

Significance
The flagellum is a highly sophisticated organelle rotated by a motor that confers swarming motility to bacterial cells. Such motility is essential for the full pathogenicity of several virulence bacteria. Several proteins are required for the assembly and operation of the flagellum. Here we report the structural characterization of FliT, a key flagellar chaperone, in the unliganded state and in complex with two substrate flagellar proteins. FliT adopts an autoinhibited structure in order to avoid futile interactions with the export gate in the absence of a substrate. Substrate binding to FliT activates complex targeting to the export gate followed by either the export of the substrate or its assembly to the export apparatus.
Keywords: flagellum, chaperones, assembly factors, NMR spectroscopy, type III secretion
Abstract
The flagellum is a complex bacterial nanomachine that requires the proper assembly of several different proteins for its function. Dedicated chaperones are central in preventing aggregation or undesired interactions of flagellar proteins, including their targeting to the export gate. FliT is a key flagellar chaperone that binds to several flagellar proteins in the cytoplasm, including its cognate filament-capping protein FliD. We have determined the solution structure of the FliT chaperone in the free state and in complex with FliD and the flagellar ATPase FliI. FliT adopts a four-helix bundle and uses a hydrophobic surface formed by the first three helices to recognize its substrate proteins. We show that the fourth helix constitutes the binding site for FlhA, a membrane protein at the export gate. In the absence of a substrate protein FliT adopts an autoinhibited structure wherein both the binding sites for substrates and FlhA are occluded. Substrate binding to FliT activates the complex for FlhA binding and thus targeting of the chaperone–substrate complex to the export gate. The activation and targeting mechanisms reported for FliT appear to be shared among the other flagellar chaperones.
The flagellum is the organelle that enables bacterial locomotion and is one of the most sophisticated protein machines (1–4). Flagella organelles may act as virulence factors because motility is crucial for the action of pathogenic bacteria (5, 6). About 25 different proteins are involved in the assembly of the flagellum, which is divided into five parts: the basal body, hook, hook–filament junction, filament, and filament cap. The basal body is a flagellum-specific type III secretion system responsible for the translocation of flagellar proteins to the distal end of the growing flagellar structure for self-assembly (7). The assembly of the hook and the filament is strictly sequential and is controlled by the export apparatus, which uses both ATP and proton motive force (PMF) to drive protein translocation (8, 9). The cytoplasmic domain of FlhA (10–12), one of the six membrane proteins of the flagellar type III export apparatus, acts as an adaptor to receive the flagella building blocks FliD and flagellin (FliC) when bound to their cognate chaperones FliT and FliS, respectively (Fig. 1) (11).
Fig. 1.
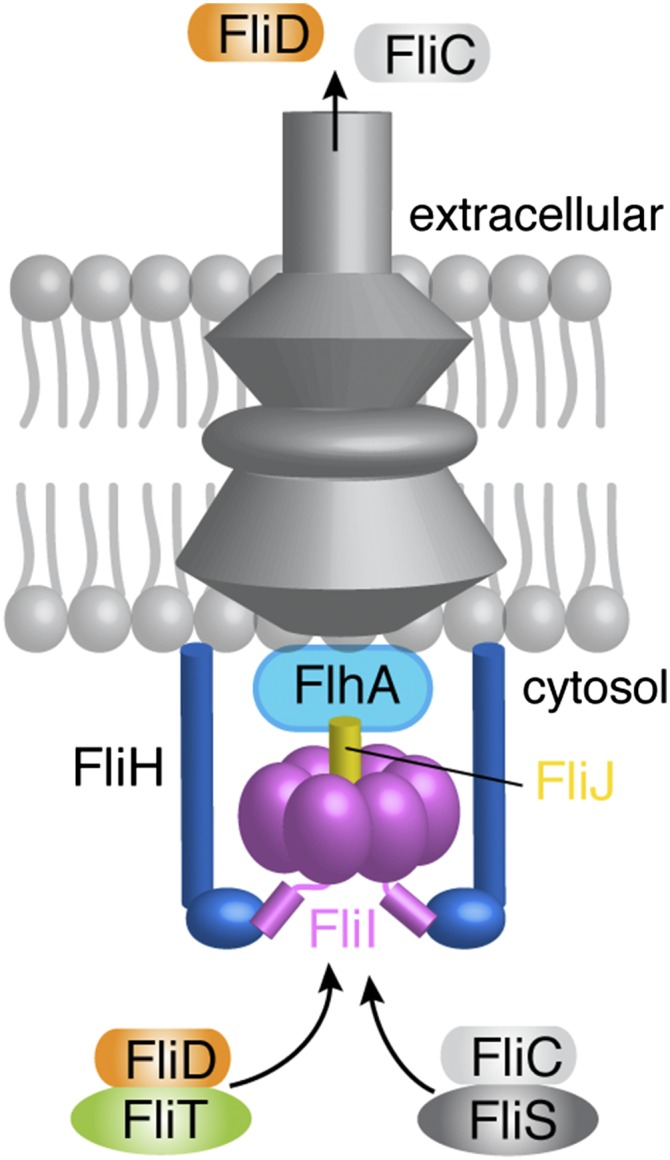
A simplified schematic of the flagellum that includes the proteins studied in this work. For a detailed view, see refs. 1–4.
The export apparatus also comprises three cytosolic proteins: FliI, FliH, and FliJ (Fig. 1). FliI (13), the only ATPase of the system, assembles to a hexameric ring (14, 15) and is located at the base of the basal body (16). The exact role of FliI is not known, but it has been implicated in powering protein export, targeting flagellar proteins to the export gate, and functioning as a sorting platform or specificity switch (17). FliI adopts a structure that is very similar to the structure of the F1 ATP synthase (13). The first 20 N-terminal residues of FliI mediate self-oligomerization (18) and are involved in binding FliH (19, 20). FliH is homologous to the β and δ subunits of the FoF1 ATP synthase (21) and is thought to provide the support point for the association of the FliI ring to the export gate (22). FliJ, which is essential for the export of flagellum building blocks, binds to the center of the hexametric FliI ring in a manner similar to the way the γ subunit binds to the β subunit of FoF1 ATP synthase (23). FliJ also was proposed to recycle substrate-free flagellar chaperones at the export gate (24). FliI, FliH, and FliJ bind to FlhA and FlhB proteins at the export gate (11, 25, 26).
Chaperones dedicated to the assembly and operation of the flagellum bind and protect their cognate substrates from aggregation or premature interactions in the cytoplasm (27–29). In addition, chaperones have been implicated in targeting flagellar proteins to the export gate (11, 30, 31). FliT is a key chaperone in the assembly and operation of the flagellum. FliT interacts with several flagellar proteins, including the exported substrate FliD and the export apparatus components FliI, FliJ, and FlhA, and negatively regulates transcription of flagellar genes by inhibiting the formation of a DNA complex with the master regulator FlhDC (11, 24, 28, 32–36). FliD is exported first during the filament assembly to form a pentameric cap that promotes self-assembly of FliC (37).
The structural details of how FliT recognizes its various partners and how it targets and escorts them to the export gate are not known. We have determined the structures of FliT in the unliganded state and in complex with its substrates FliD and FliI. The structural data show that FliT adopts an autoinhibited conformation in which both the substrate- and FlhA-binding sites are occluded. Substrate binding to FliT releases the FlhA-binding site, thereby enabling targeting of the FliT–substrate complex to the export gate.
Results
Solution Structure of the FliT Chaperone.
Salmonella enterica serovar Typhimurium FliT consists of 122 residues (∼13.7 kDa), and multiangle light scattering (MALS) analysis showed that it is a monomer in solution (Fig. S1A). At high concentrations FliT has a tendency to form a weak dimer with a dimerization dissociation constant (Kd) of ∼0.5–0.6 mM, in agreement with previously published analytical ultracentrifugation data (35). We used NMR (Fig. S1 B and C) to determine the structure of FliT in solution under conditions in which FliT is monomeric ([FliT] ∼0.08 mM). The structure shows that FliT adopts a four-helix bundle (Fig. 2A and Fig. S2A). The first three helices (α1–α3, residues 2–27, 30–49, and 58–83, respectively) are longer than the fourth (α4) helix (residues 98–112). Helices α3 and α4 are connected by a long unstructured linker. Helices α1, α2, and α3 interact intimately with each other, whereas helix α4 interacts primarily with helix α3 (Fig. 2 B and C). The positioning of helix α4 with regard to the other helices appears to be suboptimal, and the FliT structure departs from the canonical four-helix bundle. Helices α1, α2, and α3 expose a contiguous hydrophobic surface (∼1,500 Å2), and helix α4 acts as a lid to occlude it from solvent (Fig. 2D). Leu102, which docks into a hydrophobic pocket lined by nonpolar residues from the other three helices (Fig. 2 B and C), seems to be crucial in stabilizing the helix α4 position against the core of the bundle. A FliT variant in which Leu102 was replaced by Ala is prone to aggregation, and NMR spectra experience severe line broadening, especially at the interface with helix α4 (Fig. S1D). Apparently, mutation of Leu102 destabilizes the docking of helix α4, and as a result the hydrophobic core is exposed to the solvent. A FliT variant lacking helix α4 (FliTΔα4) has a strong tendency to aggregate, further highlighting the role of helix α4 in shielding the hydrophobic core of FliT.
Fig. S1.

NMR characterization of FliT. (A) MALS of FliT shows that the protein exists as a monomer in solution. (B and C) 1H-15N heteronuclear single-quantum correlation (HSQC) (B) and 1H-13C HMQC (C) spectra of U-2H–,15N Ala-13CH3–, Met-13CH3–, Ile-δ1-13CH3–, Leu,Val-13CH3/13CH3–, and Thr-13CH3–labeled FliT. Resonance assignment is shown for the NH and methyl resonances. (D) 1H-15N HSQC spectrum of FliT-L102A (orange) superimposed on the spectrum of wild-type FliT (black).
Fig. 2.

Solution structure of the FliT chaperone. (A) FliT is shown in a cartoon rendering and is colored using a continuous-gradient color scheme from the N terminus (blue) to the C terminus (red). The four helices (α1–α4) are labeled. (B and C) FliT is shown in gray cartoon with semitransparent representation of the solvent-accessible surface in two different views. The residues at the interface of the four-helix bundle are shown in ball-and-stick configuration. (D) The hydrophobic surface of FliT is colored green. Helix α4 acts as a lid to occlude the large hydrophobic surface formed by helices α1–α3 from solvent.
Fig. S2.

Ensemble of the 20 lowest-energy structural conformers of FliT (A), the FliT–FliD complex (B), and the FliT–FliI complex (C).
The FliT structure was previously determined by X-ray crystallography to be a tetramer (35). The structures in solution and in the crystal have pronounced differences. Compared with the solution structure, helix α3 in the crystal structure extends by an additional three turns, and helix α4 is detached and interacts instead with the hydrophobic core of another FliT subunit. The differences between the solution and crystal structures appear to be the result of crystal-packing effects.
Interaction Between FliT and FliD.
FliT is the chaperone for the filament-capping protein FliD, but the structural basis for their interaction is not known. To understand how FliT interacts with FliD, we used NMR spectroscopy to characterize the complex. We prepared several FliD fragments and identified the last 40 residues of the C terminus as the FliT-binding site, in agreement with previous biochemical experiments (28, 32). To identify the FliD-binding site on FliT, we titrated FliDC (the FliD fragment encompassing the last 40 C-terminal residues) to isotopically labeled FliT and monitored their interaction by NMR (Fig. S3A). Chemical shift analysis shows that the FliT residues most affected by FliD binding are located in the hydrophobic core formed by helices α1–α3 (Fig. S3B). Because the hydrophobic core in free FliT is occluded by helix α4 (Fig. 2D), FliD binding would entail displacement of helix α4. Indeed, NMR analysis shows that the helix α4 residues experience large chemical shift perturbation, and linewidth measurements suggest that helix α4 is more mobile when FliD is bound, indicating displacement of helix α4 (Fig. S3B). Taken together, the NMR data show that FliD binds to the hydrophobic surface formed by helices α1–α3 in FliT, and as a result helix α4 is displaced.
Fig. S3.

NMR studies of FliDC binding to FliT. (A) 1H-15N HSQC (Left) and 1H-13C HMQC (Right) spectra of U-2H,15N Ala-13CH3, Met-13CH3, Ile-δ1-13CH3, Leu,Val-13CH3/13CH3, and Thr-13CH3 of free FliT (blue) and of FliT in complex with FliDC (purple). FliT is in excess to ensure that all FliDC, which has a tendency to aggregate, is bound to FliT. (B) NMR analysis shows that FliDC binds to the hydrophobic surface formed by FliT helices a1–a3 (purple surface) and that helix α4 is displaced. The enhanced mobility of helix α4 in the complex was confirmed by analysis of the linewidth of the helix α4 residues (Ala105 is shown here).
Solution Structure of the FliT−FliD Complex.
To understand how FliT recognizes and binds to FliD, we sought to determine the structure of the FliT−FliD complex. The NMR spectra of FliT−FliD show significant line broadening at the interface of the complex, most likely resulting from unfavorable kinetics of complex formation and dissociation. Several different constructs were designed and prepared, and their NMR properties were tested. The best NMR spectra suitable for structure determination by NMR were provided by a construct consisting of the last 40 C-terminal residues of FliD (FliDC) fused to a FliT variant lacking the terminal helix α4 (FliTΔα4) (Fig. S4 A–C). Fusion proteins increase the effective concentration, and thus the population, of the complex and at the same time shift the kinetics of complex formation toward the slow-exchange regime. This strategy has been used extensively to determine structures of protein complexes by NMR (38). NMR analysis and comparison of the spectra of the FliTΔα4−FliDC fusion construct with the spectra of FliT−FliDC show that the molecular interface and the structural properties are essentially identical in the two complexes (Fig. S5). The solution structure of the FliTΔα4−FliDC complex was determined by NMR on the basis of a large number of intermolecular NOEs. The structure and NMR statistics are summarized in Table S1.
Fig. S4.

NMR characterization of the FliT complexes with FliD and FliI. (A and B) 1H-15N HSQC (A) and 1H-13C HMQC (B) spectra of U-2H,15N Ala-13CH3, Met-13CH3, Ile-δ1-13CH3, Leu,Val-13CH3/13CH3, and Thr-13CH3 of the FliTΔα4−FliDC complex. (C) Representative NOESY strips showing intermolecular NOEs between FliTΔα4 and FliDC. (D and E) 1H-15N HSQC (D) and 1H-13C HMQC (E) spectra of U-2H,15N Ala-13CH3, Met-13CH3, Ile-δ1-13CH3, Leu,Val-13CH3/13CH3, and Thr-13CH3 of the FliTΔα4−FliIEN complex. (F) Representative NOESY strips showing intermolecular NOEs between FliTΔα4 and FliIEN.
Fig. S5.

NMR analysis demonstrates that the protein interfaces in the FliT−FliDC and FliTΔα4−FliDC complexes are essentially identical. Cross-peaks from 1H-15N HSQC spectra of FliT−FliDC (red) and FliTΔα4−FliDC (blue) complexes show excellent correspondence.
Table S1.
NMR and structure statistics
| Protein or protein complex | FliT | FliTΔα4–FliDC | FliTΔα4–FliIEN |
| Completeness of resonance assignments* | |||
| Backbone, % | 70 | 95 | 95 |
| Methyl groups, % | 97 | 100 | 100 |
| Aromatic, % | 100 | 100 | 97 |
| Conformationally restricting restraints† | |||
| NOE | 427 | 1243 | 1114 |
| Short, |i − j| ≤ 1 | 148 | 490 | 373 |
| Medium range, 2 < |i − j| < 5 | 154 | 429 | 327 |
| Long range, |i − j| ≥ 5 | 125 | 324 | 414 |
| NOE restraints per residue | 3.8 | 8.8 | 10.6 |
| Interchain NOEs‡ | n/a | 166 | 169 |
| Hydrogen bond, long range, |i − j| ≥ 5 | 138 (0) | 212 (3) | 180 (6) |
| Dihedral angle | 170 | 289 | 214 |
| Total restraints, long range, |i − j| ≥ 5 | 735 (125) | 1,744 (282) | 1,508 (420) |
| Restraints per residue, total/long range | 6.5/1.1 | 12.4/2.3 | 14.4/4.0 |
| Residual distance restraint violations† | |||
| Average restraint violations/structure | |||
| 0.1–0.2 Å | 5.9 | 6.5 | 12.5 |
| 0.2–0.5 Å | 3.8 | 0.2 | 1.5 |
| >0.5 Å | 0.0 | 0.0 | 0.0 |
| Average rms violation/restraint, Å | 0.05 | 0.01 | 0.02 |
| Maximum distance violation, Å | 0.43 | 0.34 | 0.34 |
| Model quality† | |||
| Rmsd from average coordinates, Å | |||
| All backbone atoms, ordered/all | 0.5/1.9 | 0.5/0.5 | 0.5/1.2 |
| All heavy atoms, ordered/all | 0.9/2.1 | 0.7/0.7 | 0.9/1.6 |
| Rmsd bond lengths, Å | 0.05 | 0.019 | 0.019 |
| Rmsd bond angles, degrees | 1.1 | 1.1 | 1.1 |
| MolProbity Ramachandran plot§ | |||
| Most favored regions, 95.9% | 94.3 | 95.9 | 97.5 |
| Additionally allowed regions, % | 4.5 | 3.4 | 2.5 |
| Disallowed regions, % | 0.1 | 0.6 | 0.0 |
| Global quality scores (raw/Z-score)† | |||
| Verify3D | 0.36/1.73 | 0.36/−1.61 | 0.33/−2.09 |
| ProsaII | 0.20/1.18 | 0.80/0.62 | 0.77/0.50 |
| Procheck G-factor, ϕ/ψ§ | 0.34/−1.93 | 0.29/1.46 | 0.42/1.97 |
| Procheck G-factor, all dihedrals§ | 0.64/−0.04 | 0.11/0.65 | 0.21/1.24 |
| MolProbity clashscore | 20.94/−2.07 | 17.74/−1.52 | 16.11/−1.24 |
| Buried surface area in the complex¶ | |||
| Total/hydrophobic/polar, Å2 | n/a | 2,586.3/2,231.1/355.1 | 2,231.4/1,821.2/410.4 |
| Solvation-free energy gain/interface specificity/shape complementarity factor# | |||
| ΔiG, kcal/mol/P value/Sc | n/a | −32.8/0.028/0.718 | −24.5/0.130/0.642 |
Structural statistics were computed for the ensemble of 20 deposited structures.
Computed for each protein/complex (residues): full-length FliT (1A–122A), FliT (1A–95A):FliD (428B–467B), and FliT (1A–95A):FliI (1B–25B). Observable resonances consistent with U-[2H,15N,13C] and selective CH3 and aromatic labeling are included: Cα, Cβ, C′ (backbone), Ala(β), Ile(δ1), Leu(δ1,2), Met(ε), Thr(γ2), Val(γ1,2) (methyls), and U-[1H,13C]-Phe,Tyr, and Trp (aromatic).
Calculated for protein using the PSVS 1.5 program (1). Average distance constraints were calculated using the sum of r−6.
Interchain contacts for FliT:FliD and FliT:FliI complexes.
Ordered residue ranges [S(ϕ) + S(ψ) > 1.8] for FliT (1–122): 2–55, 58–82, 98–115. Secondary structure elements: 2–26 (α1), 30–48 (α2), 58–83 (α3), and 97–111 (α4). Ordered residue ranges [S(ϕ) + S(ψ) > 1.8] for FliT (1A–95A):FliD (428B–467B): 1–50, 53–95 (chain A), 428–466 (chain B). Secondary structure elements (chain A): 2–27 (α1), 30–48 (α2), 58–94 (α3). Secondary structure elements (chain B): 428–466 (α1). Ordered residue ranges [S(ϕ) + S(ψ) > 1.8] for FliT (1A–95A):FliI (1B–25B): 2–93 (chain A), 1–23 (chain B). Secondary structure elements (chain A): 2–27 (α1), 31–48 (α2), 58–93 (α3). Secondary structure elements (chain B): 2–20 (α1).
Calculated with NACCESS 2.1.1.
Computed with ePISA 1.51 and CCP4 6.5.
FliDC binds to FliTΔα4 by forming a long amphipathic helix running from residue Ser428 through Ser467 (Fig. 3 and Fig. S2B), which juxtaposes with the three-helix bundle (α1–α3) of FliT. A continuous string of hydrophobic residues, notably Tyr436, Leu443, Met446, Leu450, Thr453, Tyr456, Leu457, Phe461, and Met464, run along the 37-Å–long FliD helix. Key contacts are between FliT residues Trp11 and Tyr40 and between FliD residues Leu457 and Leu450, respectively. The only polar contacts appear to be hydrogen bonds between FliT Lys79 and FliD Ser454 and between FliT Glu45 and FliD Lys449. The face of the FliDC helix that is exposed to the solvent is decorated almost exclusively with polar amino acids. Mutation of residues at the interface between FliT and FliDC has a strong effect on the stability of the complex (Fig. S6A).
Fig. 3.

Solution structure of the FliTΔα4−FliDC complex. FliTΔα4 is colored green and FliDC is colored orange. The solvent-exposed surface of FliT is represented in semitransparent light gray. Residues are shown in ball-and-stick configuration. The dashed lines denote hydrogen bonds. Two views are shown related by a 90° rotation about the x axis.
Fig. S6.

Thermodynamics of FliT binding to FliD and FliI. (A and B) Effect of mutations at the protein interface and their effect on the affinity for the FliT–FliD (A) and FliT–FliI (B) complexes. The mutated residues are labeled, and the decrease in the measured affinity is indicated. All affinities were measured by ITC. (C–F) ITC profiles of FliD binding to FliT (C) and FliTΔα4 (D) and FliI binding to FliT (E) and FliTΔα4 (F).
FliD binding elicits drastic conformational changes in FliT. Superposition of the apo FliT structure with the structure of the FliT−FliDC complex reveals that the C-terminal FliD helix (residues Asn451–Ser467) occupies the same space as helix α4 in the free FliT (Fig. 4A). Thus, the structure explains why FliD binding entails displacement of FliT helix α4. In addition, the long FliDC helix pairs with FliT helix α3, resulting in the elongation of the latter by three turns of a helix. As a result, FliT−FliDC complex formation buries a total of ∼2,500 Å2, with the majority of the surface (∼2,060 Å2) consisting of nonpolar atoms and only ∼440 Å2 consisting of polar atoms.
Fig. 4.

FliD binding releases FliT helix α4 and activates the complex for FlhA binding. (A) Superposition of free FliT on the FliT−FliDC complex structure. The FliT helix α4, which our NMR data show is released upon FliD binding, is modeled in the structure. (B) 1H-13C heteronuclear multiple quantum coherence (HMQC) spectra of FliT−FliDC (blue) and in the presence of FlhAC (orange). The data show that FliT−FliDC binds to FlhAC and that the interaction is mediated primarily by FliT helix α4. (C) 1H-13C HMQC spectra of FliTΔα4−FliDC (blue) and in the presence of FlhAC (orange). The data show that truncation of FliT helix α4 abrogates binding to FlhAC.
Of note, the structural data show that FliT adopts an autoinhibited conformation in its free state. Indeed, isothermal titration calorimetry (ITC) shows that FliD binds with a 10-fold higher affinity to FliTΔα4 than to FliT (Fig. S6 C and D), confirming that helix α4 acts as an autoinhibitory element in FliT.
Targeting of FliT−FliD to the Export Gate.
FliT was shown by biochemical assays to deliver FliD to the export gate by interacting with the cytoplasmic domain of FlhA (FlhAC) (11, 36). To understand the targeting process, we characterized the interaction of FliT and FliD with FlhAC. NMR shows that free FliT does not bind to FlhAC (Fig. S7), in agreement with surface plasmon resonance (SPR) data (36). In contrast, the FliT−FliDC complex binds strongly to FlhAC (Fig. 4B). NMR analysis points to the FliT helix α4 as the primary binding site mediating the interaction between FliT−FliDC and FlhAC. Indeed, the FliTΔa4−FliDC variant, which lacks the FliT α4 helix, does not interact with FlhAC (Fig. 4C). Taken together, the data demonstrate that free FliT does not interact with FlhA because the α4 helix, which constitutes the primary FlhA-binding site, is inaccessible (Fig. 2). FliD binding displaces the FliT α4 helix, thereby poising the FliT−FliD complex for binding to FlhA.
Fig. S7.

1H-13C HMQC spectra of U-2H,15N Ala-13CH3, Met-13CH3, Ile-δ1-13CH3, Leu,Val-13CH3/13CH3, and Thr-13CH3 of free FliT (blue) and in the presence of FlhAC (magenta) show that free FliT does not interact with FlhAC.
Interaction Between FliT and FliI.
It has been shown previously by pull-down assays that FliT binds to the FliI ATPase (39). To understand how FliT interacts with FliI, we used NMR spectroscopy to characterize their complex. To identify the binding site of FliI on FliT, we added unlabeled FliI (∼50 kDa) in a stepwise manner (Fig. S8A) to isotopically labeled FliT and monitored their interaction by NMR (Fig. S8B). Differential broadening analysis shows that the FliT residues most affected by FliI binding are located in the hydrophobic core formed by helices α1–α3 (Fig. S8C), the FliT site to which FliD binds.
Fig. S8.

Interaction between FliT and FliI. (A) Structure of FliI (PDB ID code 2DPY). The extreme N-terminal region was not crystallographically resolved and is modeled here as an unstructured region. (B) Stepwise titration of unlabeled FliI to U-2H–,15N Ala-13CH3–, Met-13CH3–, Ile-δ1-13CH3–, Leu,Val-13CH3/13CH3–labeled FliT. (C) The FliT residues affected by FliI binding are colored red on the FliT surface. (D) NMR spectra show that the FliIΔ17 variant does not bind to FliT. (E) ATPase activity of FliI, FliIΔ17, and FliI in complex with FliT. The ATPase activity is expressed in nanomoles of hydrolyzed ATP per microgram of protein. (F) 1H-15N HSQC spectra of FliT−FliIEN (orange) and FliTΔα4−FliIEN (blue) complexes show very similar chemical shifts.
To determine the FliT-binding site in FliI, we prepared several FliI constructs and tested their binding to FliT. A FliI construct lacking the extreme N-terminal region (EN, residues 1–17) (FliIΔEN) showed no evidence of binding to FliT (Fig. S8D). Therefore the NMR data point to the FliI EN region as the binding site for the FliT chaperone, in agreement with previously reported pull-down assays (35). The FliI EN region also mediates self-oligomerization and stimulates ATPase activity (18), thus explaining why FliT binding to FliI inhibits its ATPase activity (Fig. S8E). FliT binds to FliI with moderate affinity (Kd ∼5 μM) as measured by ITC. As with FliD, FliI binding displaces FliT helix α4, and FliI binds with a 10-fold higher affinity to FliTΔα4 (Fig. S6 E and F).
Solution Structure of the FliT−FliI Complex.
To determine the structure of the FliT−FliI complex, we followed an approach similar to the one described above for the FliT−FliD complex. The FliTΔα4 variant fused to the FliI EN (FliTΔα4−FliIEN) yielded NMR spectra of high quality (Fig. S4 D–F). Superposition of the FliTΔα4 −FliIEN and FliT−FliIEN spectra showed that the chemical shifts are very similar (Fig. S8F), in agreement with the observation that helix α4 does not interact with FliIEN. The solution structure of the FliTΔα4−FliIEN complex was determined by NMR on the basis of a large number of intermolecular NOEs. The structure and NMR statistics are summarized in Table S1.
The FliTΔα4−FliIEN complex (Fig. 5 and Fig. S2C) forms a four-helix bundle, with FliIEN adopting an amphipathic helix extending from residues Met1 to Val25 that juxtaposes with the three-helix bundle (α1–α3) of the FliT chaperone. Complex formation buries a total of ∼2,230 Å2. The majority of the surface (∼1,820 Å2) consists of nonpolar atoms, and ∼410 Å2 consists of polar atoms. Two notable salt bridges are formed between Lys79 of FliT and Asp13 and Glu16 of FliI and between Arg4 of FliI and Glu37 of FliT. Several bulky nonpolar residues (Met1, Leu5, Trp8, Leu12, Phe15, and Met19) run along the interacting face of the FliI EN helix and insert into pockets lined by nonpolar residues in FliT. Residues Trp8 and Phe15 of FliI make key hydrophobic contacts with Tyr40 and Trp11, respectively, of FliT. Mutation of residues at the interface between FliT and FliI has a strong effect on the stability of the complex (Fig. S6B).
Fig. 5.

Solution structure of the FliTΔα4−FliIEN. FliTΔα4 is colored green, and FliIEN is colored pink. The solvent-exposed surface of FliT is represented in semitransparent light gray. Residues are shown in ball-and-stick configuration. The dashed lines denote hydrogen bonds. Two views are shown related by a 90° rotation about the x axis.
FliT Uses the Same Binding Site to Interact with Its Substrates.
Comparison of the structures of the FliT−FliI and FliT−FliD complexes demonstrates that FliD and FliI interact with essentially the same surface of FliT (Fig. S9A). To determine the site that FliT uses to interact with FliJ, we monitored the binding of unlabeled FliJ to isotopically labeled FliT (Fig. S9B). Analysis of the NMR data showed that FliJ binds to the same surface of FliT to which FliD and FliI bind. Therefore, FliT uses the same binding site to recognize and interact with its three partner proteins: FliD, FliJ, and FliI. Binding of any of the partners displaces the FliT α4 helix, and thus FliT may target these three substrates to the export gate by its direct interaction with FlhA (Fig. 4B).
Fig. S9.

(A) Superposition of the FliT−FliIEN and FliT−FliDC structures demonstrates that FliI and FliD bind to the same surface on FliT. (B) 1H-15N HSQC (Left) and 1H-13C HMQC (Right) spectra of U-2H,15N Ala-13CH3, Met-13CH3, Ile-δ1-13CH3, Leu,Val-13CH3/13CH3, Thr-13CH3 of free FliT (blue) and in complex with FliJ (purple). NMR analysis shows that FliJ, FliD, and FliI bind to the same surface on FliT.
Discussion
Here we present the solution structures of the FliT chaperone and its complexes with the cognate substrate FliD and the FliI ATPase. FliT is a key protein in the assembly and operation of the flagellum because it binds to several flagellar proteins in the cytoplasm (FliD, FliJ, and FliI) and ushers them to the export gate by interacting with the FlhA membrane protein. We show that FliT adopts an autoinhibited structure in which both the binding site for the partner proteins (the hydrophobic surface formed by helices α1–α3) and the binding site for FlhA (helix α4) are occluded. The autoinhibited structure of FliT may serve two goals: First, it occludes from the solvent, and thus protects, the hydrophobic substate-binding surface in the absence of substrates. Indeed, our data show that FliT variants in which the docking of helix α4 to the hydrophobic core is compromised are unstable and prone to aggregation. Second, the autoinhibited structure serves to mask the binding site for FlhA, which is activated for binding only when the substrate protein is to be targeted to the export gate.
Interestingly, the autoinhibition/activation and targeting mechanism presented here for FliT appears to be shared among the other flagellar proteins (Fig. 6). FlgN, the chaperone dedicated to the binding and targeting of the FlgK and FlgL hook proteins (32), likely adopts an autoinhibited structure. The overall structure of Pseudomonas aeruginosa FlgN [Protein Data Bank (PDB) ID code 2FUP] (Fig. 6B) resembles the structure of FliT (Fig. 6A), with the FlgN C-terminal helix packing against a hydrophobic surface formed by the first three helices that likely form the substrate-binding site. Although the structure of the complex between FlgN and its substrates is not known, biochemical data showed that truncation of the terminal C helix in FlgN increases the binding of substrates (30). The C-terminally truncated FlgN does not interact with FlhA (31), suggesting that, as in FliT, the C-terminal helix in FlgN serves as the FlhA-binding site. In contrast to FliT, substrate-free FlgN does interact with FlhA, albeit with lower affinity than in the complex with its substrate FlgK, probably because the FlhA-binding site in FlgN is only partially occluded (Fig. 6B). Similarly, FliS, the chaperone for FliC, adopts an autoinhibited structure (Fig. 6C), with the inhibitory structural element, located in the N terminus, released upon FliC binding (40). The N terminus of FliS serves as the FlhA-binding site, and in all three chaperones a highly conserved Tyr residue appears to be essential for efficient FlhA binding (Fig. 6) (36). Thus, the three flagellar chaperones may use similar strategies for substrate binding and activation of the complexes for binding to FlhA and thus for targeting to the export gate.
Fig. 6.

Structures of the three flagellar chaperones: FliT (this work) (A), FlgN (PDB ID code 2FUP) (B), and FliS (40) (C). The highly conserved Tyr residues that are crucial for the interaction of the substrate-bound chaperones with FlhA are labeled. The FlgN structure was resolved only up to residue Arg106 (Salmonella numbering), and the remaining C-terminal region was modeled in the structure.
We propose, based on the current results, a simplified model of how FliT may assist with the assembly and operation of the flagellum (Fig. 7). FliT binds to FliJ and ushers it to the export gate by means of the interaction between the FliT α4 helix and FlhA (Fig. 7i). Then, FliT transports FliI at the membrane (Fig. 7ii), where FliH takes over FliI from FliT (41), and FliI, with the assistance of FliJ, forms a hexamer (Fig. 7iii). When it is time for FliD to be exported, FliT binds and escorts FliD to the membrane for its export and the assembly of the filament-capping structure (Fig. 7iv). FlgN also binds to FliJ and FliI, and thus FliT and/or FlgN may escort these proteins to the export gate.
Fig. 7.

A cartoon showing how FliT may assist in the assembly of the cytoplasmic components by binding and targeting FliJ (i), FliI (ii), and FliD (iii) to the export gate (iv). See text for details.
Materials and Methods
The materials and methods used in this work are described in SI Materials and Methods.
SI Materials and Methods
Expression and Preparation of Proteins.
All constructs of Salmonella enterica FliT, FliI, FliD, FliJ, and FlhA were cloned into pET16b vector (Novagen). FliT and FliTΔα4 (FliT residues 1–95) constructs were prepared as fusions to His10-GB1 with a tobacco etch virus (TEV) protease cleavage site in between. The FliD construct FliD341–467 was prepared and fused to His10-GB1 with a TEV protease cleavage site. The FliI constructs FliI and FliIΔ17 were fused to His10-MBP with a TEV protease cleavage site. The cytoplasmic domain of FlhA328-692, FlhAC, was used. FlhAC and FliJ were fused to His10-MBP with a TEV protease cleavage site. The fusion constructs were prepared by fusing FliIEN (FliI1–25) and FliD428–467 (FliDC) to the C terminus of FliT1–95. Fusion constructs with varied linker lengths were prepared to ensure that the fusion process does not bias the conformation of the complex in any way. A 20-residue-long linker (VLFQGPSAGLVPRGSGGIEG) was selected from the pCold vector (Takara Bio). Both fusion constructs were fused to His10-GB1 including a TEV protease cleavage site. All mutants were constructed by site-directed mutagenesis using PfuTurbo High-Fidelity DNA polymerase (Agilent). All constructs were transformed into BL21(DE3) cells. For the unlabeled samples, cells were grown in Luria–Bertani (LB) medium at 37 °C in the presence of ampicillin (100 μg/mL). Protein expression was induced by the addition of 50–500 μM isopropyl-b-d-1-thiogalactopyranoside (IPTG) at OD600 ∼0.5, followed by 12–16 h of incubation at 18 °C. Cells were harvested at OD600 ∼1.5 and were resuspended in lysis buffer containing 50 mM Tris⋅HCl (pH 8.0), 500 mM NaCl, 10 mM imidazole, 1 mM β-mercaptoethanol (βME), and 5% glycerol. Cells were disrupted by a high-pressure homogenizer or sonicator and centrifuged at 50,000 × g. Proteins were purified using Ni Sepharose 6 Fast Flow resin (GE Healthcare), followed by tag removal by TEV protease at 4 °C (incubation for 12 h) and gel filtration using Superdex 75 16/60 or 200 16/60 columns (GE Healthcare). Protein concentration was determined spectrophotometrically at 280 nm using the corresponding extinction coefficient.
MALS Experiments.
MALS was measured using DAWN HELEOS-II (Wyatt Technology Corporation) downstream of a Shimadzu liquid chromatography system connected to a Superdex 200 10/300 GL (GE Healthcare) gel-filtration column. The following buffers were used: (i) 20 mM KPi (pH 7.0), 100 mM KCl, 5 mM βME, 0.5 mM EDTA, and 0.05% NaN3, and (ii) 20 mM Tris (pH 7.0), 100 mM NaCl, 5 mM βME, 0.5 mM EDTA, and 0.05% NaN3. Two hundred microliters of the sample with a concentration of 25 μM to 1 mM was injected for each run. The data were analyzed with ASTRA version 6.0.5 (Wyatt Technology Corporation).
ITC Experiments.
Calorimetric titrations were carried out on an iTC200 microcalorimeter (GE Healthcare) at 25 °C. The following ITC buffers were used: (i) 20 mM KPi (pH 8.0), 100 mM KCl, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 0.5 mM EDTA, and 0.05% NaN3 and (ii) 20 mM Hepes (pH 8.0), 100 mM NaCl, 1 mM TCEP, 0.5 mM EDTA, and 0.05% NaN3. All protein samples were extensively dialyzed against ITC buffer. All solutions were filtered using membrane filters (pore size, 0.45 μm) and thoroughly degassed for 20 min before the titrations. The sample cell (200 μL) was filled with ∼50–100 μM protein (FliI, FliT, and GB1-FliT1–95), and the ∼20- to 50-μL injection syringe was filled with an ∼200–550 μM solution of protein (GB1-FliT1–95, FliD341–467). The titrations were carried out with a preliminary 0.2-μL injection, followed by 10–16 injections of 2.5–3.8 μL each at time intervals of 3 min. The solution was stirred at 1,000 rpm. Data for the preliminary injection, which are affected by diffusion of the solution from and into the injection syringe during the initial equilibration period, were discarded. Binding isotherms were generated by plotting heats of reaction normalized by the modes of injectant versus the ratio of total injectant to total protein per injection. The data were fitted with Origin 7.0 (OriginLab Corporation).
ATPase Assays.
ATPase activity of FliI constructs was measured using the ADP-Glo Kinase Assay kit (V9101; Promega). The luminescence was measured in a Tecan Infinite M200 pro plate reader using a 96-well plate and was correlated using an ADP-to-ATP conversion curve. The reaction buffer for all ATPase reactions consisted of 20 mM Hepes, 100 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 5 mM MgCl2, 0.1 mg/mL BSA, pH 8.0. Twenty-five microliters of reaction volume were used with 1 mM [ATP] at 25 °C. ATPase activity was carried out using 1 μM FliI (or FliIΔ17) and a mixture of 1 μM FliI and 50 μM FliT. The reaction was stopped at 180 min by adding 25 μL of ADP-Glo reagent (Promega). After the reaction mixtures were incubated at room temperature for 40 min, kinase detection reagent was added, and luminescence was recorded after 60 min.
Protein Isotope Labeling for NMR Studies.
The isotope-labeled samples for NMR studies were prepared by growing the cells in minimal (M9) medium. Cells were typically harvested at OD600 ∼1.0–1.2. U-[13C,15N]– or U-[2H,13C,15N] –labeled samples were prepared by supplementing the growth medium with 1 g/L of 15NH4Cl and 2 g/L of [13C6]- or [2H7,13C6]-glucose in H2O or 99.9%-2H2O (Cambridge Isotope Laboratories and Isotec). Methyl-protonated samples were prepared as described previously (42, 43) using 50 mg/L alpha-ketobutyric acid, 85 mg/L alpha-ketoisovaleric acid, 50 mg/L of 13CH3-Met, 50 mg/L 2H2,13CH3-Ala, and 50 mg/L U-2H, Thr-γ2[13CH3]. For selective labeling of Phe and Tyr residues, 50 mg/L of U-[13C,15N]-Phe and U-[13C,15N]-Tyr were added to the cell culture.
NMR Spectroscopy.
All NMR data were collected on Bruker AVANCE III 600-, 700-, and 900-MHz devices equipped with 5-mm cryogenic probes. All recorded spectra were processed with NMRPipe (44) and analyzed using NMRView (45). NMR samples were prepared in 20 mM KPi, 100 mM KCl, 0.05% NaN3, 0.5 mM EDTA, 5 mM βME, pH 7.0. Spectra were recorded at 25 or 32 °C. Transverse relaxation optimized spectroscopy (TROSY)-based triple-resonance experiments were recorded for backbone resonance assignment. 13Cα, 13Cβ, 13C′, and backbone 1H and 15N chemical shifts were used to compute dihedral angle restraints using TALOSN (46). Assignments for selectively [1H,13C]-labeled methyl-bearing and aromatic (Phe, Trp, and Tyr) residues were obtained using a combination of 3D 13C,15N-NOESY-HMQC and simultaneous 13C,15N-HMQC-NOESY-HMQC with 300-ms mixing time, following a previously published strategy (47). Assignment was verified and augmented using through-bond 3D HCCH-COSY and total correlation spectroscopy (TOCSY) experiments acquired on the U-[13C,15N]–labeled sample.
Structure Calculation.
Structures were obtained from experimentally derived NOEs, dihedral angles, and hydrogen bond restraints. NOE restraints were obtained using automated NOESY cross-peak assignment and initial structure calculations using CYANA 3.96 (48). NOE peak assignments were further refined manually and iteratively by additional CYANA runs. The lowest target function CYANA-derived structure ensemble was subjected to restrained molecular dynamics energy refinement in a water bath using CNS (49). Structure statistics are presented in Table S1.
Acknowledgments
This work was supported by NIH Grant AI094623 (to C.G.K.) and Fonds Wetenschappelijk Onderzoek Grant T3RecS G002516N (to A.E.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates reported in this paper have been deposited in the Protein Data Bank, www.wwpdb.org (PDB ID codes 5KS6, 5KRW, and 5KP0). The NMR chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank, www.bmrb.wisc.edu (accession nos. 30136, 30134, and 30127).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1607845113/-/DCSupplemental.
References
- 1.Chevance FF, Hughes KT. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol. 2008;6(6):455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minamino T. Protein export through the bacterial flagellar type III export pathway. Biochim Biophys Acta. 2014;1843(8):1642–1648. doi: 10.1016/j.bbamcr.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Altegoer F, Bange G. Undiscovered regions on the molecular landscape of flagellar assembly. Curr Opin Microbiol. 2015;28:98–105. doi: 10.1016/j.mib.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Diepold A, Armitage JP. 2015. Type III secretion systems: The bacterial flagellum and the injectisome. Philos Trans R Soc Lond B Biol Sci 370:20150020.
- 5.Haiko J, Westerlund-Wikström B. The role of the bacterial flagellum in adhesion and virulence. Biology (Basel) 2013;2(4):1242–1267. doi: 10.3390/biology2041242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaban B, Hughes HV, Beeby M. The flagellum in bacterial pathogens: For motility and a whole lot more. Semin Cell Dev Biol. 2015;46:91–103. doi: 10.1016/j.semcdb.2015.10.032. [DOI] [PubMed] [Google Scholar]
- 7.Blocker A, Komoriya K, Aizawa S. Type III secretion systems and bacterial flagella: Insights into their function from structural similarities. Proc Natl Acad Sci USA. 2003;100(6):3027–3030. doi: 10.1073/pnas.0535335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minamino T, Namba K. Distinct roles of the FliI ATPase and proton motive force in bacterial flagellar protein export. Nature. 2008;451(7177):485–488. doi: 10.1038/nature06449. [DOI] [PubMed] [Google Scholar]
- 9.Paul K, Erhardt M, Hirano T, Blair DF, Hughes KT. Energy source of flagellar type III secretion. Nature. 2008;451(7177):489–492. doi: 10.1038/nature06497. [DOI] [PubMed] [Google Scholar]
- 10.Minamino T, Macnab RM. Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol. 1999;181(5):1388–1394. doi: 10.1128/jb.181.5.1388-1394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bange G, et al. FlhA provides the adaptor for coordinated delivery of late flagella building blocks to the type III secretion system. Proc Natl Acad Sci USA. 2010;107(25):11295–11300. doi: 10.1073/pnas.1001383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saijo-Hamano Y, et al. Structure of the cytoplasmic domain of FlhA and implication for flagellar type III protein export. Mol Microbiol. 2010;76(1):260–268. doi: 10.1111/j.1365-2958.2010.07097.x. [DOI] [PubMed] [Google Scholar]
- 13.Imada K, Minamino T, Tahara A, Namba K. Structural similarity between the flagellar type III ATPase FliI and F1-ATPase subunits. Proc Natl Acad Sci USA. 2007;104(2):485–490. doi: 10.1073/pnas.0608090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claret L, Calder SR, Higgins M, Hughes C. Oligomerization and activation of the FliI ATPase central to bacterial flagellum assembly. Mol Microbiol. 2003;48(5):1349–1355. doi: 10.1046/j.1365-2958.2003.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazetani K, Minamino T, Miyata T, Kato T, Namba K. ATP-induced FliI hexamerization facilitates bacterial flagellar protein export. Biochem Biophys Res Commun. 2009;388(2):323–327. doi: 10.1016/j.bbrc.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, et al. Structural diversity of bacterial flagellar motors. EMBO J. 2011;30(14):2972–2981. doi: 10.1038/emboj.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stafford GP, et al. Sorting of early and late flagellar subunits after docking at the membrane ATPase of the type III export pathway. J Mol Biol. 2007;374(4):877–882. doi: 10.1016/j.jmb.2007.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minamino T, et al. Oligomerization of the bacterial flagellar ATPase FliI is controlled by its extreme N-terminal region. J Mol Biol. 2006;360(2):510–519. doi: 10.1016/j.jmb.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Lane MC, O’Toole PW, Moore SA. Molecular basis of the interaction between the flagellar export proteins FliI and FliH from Helicobacter pylori. J Biol Chem. 2006;281(1):508–517. doi: 10.1074/jbc.M507238200. [DOI] [PubMed] [Google Scholar]
- 20.McMurry JL, Murphy JW, González-Pedrajo B. The FliN-FliH interaction mediates localization of flagellar export ATPase FliI to the C ring complex. Biochemistry. 2006;45(39):11790–11798. doi: 10.1021/bi0605890. [DOI] [PubMed] [Google Scholar]
- 21.Pallen MJ, Bailey CM, Beatson SA. Evolutionary links between FliH/YscL-like proteins from bacterial type III secretion systems and second-stalk components of the FoF1 and vacuolar ATPases. Protein Sci. 2006;15(4):935–941. doi: 10.1110/ps.051958806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hara N, Morimoto YV, Kawamoto A, Namba K, Minamino T. Interaction of the extreme N-terminal region of FliH with FlhA is required for efficient bacterial flagellar protein export. J Bacteriol. 2012;194(19):5353–5360. doi: 10.1128/JB.01028-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ibuki T, et al. Common architecture of the flagellar type III protein export apparatus and F- and V-type ATPases. Nat Struct Mol Biol. 2011;18(3):277–282. doi: 10.1038/nsmb.1977. [DOI] [PubMed] [Google Scholar]
- 24.Evans LD, Stafford GP, Ahmed S, Fraser GM, Hughes C. An escort mechanism for cycling of export chaperones during flagellum assembly. Proc Natl Acad Sci USA. 2006;103(46):17474–17479. doi: 10.1073/pnas.0605197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMurry JL, Van Arnam JS, Kihara M, Macnab RM. Analysis of the cytoplasmic domains of Salmonella FlhA and interactions with components of the flagellar export machinery. J Bacteriol. 2004;186(22):7586–7592. doi: 10.1128/JB.186.22.7586-7592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minamino T, et al. Role of the C-terminal cytoplasmic domain of FlhA in bacterial flagellar type III protein export. J Bacteriol. 2010;192(7):1929–1936. doi: 10.1128/JB.01328-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auvray F, Thomas J, Fraser GM, Hughes C. Flagellin polymerisation control by a cytosolic export chaperone. J Mol Biol. 2001;308(2):221–229. doi: 10.1006/jmbi.2001.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett JC, Thomas J, Fraser GM, Hughes C. Substrate complexes and domain organization of the Salmonella flagellar export chaperones FlgN and FliT. Mol Microbiol. 2001;39(3):781–791. doi: 10.1046/j.1365-2958.2001.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aldridge P, Karlinsey J, Hughes KT. The type III secretion chaperone FlgN regulates flagellar assembly via a negative feedback loop containing its chaperone substrates FlgK and FlgL. Mol Microbiol. 2003;49(5):1333–1345. doi: 10.1046/j.1365-2958.2003.03637.x. [DOI] [PubMed] [Google Scholar]
- 30.Thomas J, Stafford GP, Hughes C. Docking of cytosolic chaperone-substrate complexes at the membrane ATPase during flagellar type III protein export. Proc Natl Acad Sci USA. 2004;101(11):3945–3950. doi: 10.1073/pnas.0307223101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minamino T, et al. Interaction of a bacterial flagellar chaperone FlgN with FlhA is required for efficient export of its cognate substrates. Mol Microbiol. 2012;83(4):775–788. doi: 10.1111/j.1365-2958.2011.07964.x. [DOI] [PubMed] [Google Scholar]
- 32.Fraser GM, Bennett JC, Hughes C. Substrate-specific binding of hook-associated proteins by FlgN and FliT, putative chaperones for flagellum assembly. Mol Microbiol. 1999;32(3):569–580. doi: 10.1046/j.1365-2958.1999.01372.x. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto S, Kutsukake K. FliT acts as an anti-FlhD2C2 factor in the transcriptional control of the flagellar regulon in Salmonella enterica serovar typhimurium. J Bacteriol. 2006;188(18):6703–6708. doi: 10.1128/JB.00799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aldridge C, et al. The interaction dynamics of a negative feedback loop regulates flagellar number in Salmonella enterica serovar Typhimurium. Mol Microbiol. 2010;78(6):1416–1430. doi: 10.1111/j.1365-2958.2010.07415.x. [DOI] [PubMed] [Google Scholar]
- 35.Imada K, Minamino T, Kinoshita M, Furukawa Y, Namba K. Structural insight into the regulatory mechanisms of interactions of the flagellar type III chaperone FliT with its binding partners. Proc Natl Acad Sci USA. 2010;107(19):8812–8817. doi: 10.1073/pnas.1001866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinoshita M, Hara N, Imada K, Namba K, Minamino T. Interactions of bacterial flagellar chaperone-substrate complexes with FlhA contribute to co-ordinating assembly of the flagellar filament. Mol Microbiol. 2013;90(6):1249–1261. doi: 10.1111/mmi.12430. [DOI] [PubMed] [Google Scholar]
- 37.Ikeda T, Oosawa K, Hotani H. Self-assembly of the filament capping protein, FliD, of bacterial flagella into an annular structure. J Mol Biol. 1996;259(4):679–686. doi: 10.1006/jmbi.1996.0349. [DOI] [PubMed] [Google Scholar]
- 38.Krois AS, Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc Natl Acad Sci USA. 2016;113(13):E1853–E1862. doi: 10.1073/pnas.1602487113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minamino T, Kinoshita M, Imada K, Namba K. Interaction between FliI ATPase and a flagellar chaperone FliT during bacterial flagellar protein export. Mol Microbiol. 2012;83(1):168–178. doi: 10.1111/j.1365-2958.2011.07924.x. [DOI] [PubMed] [Google Scholar]
- 40.Evdokimov AG, et al. Similar modes of polypeptide recognition by export chaperones in flagellar biosynthesis and type III secretion. Nat Struct Biol. 2003;10(10):789–793. doi: 10.1038/nsb982. [DOI] [PubMed] [Google Scholar]
- 41.Sajó R, et al. Soluble components of the flagellar export apparatus, FliI, FliJ, and FliH, do not deliver flagellin, the major filament protein, from the cytosol to the export gate. Biochim Biophys Acta. 2014;1843(11):2414–2423. doi: 10.1016/j.bbamcr.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Saio T, Guan X, Rossi P, Economou A, Kalodimos CG. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science. 2014;344(6184):1250494. doi: 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monneau YR, et al. Exploiting E. coli auxotrophs for leucine, valine, and threonine specific methyl labeling of large proteins for NMR applications. J Biomol NMR. 2016;65(2):99–108. doi: 10.1007/s10858-016-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 45.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 46.Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR. 2013;56(3):227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi P, et al. A hybrid NMR/SAXS-based approach for discriminating oligomeric protein interfaces using Rosetta. Proteins. 2015;83(2):309–317. doi: 10.1002/prot.24719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273(1):283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 49.Brunger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc. 2007;2(11):2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]