the base of the hydrogel. After uncaged thiols react with the Alexa Fluor reagent, the patterns were imaged and quantified by confocal microscopy. Images from those experiments (Figure 5B) illustrate how clean patterns can be prepared using this approach. As expected, the intensity of thiol labeling was greater in hydrogels prepared using mBhc compared with Bhc due to the greater uncaging efficiency of the former. Quantitative image analysis of the immobilized Alexa Fluor 546 dye (Figure 5C) shows that the uncaging efficiency of the mBhc-functionalized hydrogel is approximately 4-fold higher than that obtained using the Bhc-containing material. Overall, these results further highlight the utility of the mBhc group for thiol protection.
Scheme 5.

Schematic representation of (A) synthesis of mBhc and Bhc protected cysteamine, followd by (B) conjugation to HA-carboxylic acids using DMT-MM prior to crosslinking HA-furan with PEG-bismaleimide.
Figure 5.

Comparison of two-photon patterning using Bhc- and mBhc-caged thiol. (A) Bhc or mBhc to is conjugated to HA-carboxylic acids using DMT-MM prior to crosslinking HA-furan with PEG-bismaleimide. (B) Schematic representation of two-photon patterning in Bhc or mBhc-conjugated HA hydrogels. A 3D hydrogel scaffold (i) is formed when Bhc/mBhc-modified HA-furan is chemically crosslinked with PEG-bismaleimide (ii).. The resulting photo-labile hydrogel undergoes photolysis of the Bhc/mBhc groups using two-photon irradiation to liberate free thiols in discrete regions of the hydrogel, which then react with maleimide-bearing Alexa Fluor 546 (mal-546) (iii) (C) Visualization of mal-546 patterns in the x-y plane and z-dimension in mBhc and Bhc conjugated HA hydrogels. Regions of interest were scanned 5 to 20 times at a fixed z-dimension. The concentrations of mBhc and Bhc were matched based on UV absorbance. Patterns in mBhc and Bhc conjugated HA hydrogels were imaged at different confocal settings due to Bhc patterns being so faint in comparison to mBhc patterns. (D) The z-axis profile of immobilized mal-546 in mBhc and Bhc conjugated HA hydrogels was quantified with the maximum intensity was centered at 0 µm.
Conclusion
In this work, we have developed mBhc as an alternative, coumarin-based caging group capable of mediating thiol photo-release through both one- and two-photon irradiation. The design of mBhc was guided by recently reported mechanistic experiments that showed that photo-isomerization of Bhc-caged thiols leads to a low uncaging yield. Studies of the spectral properties of mBhc show minimal variations from those of Bhc suggesting that mBhc should be a useful chromophore with high fluorogenic character. A form of mBhc [Fmoc-Cys(MOM-mBhc)-OH] suitable for solid phase synthesis was prepared and used to assemble a K-Ras derived peptide incorporating a caged cysteine residue. One-photon photolysis of the caged peptide at 365 nm resulted in clean conversion to the free peptide with a photolysis quantum yield of 0.01. The two-photon action cross-section of the caged peptide was also measured to be 0.13 GM at 800 nm, comparable to that of Bhc-OAc. The one-and two-photon uncaging of the caged peptide in the presence of PFTase and FPP generated a farnesylated peptide indicating that the free peptide which resulted from photolysis can be recognized by PFTase and become enzymatically modified. The high two-photon uncaging efficiency of mBhc protected thiols was also harnessed to create 3D patterns of thiols inside hydrogels for material science applications. Overall, this work sets the stage for future work requiring caged sulfhydryl groups. Given the unique reactivity of thiols, the mBhc protecting group developed here should be useful for a variety of applications in biology and material science.
Experimental Section
General
All reagents needed for solid phase peptide synthesis were purchased from Peptide International (Louisville, KY). All other solvents and reagents used for synthesis and other experiments were purchased from Sigma Aldrich (St. Louis, MO) or Caledon Laboratory Chemicals (Georgetown, ON, Canada). Lyophilized sodium hyaluronate (HA) was purchased from Lifecore Biomedical (2.15 × 105 amu) (Chaska, MN, USA). Dimethyl sulfoxide (DMSO), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM), 1-methyl-2-pyrrolidinone (NMP), furfurylamine, and Dulbecco’s phosphate buffered saline (PBS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-(2-hydroxyethyl)maleimide was purchased from Strem Chemicals (Newburyport, MA, USA). 2-(N-mortholino)ethanesulfonic acid (MES) were purchased from BioShop Canada Inc. (Burlington, ON, Canada). Dialysis membranes were purchased from Spectrum Laboratories (Rancho Dominguez, CA, USA). Alexa Fluor 546 maleimide (mal-546) was purchased from Thermo Scientific (Waltham, MA, USA). HPLC analysis (analytical and preparative) was performed using a Beckman model 125/166 instrument, equipped with a UV detector and C18 columns (Varian Microsorb-MV, 5 µm, 4.6 × 250 mm and Phenomenex Luna, 10 µm, 10 × 250 mm respectively). 1H NMR data of synthetic compounds were recorded at 500 MHz on a Varian Instrument at 25 °C, unless noted. 13C NMR data of synthetic compounds were recorded at 125 MHz on a Varian Instrument at 25 °C, unless noted.
Procedure for solid phase peptide synthesis
Peptides were synthesized using an automated solid-phase peptide synthesizer (PS3, Protein Technologies Inc., Memphis, TN) employing Fmoc/HCTU based chemistry. The synthesis started by transferring Fmoc-Met-Wang resin (0.25 mmol) into a reaction vessel followed 45 min swelling in DMF. Peptide chain elongation was performed using HCTU and N-methylmorpholine. Standard amino acid coupling was carried out by incubation of 4 equiv of both HCTU and the Fmoc protected amino acid with the resin for 30 min. Coupling of Fmoc-Cys(MOM-mBhc)-OH (17) was performed by incubation of 1.5 equiv of both the amino acid and HCTU with the resin for 6 h. Peptide chain elongation was completed by N-terminus deprotection using 10% piperidine in DMF (v/v). Global deprotection and resin cleavage was accomplished via treatment with Reagent K. Peptides were then precipitated with Et2O, pelleted by centrifugation and the residue rinsed twice with Et2O. The resulting crude peptide was dissolved in CH3OH and purified by preparative RP-HPLC. KKKSKTCC(mBhc)IM (18). ESI-MS: calcd for [C67H115BrN16O17S2 + 2H]2+ 780.3698, found 780.3777. 5-Fam-KKKSKTKC(mBhc)VIM calcd for [C88H125BrN16O23S2+2H]2+ 959.3937, found 959.3782.
Hc-Boc-cysteamine (7)
7-hydroxycoumarin bromide (1a, 1 g, 3.9 mmol), N-(tert-butoxycarbonyl)aminoethanethiol (10, 0.86 mL, 5.1 mmol) and 1,8-diazabicyloundec-7-ene (0.76 mL, 5.1 mmol) were dissolved in 70 mL of THF and refluxed for 4 h. The reaction was judged completed by TLC (2:3, Hexanes/EtOAc). Solvent was removed in vacuo, and the crude mixture was diluted in 75 mL EtOAc. The organic layer was washed with 50 mL of 0.1 M NH4Cl(aq), brine, and then dried over Na2SO4. The solvent was removed in vacuo and the crude mixture was purified via silica gel chromatography (2:1 Hexanes/EtOAc) to give 836 mg of 7 as a yellow oil (61 % yield). 1H NMR (d6-acetone) δ 7.79 (1H, d, J = 8.4 Hz), 6.95 (1H, dd, J = 8.75, 2.8 Hz), 6.32 (1H, s), 3.95 (2H, s), 3.37 (2H, q, J = 6.3 Hz), 2.74 (2H, t, J = 7 Hz), 1.54 (9H, s).
Ethyl 4-bromo-2-methyl acetoacetate (14)
Compound 14 was prepared by minor modification of a published procedure.23 To an ice cold solution of ethyl 2-methylacetoacetate (2 mL, 14 mmol) in 50 mL CHCl3 was added a solution of Br2 (0.68 mL, 14 mmol) in 10 mL CHCl3 over 15 min. The mixture was then warmed to rt and stirred overnight. The organic layer was then washed with 50 mL solution of 0.1 M sodium thiosulfate, brine and then dried over Na2SO4. The solvent was evaporated in vacuo to yield 2.34 g of 14 as a pale orange oil (75 % yield). The resulting material was directly used for the next step without further purification.
6-Bromo-7-hydroxy-3-methylcoumarin-4-ylmethyl bromide (mBhc-Br, 3a)
A solution of 4-bromoresrocinol (1 g, 5.3 mmol) and ethyl 4-bromo-2-methyl acetoacetate 14 (2.3 g, 10.4 mmol) in 30 mL of CH3SO3H was stirred at rt overnight. The mixture was then fractionated between 100 mL H2O and 100 mL EtOAc. The organic layer was separated, washed with brine and dried over Na2SO4. Solvent was removed in vacuo and the resulting crude mixture was purified via silica gel chromatography (2:1, Hexanes/EtOAc) to give 645 mg of 3 as a pale yellow solid. 1H NMR (d6-acetone) δ 8.01 (1H, s), 6.95 (1H, s), 4.85 (2H, s), 2.21 (3H, s). 13C NMR (d6-acetone) δ 160.81, 156.32, 153.12, 143.73, 128.59, 121.46, 111.94, 106.00, 103.45, 24.14, 11.98. HR-MS (ESI) m/z calcd for [C11H7Br2O3]− 346.8720, found 346.8720.
MOM-mBhc-Br (15)
To a stirred solution of 3a (400 mg, 1.15 mmol) and chloromethyl methyl ether (MOM-Cl, 0.13 mL, 1.72 mmol) was added 1,8-diazabicyloundec-7-ene (0.19 mL, 1.3 mmol). The mixture was stirred for about 2 h until complete as judged by TLC (in CH2Cl2). The solvent was evaporated in vacuo. The resulting crude material was dissolved in a small amount of CH2Cl2 which was then loaded onto a silica gel column and purified to give 428 mg of 3 as pale yellow oil (95 % yield). 1H NMR (CDCl3) δ 7.82 (1H, s), 7.15 (1H, s), 5.31, (2H, s), 4.62 (2H, s), 3.52 (3H, s), 2.28 (3H, q, J = 6.3 Hz), 13C NMR (CDCl3) δ 161.40, 155.55, 152.80, 142.50, 128.03, 123.26, 113.33, 108.66, 103.96, 95.21, 56.64, 37.09, 12.91. HR-MS (ESI) m/z calcd for (C13H12Br2O4 + Na)+ 414.8980, found 414.9001.
mBhc-cysteamine (23)
mBhc-Br (3a, 0.8 g, 2.3 mmol), N-(tert-butoxycarbonyl)aminoethanethiol 10 (0.51 mL, 3.0 mmol) and 1,8-diazabicyloundec-7-ene (0.45 mL, 3.0 mmol) were dissolved in 50 mL of THF and refluxed for 4 h. The reaction was judged completed by TLC (2:3, Hexanes/EtOAc). The solvent was removed in vacuo, and the crude mixture was diluted in 50 mL EtOAc. The organic layer was washed with 50 mL of 0.1 M NH4Cl(aq), brine, and then dried over Na2SO4. The solvent was removed in vacuo and the crude mixture was purified via silica chromatography (2:1 Hexanes/EtOAc) to give 664 mg of mBhc-Boc-cysteamine as a yellow oil. The purified compound was dissolved in 10 mL solution of CH2Cl2:TFA (1:1) and stirred for 30 min. The mixture was evaporated and purified via silica (1:1 Hex/EtOAc) to give 514 mg (65 % yield) of the desired free amine as white solid. 1H NMR (d6-acetone) δ 7.92 (1H, s), 6.77 (1H, s), 6.32 (1H, s), 4.06 (2H, s), 3.29 (2H, q, J = 6.3 Hz), 3.02 (2H, t, J = 7 Hz), 2.19 (3H, s), 13C NMR (D2O) δ 163.53, 155.54, 151.78, 139.08, 128.88, 124.15, 113.36, 106.72, 103.38, 49.95, 48.31, 34.24, 13.52. HR-MS (ESI) m/z calcd for (C13H14BrNO3 + H)+ 343.9951, found 414.9827.
Fmoc-Cys(MOM-mBhc)-OCH3 (16)
Bromide 15 (400 mg, 1 mmol) and Fmoc-Cys-OCH3 (714 mg, 2 mmol) were dissolved in 10 mL of a solution of 2:1:1 DMF/CH3CN/H2O/0.1% TFA (v/v/v/v). Zn(OAc)2 was then added (550 mg, 2.5 mmol) and the reaction monitored by TLC (1:1 Hexanes/EtOAc). After two days, the solvent was removed and the reaction purified via column chromatography (1:1 Hexanes/EtOAc) to give 530 mg of 16 as yellow powder (81% yield). 1H NMR (CDCl3) 7.80 (1H, s), 7.72 (2H, t, J=8), 7.59 (2H, t, J=7), 7.32–7.40 (2H, m), 7.28 (2H, t, J=7), 7.11 (1H, s), 5.61 (1H, d, J=6.5), 5.28 (2H, s), 4.53 (1H, t, J=7), 4.45 (1H, t, J=7), 4.22 (1H, t, J=6.5), 3.70–3.80 (6H, m), 3.51 (3H, s), 3.16 (1H, q),, 2.20 (3H, s). 13C NMR (CDCl3) δ 170.81, 161.41, 155.74, 155.32, 152.69, 143.67, 143.59, 143.53, 141.35, 141.31 128.57, 127.79, 127.76 127.11, 127.05, 125.01, 124.96, 122.31, 120.02, 120.01, 114.16, 108.41, 103.79, 95.18, 67.07, 53.87, 52.97, 47.19, 35.62, 29.83, 13.27. HR-MS (ESI) m/z calcd for [C32H30BrNNaO8S + Na]+ 690.0773, found 690.0720.
Fmoc-Cys(MOM-mBhc)-OH (17)
Ester 16 (200 mg, 0.30 mmol) and (CH3)3SnOH (135 mg, 0.75 mmol) were dissolved in CH2Cl2 (5 mL) and brought to reflux. After 7 h, the reaction was judged complete by TLC (1:1 Hexanes/EtOAc), the solvent removed in vacuo and the resulting oil redissolved in EtOAc (20 mL). The organic layer was washed with 5% HCl (3 × 10 mL) and brine (3 × 10 mL), dried with Na2SO4 and evaporated to give 173 mg of 17 as a yellow powder (90% yield). 1H NMR (CDCl3) δ 9.68 (1H, s), 7.73 (1H, s), 7.64 (2H, t, J=7), 7.55 (2H, t, J=7), 7.32 (2H, t, J=7.5), 7.23 (2H, m), 6.98 (1H, s), 5.93 (1H, d, J=7.5), 5.19 (2H, s), 4.72 (1H, m), 4.65 (1H, t, J=7), 4.41 (1H, t, J=7), 4.16 (1H, t, J=7), 3.70–3.80 (2H, m), 3.45 (3H, s), 3.20 (1H, m), 3.06 (1H, q), 2.14 (3H, s). 13C NMR (CDCl3) δ 173.41, 161.83, 156.20, 155.21, 152.43, 144.06, 143.63, 143.48, 141.26, 141.22, 128.58, 127.11, 127.06, 125.02, 119.98, 114.09, 108.52, 103.58, 95.06, 67.26, 56.63, 53.72, 47.09, 35.51, 29.79, 14.18, 13.24, 31.07, 14.07. HR-MS (ESI) m/z calcd for [C31H28BrNNaO8S + Na]+ 676.0601, found 676.0601.
General procedure for UV photolysis of caged molecules
Solutions of caged compound were prepared in photolysis buffer (50 mM phosphate buffer, pH 7.4 containing 1 mM DTT) at a final concentration of 25–250 µM. Aliquots (100 µL) of caged compound solutions were transferred into quartz cuvettes (10 × 50 mm) and irradiated for varying amounts of time with 365 nm UV light using a Rayonet reactor (2 × 14 watt RPR-3500 bulbs). After different irradiation times, the samples were analyzed by RP-HPLC or LC-MS.
General procedure for two-photon photolysis of caged molecules
Solutions of caged compounds were prepared in photolysis buffer (50 mM phosphate buffer, pH 7.4 containing 1 mM DTT) at a final concentration of 300 µM. Aliquots (15 µL) of caged compound solutions were transferred into 15 µL quartz cuvettes (Starna Cells Corp. dimensions: 1mm × 1mm) and irradiated using two-photon laser apparatus at 800 nm for varying amount of time. After each reaction the samples were analyzed by RP-HPLC or LC-MS. The light source utilized for two-photon irradiation was a homebuilt, regeneratively amplified Ti:sapphire laser system. This laser operates at 1 kHz with 210 mw pulses centered at a wavelength of 800 nm. The laser pulses have a Gaussian full width at half maximum of 80 fs.
One-photon quantum yield (Qu) and two-photon uncaging cross-section (δu) of 18
Qu and δu for 18 were measured by comparing its photolysis rate with Bhc-OAc as a reference (Qu = 0.04 at 365 nm, δu = 0.45 at 800 nm). As described above, aliquots containing 18 were irradiated with either a 365 nm lamp or an 800 nm laser for varying amounts of time. Each sample was analyzed by RP-HPLC to monitor the disappearance of the starting caged compound over time. Similar photolysis experiments were conducted with Bhc-OAc solutions (100 µM for UV and 300 µM for IR) in 50 mM phosphate buffer, pH 7.2. Photolyzed Bhc-OAc solutions were also analyzed by RP-HPLC. The compounds were eluted with a gradient of Solvent A and Solvent B (gradient of a 1% increase in Solvent B/min, flow rate 1 mL/min) monitored by absorbance at 220 nm. Reaction progress data was analyzed as described above and the first order decay constants for the two compounds were used in the formula Φu or δu (18) = Φu or δu (reference) × Kobs (18)/Kobs(reference) to calculate the value of δu for 18 where Φu (reference) = 0.04 and δu (reference) = 0.42 GM.
UV and two-photon triggered farnesylation of 18
A 7.5 µM solution of compound 18 was prepared in prenylation buffer (15 mM DTT, 10 mM MgCl2, 50 µM ZnCl2, 20 mM KCl and 22 µM FPP, 50 mM PB buffer) and divided into three 100 µL aliquots. Yeast PFTase was added to the first aliquot to give a final concentration of 30 nM but the resulting sample was not subjected to photolysis; the second aliquot was irradiated in absence of yeast PFTase while the third sample was supplemented with yeast PFTase (50 nM) and then photolyzed with either UV or laser light. UV photolysis was conducted for 1 min at 365 nm while two-photon irradiation was performed for 5 min at 800 nm. Each sample was incubated for 30 min at rt and then analyzed by LC-MS as described above.
General procedure for LC-MS analysis
Aliquots (100 µL) of caged compound solutions which were diluted down to 5–20 µM were analyzed by LC-MS. The general gradient for LC-MS analysis was 0–100% H2O/0.1% HCO2H (v/v) to CH3CN/0.1% HCO2H (v/v) in 25 min.
Synthesis of mBhc- and Bhc-modified HA-furan (25 a,b)
HA-furan was prepared as previously described.28 To synthesize HA-furan (24) modified with mBhc or Bhc, HA-furan was dissolved in NMP:MES (100 mM, pH 5.5) at a ratio of 1:1 to achieve 0.50% w/v HA-furan. DMT-MM was then added (5 equiv. relative to free carboxylic acids) followed by the dropwise addition of a solution of mBhc or Bhc in DMSO (1 equiv. relative to free carboxylic acids). The reaction was stirred at rt in the dark for 24 h and then dialyzed against H2O:NMP:DMSO (2:1:1, v/v/v) for 1 d. The organic fraction of the solution was halved every 24 h for 3 days before being replaced with only H2O for the final 2 days and then lyophilized. The resulting HA-furan modified with either Bhc or mBhc was then dissolved in K2CO3 (1.0% w/v, 10 eq relative to furans) for 24 h, dialyzed against H2O for 3 days, and lyophilized.
Preparation of HA Hydrogels for Photopatterning
HA-furan-(mBhc or Bhc) was dissolved overnight in MES (100 mM, pH 5.5):DMSO (3:1, v/v) and mixed with an equal volume of a solution of bis-maleimide-poly(ethylene glycol) (mal2-PEG) dissolved in MES buffer (100 mM, pH 5.5). The mixture was pipetted into 96-well glass bottom plates and allowed to react overnight at 37 °C to form hydrogels with a final concentration of 2.00% HA-furan-(mBhc or Bhc) and a 1:1 ratio of furan:maleimide. The mBhc and Bhc concentrations in the hydrogels were matched based on their UV absorbance at their maximum peak intensity at 365 nm. The unreacted furans in the hydrogel were quenched with 30 mM N-(2-hydroxyethyl)maleimide in MES buffer (100 mM, pH 5.5) for 24 h at rt. The N-(2-hydroxyethyl)maleimide was washed from the gel with borate buffer (100mM, pH 9.0) to hydrolyze any remaining maleimides, followed by extensively washing of the hydrogel with PBS (pH 6.8). A solution of Alexa Fluor 546 maleimide (100 µM in PBS pH 6.8) was then soaked into the hydrogel overnight at 4 °C and excess supernatant was removed prior to photopatterning. The resulting HA-furan-(mBhc or Bhc)/mal2-PEG hydrogels are herein described as HAmBhc/PEG and HABhc/PEG, respectively.
Photopatterning of hydrogels
HAmBhc/PEG and HABhc/PEG hydrogels were photopatterned using a Zeiss LSM710 META confocal microscope equipped with a Coherent Chameleon two-photon laser and a 10× objective. For patterning experiments, the two-photon laser was set to 740 nm with 38% power (1660 mW max power) and a scan dwell time of 106.83 µs/µm. Due to high non-specific binding of Alexa Fluor 546 maleimide in the HABhc/PEG hydrogels compared to HAmBhc/PEG hydrogels, unreacted Alexa Fluor 546 maleimide was not washed from the hydrogels. This maintained the same background fluorescence in the HAmBhc/PEG and HABhc/PEG hydrogels, allowing the patterns to be directly compared. The concentration of Alexa Fluor 546 maleimide immobilized in the patterns exceeded the bulk unreacted Alexa Fluor 546 maleimide solution, permitting the visualization of the patterns. Alexa Fluor 546 maleimide reacted with the uncaged thiols for 4 h prior to imaging. Patterns were imaged on an Olympus Fluoview FV1000 confocal microscope with x-y scans every 5 µm in the z direction. Imaged photopatterns were quantified using ImageJ against a standard curve of HA/PEG gels containing Alexa Fluor 546 maleimide at different concentrations. The background concentration of unreacted Alexa Fluor 546 maleimide was subtracted from the concentration immobilized in the patterns.
Supplementary Material
Figure 4.

Photo-triggered farnesylation of an mBhc-protected K-Ras peptide. (A) Structures of peptides and products relevant to this study. (B) EIC chromatogram (m/z = 520.65, calcd for [M + 3H]3+= 520.58) of a 7.5 µM solution of 18 in a prenylation buffer containing PFTase with no irradiation. (C) EIC chromatogram (m/z = 647.45, calcd for [M + 2H]2+= 647.39) of a 7.5 µM solution of 18 after 60 s irradiation at 365 nm in prenylation buffer without PFTase showing the formation of free peptide 19. (D) EIC chromatogram (m/z = 500.06, calcd for [M + 3H]3+= 499.99) of a 7.5 µM of 18 after 60 s irradiation at 365 nm in presence of PFTase showing the formation of farnesylated peptide 21.
Acknowledgments
Financial support for these studies through National Institutes of Health Grant GM084152, National Science Foundation Grant CHE-1308655, the University of Minnesota Supercomputer Institute, and the University of Minnesota Graduate School, the Canadian Institutes of Health Research (CIHR) and the Natural Sciences and Engineering Research Council (NSERC) through the Collaborative Health Research Program is gratefully acknowledged.
References
- 1.Klán P, Šolomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J. Chem. Rev. 2013;113:119–191. doi: 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pelliccioli AP, Wirz J. Photochem. Photobiol. Sci. 2002;1:441–458. doi: 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]
- 3.Ellis-Davies GCR. Nat. Methods. 2007;4:619–628. doi: 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee H-M, Larson DR, Lawrence DS. ACS Chem. Biol. 2009;4:409–427. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bort G, Gallavardin T, Ogden D, Dalko PI. Angew. Chem. Int. Ed. 2013;52:4526–4537. doi: 10.1002/anie.201204203. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Tang S, Zheng J-S, Zhao R, Wang ZP, Shao W, Chang H-N, Cheng JY, Zhao H, Liu L, Qi H. Nat. Commun. 2015;6:7220. doi: 10.1038/ncomms8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang X, Shestopalov IA, Sinha S, Zheng G, Pitt CLW, Li W-H, Olson AJ, Chen JK. J Am. Chem. Soc. 2009;131:13255–13269. doi: 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatterdam V, Ramadass R, Stoess T, Fichte MAH, Wachtveitl J, Heckel A, Tampé R. Angew. Chem. Int. Ed. 2014;53:5680–5684. doi: 10.1002/anie.201309930. [DOI] [PubMed] [Google Scholar]
- 9.Wylie RG, Shoichet MS. Biomacromolecules. 2011;12:3789–3796. doi: 10.1021/bm201037j. [DOI] [PubMed] [Google Scholar]
- 10.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo Y, Shoichet MS. Nat. Mater. 2004;3:249–253. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 12.Chapman HA, Riese RJ, Shi G-P. Annu. Rev. Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- 13.Haugaard N. Ann. N. Y. Acad. Sci. 2000;899:148–158. doi: 10.1111/j.1749-6632.2000.tb06183.x. [DOI] [PubMed] [Google Scholar]
- 14.Uprety R, Luo J, Liu J, Naro Y, Samanta S, Deiters A. Chem Bio Chem. 2014;15:1793–1799. doi: 10.1002/cbic.201400073. [DOI] [PubMed] [Google Scholar]
- 15.Philipson KD, Gallivan JP, Brandt GS, Dougherty DA, Lester HA. Am. J. Physiol. Cell Physiol. 2001;281:C195–C206. doi: 10.1152/ajpcell.2001.281.1.C195. [DOI] [PubMed] [Google Scholar]
- 16.Capozzi G, Modena G. In: The Thiol Group: Vol. 2 (1974) Patai S, editor. John Wiley & Sons, Ltd.; Chichester, UK: 1974. pp. 785–839. [Google Scholar]
- 17.DeGraw AJ, Hast MA, Xu J, Mullen D, Beese LS, Barany G, Distefano MD. Chem. Biol. Drug Des. 2008;72:171–181. doi: 10.1111/j.1747-0285.2008.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo J, Uprety R, Naro Y, Chou C, Nguyen DP, Chin JW, Deiters A. J Am. Chem. Soc. 2014;136:15551–15558. doi: 10.1021/ja5055862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kotzur N, Briand B, Beyermann M, Hagen V. J Am. Chem. Soc. 2009;131:16927–16931. doi: 10.1021/ja907287n. [DOI] [PubMed] [Google Scholar]
- 20.Abate-Pella D, Zeliadt NA, Ochocki JD, Warmka JK, Dore TM, Blank DA, Wattenberg EV, Distefano MD. Chem Bio Chem. 2012;13:1009–1016. doi: 10.1002/cbic.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahmoodi MM, Abate-Pella D, Pundsack TJ, Palsuledesai CC, Goff PC, Blank DA, Distefano MD. J Am. Chem. Soc. 2016;18:5848–5859. doi: 10.1021/jacs.5b11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuta T, Wang SS-H, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY. Proc. Natl. Acad. Sci. 1999;96:1193–1200. doi: 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svendsen A, Boll PM. Tetrahedron. 1973;29:4251–4258. [Google Scholar]
- 24.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angew. Chem. Int. Ed. 2005;44:1378–1382. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- 25.Palsuledesai CC, Distefano MD. ACS Chem. Biol. 2015;10:51–62. doi: 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochocki JD, Distefano MD. Med Chem Commun. 2013;4:476–492. doi: 10.1039/C2MD20299A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nat. Mater. 2011;10:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 28.Nimmo CM, Owen SC, Shoichet MS. Biomacromolecules. 2011;12:824–830. doi: 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.