

Significance
Darwinian life requires the ability to replicate genotypes and express phenotypes. Although all extant life relies on protein enzymes to accomplish these tasks, life in the ancestral RNA world would have used only RNA enzymes. Here, we report the in vitro evolution of an improved RNA polymerase ribozyme that is able to synthesize structured functional RNAs, including aptamers and ribozymes, and replicate short RNA sequences in a protein-free form of the PCR. Thus, the replication of RNA and the expression of functional RNA can be accomplished with RNA alone. Combining and improving these activities may enable the self-sustained evolution of RNA and offers a potential route to a synthetic form of RNA life.
Keywords: self-replication, ribozyme, RNA enzyme, PCR, in vitro evolution
Abstract
In all extant life, genetic information is stored in nucleic acids that are replicated by polymerase proteins. In the hypothesized RNA world, before the evolution of genetically encoded proteins, ancestral organisms contained RNA genes that were replicated by an RNA polymerase ribozyme. In an effort toward reconstructing RNA-based life in the laboratory, in vitro evolution was used to improve dramatically the activity and generality of an RNA polymerase ribozyme by selecting variants that can synthesize functional RNA molecules from an RNA template. The improved polymerase ribozyme is able to synthesize a variety of complex structured RNAs, including aptamers, ribozymes, and, in low yield, even tRNA. Furthermore, the polymerase can replicate nucleic acids, amplifying short RNA templates by more than 10,000-fold in an RNA-catalyzed form of the PCR. Thus, the two prerequisites of Darwinian life—the replication of genetic information and its conversion into functional molecules—can now be accomplished with RNA in the complete absence of proteins.
The core informational processes of Darwinian evolution are the replication of genes and their expression as functional molecules, which in modern biology require the action of protein enzymes. Features common to all extant life suggest the existence of an RNA world where the replication and expression of genetic information depended on RNA enzymes rather than genetically encoded proteins (1, 2). Accordingly, substantial efforts have been directed toward reconstructing RNA life via the protein-free replication of RNA (3, 4). RNA-joining ribozymes have been modified to assemble new copies of themselves from smaller RNA substrates (5), in one case achieving self-replication with exponential growth (6). However, these self-replicating ribozymes require complex oligonucleotide substrates, which limits their ability to synthesize functional RNA molecules other than additional copies of themselves, which in turn limits their ability to express and evolve functions beyond self-replication.
A more general solution is offered by the template-directed polymerization of RNA monomers, the mechanism used in modern biology for the synthesis of DNA and RNA. RNA-templated RNA polymerization has been demonstrated both nonenzymatically (7, 8) and with a variety of natural and synthetic ribozymes (9–11). Although none of these systems have been able to replicate RNA exponentially, extensive copying of RNA templates has been achieved with evolved variants of the class I RNA polymerase ribozyme. This ribozyme can synthesize tandem RNA repeats over 100 nt long and even complete a substantial portion of a small endonuclease ribozyme (12–15). However, the polymerase strongly prefers cytidine-rich templates that lack any secondary structure and has much more limited activity in other contexts (15). These limitations preclude the synthesis of most functional RNAs, which often are highly structured, and the replication of RNA, which requires the reciprocal synthesis of both an RNA template and its complement.
Template restrictions might be eased by using in vitro evolution to select directly for the synthesis of functional RNAs from “difficult” templates. In the present study, an engineered form of the class I polymerase was selected for its ability to synthesize functional RNA aptamers from complementary templates. In contrast to previous in vitro evolution efforts directed at RNA polymerization, this approach does not select directly for chemical bond formation, but rather for the efficient and accurate transfer of functional information from template to cRNA. A polymerase variant isolated after 24 rounds of evolution exhibited dramatically improved activity and template generality, enabling both the synthesis of structurally complex RNAs and the replication of short RNA sequences in a protein-free form of the PCR. This work demonstrates that the replication and expression of genetic material can be accomplished with RNA alone. The prospect for further improvement of these activities suggests a practical route toward the development of RNA-based life in the laboratory.
Results
Evolution Based on Aptamer Completion.
An engineered form of the class I polymerase ribozyme [wild type (WT); Fig. S1] was constructed by combining known advantageous features, including a large deletion within the 3′-terminal domain; a 5′-terminal sequence tag that binds to a complementary region of the template; and a set of activity-enhancing mutations (14, 16). Random mutations then were introduced throughout the molecule at a frequency of 10% per nucleotide position to generate a population of 1014 distinct variants to initiate the in vitro evolution process. During each round of evolution (Fig. 1A), an RNA primer was covalently attached to the 5′ end of the polymerase and nucleotide extension reactions were performed using a separate RNA template and the four nucleoside triphosphates (NTPs).
Fig. S1.

Sequence and secondary structure of the WT polymerase ribozyme. Changes relative to the original class I polymerase (12) are highlighted, with the 5′-terminal tag that binds to a complementary region of the template (14) shown in purple, a set of activity-enhancing mutations derived from the tC19Z ribozyme (14) shown in orange, and the site of a large deletion within the 3′-terminal domain (13) shown in red. Nucleotides that were added to enable primer binding for mutagenic PCR are shown in green.
Fig. 1.

In vitro evolution of RNA polymerase ribozymes. (A) Selective amplification of ribozymes that extend a tethered RNA primer (magenta) on a separate RNA template (brown) to complete a 3′-truncated aptamer. (1) Attachment of the primer to the ribozyme via a photocleavable linker; (2) hybridization of the primer to the template; (3) extension of the primer by polymerization of NTPs (cyan); (4) capture of full-length materials by binding the aptamer portion to its immobilized ligand (green); (5) photocleavage to release the ribozyme portion; (6) reverse transcription and PCR amplification of the released ribozyme; and (7) transcription to generate progeny ribozymes. (B) Sequence and secondary structure of RNA aptamers that bind either cyanocobalamin (Left) or GTP (Right). (C) Sequence and secondary structure of the 24-3 polymerase. Red circles indicate mutations relative to WT, with color intensity indicating mutations that were universal (dark), common (medium), or sporadic (light) among the clones that were sequenced.
Two forms of selection pressure were applied to the population to obtain more active polymerases. First, the polymerase was challenged to extend the attached primer to complete a 3′-truncated RNA aptamer, enabling selection based on binding of the completed aptamer to its cognate ligand. Both the cyanocobalamin (17) and GTP (18) aptamers were used to provide varied sequence contexts that are largely intolerant of mutation (Fig. 1B), thus imposing selection pressure for both sequence generality and accuracy. Second, a gel-shift selection procedure was used to ensure the polymerase extension products had reached full length, as reflected by their corresponding mobility in a denaturing polyacrylamide gel.
Twenty-four rounds of in vitro evolution were carried out (Table S1), progressively increasing the selection stringency by increasing the length of RNA to be synthesized and decreasing the time allowed for polymerization. By the 24th round, the population could readily complete a 30-nt truncation of the GTP aptamer and copy purine-rich templates. Analysis of cloned individuals from the evolved population revealed that 11 mutations had swept to fixation and 10 additional mutations were present in many of the clones. A screen of 10 specific individuals showed that all had dramatically improved activity compared with WT, the most active of which (designated 24-3) contained 17 mutations (Fig. 1C).
Table S1.
Parameters for each round of in vitro evolution
| Round | Method | Nt added | Time (h) | Primer | Template | Splint | Mut |
| 1 | gel | 20 | 24 | SP1 | T-R0 | Spl1 | |
| 2 | gel | 20 | 24 | SP1 | T-R0 | Spl1 | |
| 3 | gel | 20 | 24 | SP1 | T-R0 | Spl1 | |
| 4 | gel | 20 | 24 | SP1 | T-R0 | Spl1 | + |
| 5 | apt | 12 | 4 | SP2 | T-B12 | Spl2 | |
| 6 | apt | 12 | 4 | SP2 | T-B12 | Spl2 | |
| 7 | apt | 12 | 4 | SP2 | T-B12 | Spl2 | |
| 8 | gel | 18 | 24 | SP3 | T-B12 | Spl2 | |
| 9 | gel | 18 | 24 | SP3 | T-B12 | Spl2 | |
| 10 | gel | 18 | 24 | SP3 | T-B12 | Spl2 | |
| 11 | gel | 18 | 24 | SP3 | T-B12 | Spl2 | |
| 12 | gel | 18 | 24 | SP3 | T-B12 | Spl2 | + |
| 13 | gel | 30 | 2 | SP4 | T-GTP | Spl3 | |
| 14 | gel | 30 | 2 | SP4 | T-GTP | Spl3 | |
| 15 | gel | 30 | 2 | SP4 | T-GTP | Spl3 | |
| 16 | gel | 30 | 2 | SP4 | T-GTP | Spl3 | |
| 17 | gel+apt | 30 | 2 | SP4 | T-GTP | Spl3 | + |
| 18 | gel | 32 | 6 | SP1 | T-R12 | Spl1 | |
| 19 | gel | 32 | 1 | SP1 | T-R12 | Spl1 | |
| 20 | gel | 32 | 0.25 | SP1 | T-R12 | Spl1 | |
| 21 | gel | 40 | 0.25 | SP1 | T-R20 | Spl1 | |
| 22 | gel+apt | 30 | 0.25 | SP4 | T-GTP | Spl3 | + |
| 23 | gel+apt | 30 | 0.25 | SP4 | T-GTP | Spl3 | |
| 24 | gel+apt | 30 | 0.25 | SP4 | T-GTP | Spl3 |
The selection method involved gel-shift (gel) and/or aptamer capture (apt), following polymerase extension of 12–40 nt for 0.25–24 h. See Table S4 for sequences of primers, templates, and splints. Mutagenic PCR was performed after certain rounds, as indicated by +.
Properties of the 24-3 Polymerase.
The rate of polymerase-catalyzed nucleotide addition to a template-bound primer was measured using a cytidine-rich template of 11 nt, which has been used extensively to demonstrate the prowess of the class I polymerase (12–15). The average rate of primer extension by 24-3 is 1.2 nt/min, which is ∼100-fold faster than that of the WT or previously reported tC19Z polymerase (14) (Fig. 2A). The fidelities of the WT and 24-3 polymerases on this template, at comparable yields of product, are 96.6% and 92.0%, respectively (Table S2). The higher error rate of 24-3 is due primarily to an increased tendency for G•U wobble pairing. Excluding these mutations, WT and 24-3 have comparable fidelities. It appears that selection for aptamer completion was tolerant of occasional wobble mutations relative to the demands for increased rate and improved sequence generality.
Fig. 2.

Improved activity of the 24-3 polymerase. (A) Primer extension on an 11-nt C-rich template catalyzed by the 24-3 (black), WT (gray), or tC19Z (white) polymerase. Data represent the average of three replicates, with SDs in the range of 0.03–0.09 nt/min and not visible in the figure. Initial rates based on a linear fit of the data (at right, with replicates shown) were 1.2, 0.012, and 0.005 nt/min, respectively. Reaction conditions: 1 μM polymerase, 0.4 μM primer, 0.5 μM template, 4 mM each NTP, 200 mM MgCl2, and 50 mM Tris (pH 8.3) at 17 °C. (B) Primer extension on templates containing a 20-nt C-rich sequence interrupted by a purine-rich sequence of 0, 4, 8, or 12 nt (R0, R4, R8, or R12, respectively). (C) Primer extension on templates containing stem-loops of either 4 (S4) or 6 bp (S6), in comparison with the 5′ portion of the stem alone (Y4 or Y6, respectively). Reaction conditions for B and C as above, but using 100 nM polymerase, 40 nM primer, and 50 nM template and incubating for 24 h. P, unextended primer. Dots indicate full-length extension products, with extralength material corresponding to continued extension through the spacer and portion of the template that is complementary to the sequence tag of the ribozyme.
Table S2.
Fidelity of the WT and 24-3 polymerases
| Polymerase | Expected | Observed (%) | |||||
| A | C | G | U | Del | Ins | ||
| WT | A | 91.3 | 1.0 | 1.4 | 0.1 | 6.2 | 0.0 |
| C | 0.0 | 99.1 | 0.1 | 0.8 | 0.0 | 0.0 | |
| G | 0.1 | 0.7 | 99.0 | 0.2 | 0.0 | 0.0 | |
| U | 0.7 | 0.2 | 1.6 | 97.4 | 0.1 | 0.1 | |
| 24-3 | A | 81.1 | 0.4 | 7.4 | 0.4 | 10.7 | 0.0 |
| C | 0.2 | 93.3 | 0.2 | 6.0 | 0.3 | 0.0 | |
| G | 0.3 | 0.6 | 98.2 | 0.2 | 0.6 | 0.0 | |
| U | 0.4 | 0.4 | 1.5 | 96.5 | 1.3 | 0.1 | |
Operating on the favorable template 3′-ACGCUUCGCAC-5′, the average fidelities of WT and 24-3 were 96.6% and 92.0%, respectively, calculated as the geometric mean of the fidelities for each templating base, including deletions. Excluding wobble mutations, the fidelities were 97.2% and 95.5%, respectively. Bold values indicate correct nt incorporation.
The WT and 24-3 polymerases were challenged to extend a primer through various difficult sequence features over 24 h. Both WT and 24-3 can extend readily through a 20-nt C-rich sequence, but extension by WT halts at four consecutive purines, whereas 24-3 extends readily through purine-rich sequences of 12 nt (Fig. 2B). Similarly, when presented with a 4-bp stem closed by a stable “tetraloop” structure, WT halts at the start of the stem, even though it can extend through the same sequence in single-stranded form (Fig. 2C). In contrast, 24-3 reads through the entire stem-loop in high yield. Longer stems pose a barrier to 24-3, although detectable full-length extension products are seen for stems of up to 8 bp (Fig. S2).
Fig. S2.

Primer extension by the 24-3 polymerase on templates containing stem-loop structures. The templates contained a 4-nt (S4), 6-nt (S6), or 8-nt (S8,8a–c) stem closed by a stable tetraloop (either 5′-UCCG-3′ or 5′-GCAA-3′). Reaction conditions: either 50 nM polymerase, 40 nM primer, 50 nM template, and no tetraethylammonium chloride (TEA); or 400 nM polymerase, 160 nM primer, 200 nM template, and 1 M TEA; both with 4 mM each NTP, 200 mM MgCl2, and 50 mM Tris (pH 8.3) at 17 °C for 72 h. P, unextended primer. Dots indicate full-length extension products, with extralength material corresponding to continued extension through the spacer and portion of the template that is complementary to the sequence tag of the ribozyme.
Synthesis of Functional RNAs.
The improved properties of 24-3 enable it to synthesize structured, functional RNAs that previously were inaccessible for any polymerase ribozyme. The cyanocobalamin and GTP aptamers can be synthesized from a separate primer in 47% and 18% yield, respectively, after 24 h (Fig. 3 A and B). The 24-3 polymerase also can synthesize ribozymes that have heterogeneous sequence and substantial secondary structure, such as the entire F1 ligase ribozyme (19), which is obtained in 2% yield after 24 h (Fig. 3C). Following their RNA-catalyzed synthesis, the full-length aptamers and ribozyme were gel purified and tested for their respective activities (Fig. 3 E and F). Compared with chemically synthesized controls, the aptamers synthesized by the polymerase are captured 3- to 6-fold less effectively by the corresponding immobilized ligand and the ligase has 10-fold reduced initial rate of reaction. This reduction is presumably due to mutational load, consistent with the observation that when the polymerase is required to synthesize only 12 (rather than 18) nucleotides to complete the cyanocobalamin aptamer, ligand capture is reduced by only 20%.
Fig. 3.

Synthesis of functional RNAs by the 24-3 polymerase. Synthesis of (A) the cyanocobalamin aptamer after 24 h, (B) the GTP aptamer after 24 h, (C) the F1 ligase ribozyme after 24 h, and (D) yeast phenylalanyl-tRNA after 72 h. Polymerization conditions as in Fig. 2B, but using 50 nM polymerase. Dots indicate full-length extension products. The slightly reduced mobility of full-length aptamers generated by the polymerase compared with those from chemical synthesis is due to a small difference (29 Da) in the composition of the 5′-fluorescein label. Sequence and secondary structure of the primer (magenta) and polymerized portion (cyan) of each RNA are shown below the respective gel images; substrates for the F1 ligase are shown in black. (E) Fraction of cyanocobalamin (B12) or GTP aptamers bound and eluted from corresponding ligand-derived resins, comparing synthetic (Syn) and ribozyme-synthesized (Rz) aptamers and a nonbinding control RNA (–). (F) Catalytic activity of the full-length, ribozyme-synthesized F1 ligase in a reaction mixture containing 1 μM ligase, 0.2 μM 5′ substrate, 10 μM 3′ substrate, 25 mM MgCl2, and 50 mM EPPS (pH 8.5) at 20 °C. Data were fit to the following equation: yield = yieldmax (1 – e–kobs•t), where yieldmax = 13% and kobs = 0.11 min–1.
As a final test of polymerase generality, 24-3 was used to synthesize yeast phenylalanyl tRNA from a 15-nt primer (Fig. 3D). Despite the stable and complex structure of the template, full-length tRNA was obtained in 0.07% yield after 72 h. This RNA product is close to the limit of what can be achieved with the polymerase, but is likely the first time a tRNA molecule has been synthesized by a ribozyme since the end of the RNA world, nearly four billion years ago.
Exponential Amplification of RNA.
The most commonly practiced method for amplifying nucleic acids is the PCR, which entails repeated cycles of heat denaturation and reciprocal primer extension and depends on the activity of a polymerase protein. The 24-3 RNA polymerase was used to carry out PCR-like amplification, but in an all-RNA system (riboPCR) using a starting RNA template, RNA primers, and the four NTPs. The concentration of Mg2+ was reduced to minimize spontaneous RNA cleavage, PEG8000 was used as a molecular crowding agent to improve ribozyme activity at the reduced Mg2+ concentration (20, 21), and tetrapropylammonium chloride was added to lower the melting temperature of the duplex RNA (22, 23). Under these conditions, 1 nM of a 24-nt RNA template, composed of two 10-nt primer-binding sites flanking the sequence AGAG, was driven through repeated thermal cycles, resulting in 98 nM newly synthesized template and 106 nM of its complement, corresponding to 100-fold amplification (Fig. 4A). Sequencing of the amplified products revealed that the central AGAG sequence was largely preserved, albeit with a propensity to mutate the third position from A to G, reflecting the low barrier to wobble pairing (Table S3).
Fig. 4.

RNA-catalyzed exponential amplification of RNA (riboPCR). (A) Amplification of a 24-nt template, tracking either the template (white circles) or complement (black circles) compared with the reaction with no input template (white and black squares, respectively). Reaction conditions: 0.4 µM polymerase, 0.2 µM each primer, ±1 nM starting template, 4 mM each NTP, 50 mM MgCl2, 0.9 M TPA, 6% (wt/vol) PEG8000, and 50 mM Tris (pH 8.3), with thermal cycles of 72 °C for 2 s and then 17 °C for 4 h. (B) Real-time riboPCR of a 20-nt template, monitored by FRET between two fluorescently labeled primers. Reaction conditions as above, but with starting template concentrations of 10 nM (dark red), 3.2 nM (red), 1 nM (orange), 0.32 nM (yellow), 0.1 nM (light green), 32 pM (green), 10 pM (cyan), 3.2 pM (blue), 1 pM (indigo), or 0 (violet) and with thermal cycles of 68 °C for 1 s and then 17 °C for 30 min. Dashed line indicates a threshold FRET value of 10% of maximum. For each starting template concentration, three replicates were obtained (thin lines) and averaged (thick line) to determine a cycle-to-threshold (Ct) value. (C) Semilog plot of Ct vs. starting template concentration, demonstrating a linear relationship with slope of –8.8, corresponding to per-cycle amplification of 1.3-fold.
Table S3.
Mutations during riboPCR
| Input | Output (of 27) | Consensus | ||||
| A | C | G | U | Del | ||
| A1 | 26 | 0 | 1 | 0 | 0 | A |
| G2 | 1 | 0 | 26 | 0 | 0 | G |
| A3 | 15 | 1 | 5 | 2 | 4 | A |
| G4 | 0 | 0 | 20 | 0 | 7 | G |
Observed mutations in the central AGAG region of a 24-nt template after 100-fold amplification by riboPCR. The consensus from 27 sequenced clones is shown at the right. Bold values indicate correct nt incorporation.
Amplification of a 20-nt template (without central insert) was monitored in real time, exploring input template concentrations ranging from 10 nM to 1 pM. The resulting amplification profiles are typical for real-time PCR, shifted by a constant number of cycles per log-change in starting template concentration (Fig. 4B). A plot of cycle-to-threshold vs. logarithm of template concentration is linear across the entire range of dilutions (Fig. 4C), indicating exponential amplification of the template RNA with a per-cycle amplification efficiency of 1.3-fold. This result matches the value obtained from direct measurements made after each thermal cycle starting at low template concentrations (Fig. S3A). As with conventional PCR, product inhibition at high template concentrations slows amplification, which for riboPCR occurs starting at ∼10 nM template. Below this concentration, exponential amplification is robust, causing 1 pM starting RNA to be amplified to ∼40 nM product after 24 h, which corresponds to 40,000-fold amplification (Fig. S3B). In comparison, over the same time, riboPCR without any input template yielded only ∼5 nM of similarly sized, nonspecific product RNAs that likely correspond to “primer-dimers” and materials generated by other mispriming events.
Fig. S3.

RiboPCR amplification of a 20-nt template. (A) Yield of newly synthesized template (white circles) and complement (black circles) over the first 12 cycles, starting with 200 pM template. Data were fit to a single exponential growth equation, giving growth rates of 0.27 and 0.18 per cycle for the complement and template, respectively, which corresponds to 1.2- to 1.3-fold amplification per cycle. (B) Amplification starting from low initial template concentrations to yield both complement and template. Reaction conditions: 0.4 µM polymerase, 0.2 µM each primer, 0, 0.1, 1, or 10 pM starting template, 4 mM each NTP, 50 mM MgCl2, 0.81 M tetrapropylammonium chloride, 6% (wt/vol) PEG8000, and 50 mM Tris (pH 8.3), with thermal cycles of 68 °C for 3 s and then 17 °C for 30 min.
Discussion
The RNA-templated synthesis of RNA, as catalyzed by a ribozyme, has been known for 20 y (10). All examples to date, however, have suffered from slow rates and a strong preference for unstructured, C-rich templates (12–16). In vitro evolution was used to select polymerase variants that can synthesize functional RNA aptamers, resulting in dramatically improved activity and sequence generality. The evolved 24-3 polymerase can extend through regions of secondary structure, enabling the synthesis of a variety of functional RNAs, including aptamers and ribozymes.
Previously, the strong limitations on template sequence in protein-free RNA polymerization systems excluded the possibility of carrying out cycles of replication because both the template and its complement could not conform to these limitations. Furthermore, to sustain life, RNA replication must proceed with sufficient yield to support exponential amplification. The improved properties of the 24-3 polymerase enable the residue-by-residue copying and exponential amplification of short RNAs, with a per-cycle amplification efficiency of 1.3-fold. Analogous processes have been proposed for the replication of RNA on the early Earth, with hot-cold cycles potentially driven by diurnal variation or convection in a hydrothermal vent (24).
Inefficient strand displacement and modest fidelity limit the yield and specific activity of RNAs synthesized by the 24-3 polymerase and similarly limit riboPCR amplification to templates of only 20–25 nt. Both of these properties might be improved by selecting polymerase ribozymes that complete the synthesis of more demanding functional motifs, such as other ribozymes. Alternatively, selection methods might be used that take advantage of riboPCR, selecting directly for the amplification of functional RNAs. Such methods would be powerful because differences in fitness would be reflected as an exponential rather than linear effect, which is a key feature of Darwinian evolution.
The vestiges of the late RNA world appear to be shared by all extant life on Earth, most notably in the catalytic center of the ribosome (25), but most features of RNA-based life likely were lost in the Archaean era (2). Whatever forms of RNA life existed, they must have had the ability to replicate genetic information and express it as functional molecules. The 24-3 polymerase is the first known ribozyme that is able to amplify RNA and to synthesize complex functional RNAs. To achieve fully autonomous RNA replication, these two activities must be combined and further improved to provide a polymerase ribozyme that can replicate itself and other ribozymes of similar complexity. Such a system could, under appropriate conditions, be capable of self-sustained Darwinian evolution and would constitute a synthetic form of RNA life.
Materials and Methods
Materials.
All oligonucleotides used in this study are listed in Table S4. Synthetic oligonucleotides were either purchased from Integrated DNA Technologies or prepared by solid-phase synthesis using an Expedite 8909 DNA/RNA synthesizer, with reagents and phosphoramidites purchased from Glen Research. All RNA templates used in RNA polymerization reactions contained a 5′-terminal region complementary to the 5′ end of the polymerase (14). RNA templates were prepared either synthetically or by in vitro transcription from synthetic DNA templates, as described in SI Materials and Methods. RNA polymerase ribozymes were prepared by in vitro transcription, with dsDNA templates generated by PCR from corresponding plasmid DNA using the primers listed in Table S4. All RNA templates and ribozymes were purified by denaturing (8 M urea) PAGE and ethanol precipitation before use.
Table S4.
Sequences of RNA and DNA molecules used in this study
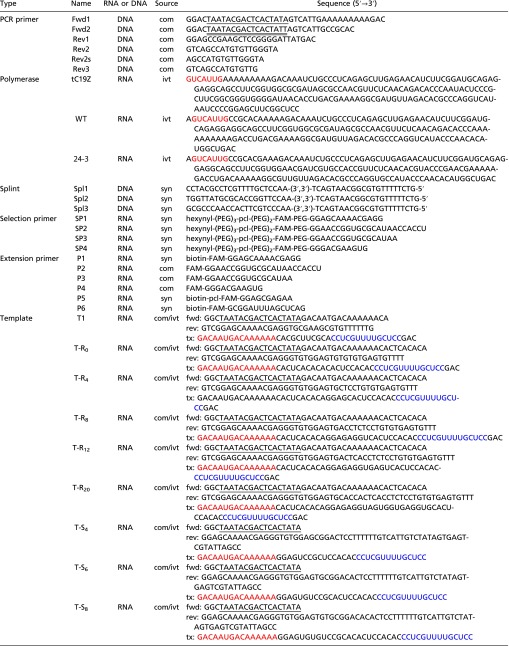 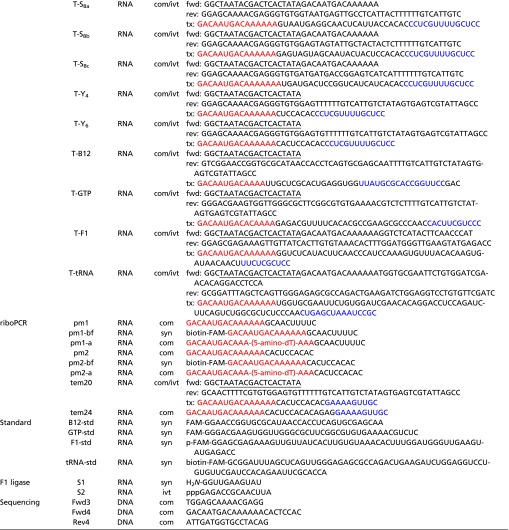
|
The molecules were synthesized in-house (syn), purchased from IDT (com), or prepared by in vitro transcription (ivt). The canonical T7 RNA polymerase promoter sequence is underlined; the ϕ2.5 promoter sequence, which initiates transcription with adenosine (41), is double underlined. Sequences in red indicate the 5′ tag used on polymerase ribozymes and templates to improve processivity. Sequences in blue are primer binding sites on templates used for polymerase extension assays. The tC19Z polymerase was prepared using PCR primers Fwd1 and Rev1, the WT polymerase using primers Fwd2 and Rev2, and the 24-3 polymerase using primers Fwd2 and Rev3. dsDNA templates were prepared by cross-extension of forward (fwd) and reverse (rev) primers, followed by in vitro transcription to generate the corresponding RNA (tx). FAM, 6-fluorescein label; pcl, photocleavable linker; PEG, polyethylene glycol spacer.
In Vitro Evolution.
A pool of synthetic oligodeoxynucleotides was prepared, encoding the WT polymerase ribozyme (Fig. S1) and randomizing all nucleotide positions between the two primer regions at a frequency of 10% per position. The DNA was made double-stranded by primer extension using SuperScript II reverse transcriptase (ThermoFisher), except that 1.5 mM MnCl2 was added to enhance extension through DNA lesions that occurred during synthesis (26). The yield of full-length dsDNA was 165 pmol (55% extension efficiency), corresponding to 1014 distinct sequences. The dsDNA was amplified by five cycles of PCR and then purified using the Qiagen PCR purification kit.
Detailed procedures for each step of selection are provided in SI Materials and Methods, and the reaction conditions used in each round of in vitro evolution are listed in Table S1. In outline, each round began with in vitro transcription of the population of ribozymes, followed by tethering of an RNA primer to the 5′ end of the ribozymes. The resulting ribozyme-primer conjugates were annealed to a template RNA, and the primer was extended by the ribozyme in the presence of the four NTPs. The extended materials were captured on streptavidin, the template RNA was removed, and the desired products were selected by either a gel-shift method or capture of a ribozyme-synthesized aptamer. For gel-shift selection, the products were separated by PAGE and the fully extended materials were eluted from the gel and reverse transcribed. For aptamer-based selection, the ribozyme portion of the products was reverse transcribed to form an RNA–cDNA heteroduplex, and then extended materials that contained a functional aptamer were captured by binding to the corresponding ligand (cyanocobalamin or GTP). In both cases, the isolated cDNA was amplified by PCR, followed by in vitro transcription to generate the progeny population of ribozymes.
RNA-Catalyzed RNA Polymerization.
RNA polymerization reactions were performed with separate ribozyme, RNA template, and 5′-biotinylated RNA primer under two different concentration regimes. For kinetic analyses and fidelity studies, the reaction mixtures contained 1 μM ribozyme, 0.5 μM template, and 0.4 μM primer; for all other studies they contained either 50 or 100 nM ribozyme, 50 nM template, and 40 nM primer. In all cases, the RNAs first were heated at 80 °C for 1 min, cooled to 17 °C over 5 min, and then added to the final mixture, which also contained 4 mM each NTP, 200 mM MgCl2, 0.05% TWEEN20, and 50 mM Tris (pH 8.3, prepared at 19 °C). Polymerization was carried out at 17 °C and quenched by adding 0.4 volumes of 500 mM EDTA. The biotinylated primers and extended products were captured on streptavidin C1 Dynabeads (ThermoFisher), washed four times with alkali (25 mM NaOH, 1 mM EDTA, and 0.05% TWEEN20) and twice with TE (1 mM EDTA, 0.05% TWEEN20, and 10 mM Tris, pH 8.0) plus 8 M urea, and then eluted with 98% formamide and 10 mM EDTA (pH 8.0) at 95 °C for 10 min. The reaction products were analyzed by PAGE to determine the average number of nucleotides added to the primer at various times. Sequencing of the extension products for fidelity studies and analysis of their aptamer binding or ligase activity are described in SI Materials and Methods.
Amplification of RNA by riboPCR.
All primers for riboPCR contained a 5′-terminal region that was complementary to the 5′ end of the polymerase, followed by a hexadenylate spacer. Reactions were performed in duplicate, with one of the two primers (for synthesis of either the template or complement) labeled with biotin and fluorescein. Amplification reactions used 400 nM 24-3 polymerase, 200 nM each primer, and varying amounts of template, which first were annealed, then added to a mixture containing 4 mM each NTP, 50 mM MgCl2, 0.9 M tetrapropylammonium chloride (TPA), 6% (wt/vol) PEG8000, 0.05% TWEEN20, and 50 mM Tris (pH 8.3). The reactions were carried out in a Bio-Rad C1000 thermocycler. Aliquots were taken at various times, and the biotinylated materials were captured on streptavidin C1 Dynabeads, washed once with alkali, and once with TE plus 8 M urea, and then eluted with formamide as described above.
Real-time tracking of riboPCR used primers that contained an internal amino-dT residue within the hexadenylate spacer that had been labeled with either fluorescein (donor) or Cy5 (acceptor) through conjugation of the corresponding N-hydroxysuccinimidyl esters (Lumiprobe). The reactions were carried out in a ThermoFisher Viaa7 thermocycler. The FRET signal was corrected for fluorescence channel cross-talk and adjusted to a common baseline. Detailed procedures are provided in SI Materials and Methods.
SI Materials and Methods
In Vitro Transcription.
RNA templates were transcribed in a mixture containing 0.5 μM dsDNA template, 15 U/μL T7 RNA polymerase, 0.002 U/μL inorganic pyrophosphatase, 5 mM each NTP (Sigma-Aldrich), 25 mM MgCl2, 2 mM spermidine, 10 mM DTT, and 40 mM Tris (pH 8.0), which was incubated at 37 °C for 2 h and then for an additional 1 h following addition of 0.1 U/μL TurboDNase (ThermoFisher). The dsDNA templates were generated by cross-extending 5 µM each of two synthetic oligodeoxynucleotides in a reaction mixture containing 5 U/μL SuperScript II reverse transcriptase (ThermoFisher), 0.5 mM each dNTP (Denville Scientific), 3 mM MgCl2, 75 mM KCl, 10 mM DTT, and 50 mM Tris (pH 8.3), which was incubated at 42 °C for 1 h and then incubated at 70 °C for 15 min to inactivate the enzyme. In vitro transcription of polymerase ribozymes was performed as described above, but using 20 μg/mL dsDNA templates generated by PCR.
Preparation of Ribozyme-Primer Conjugates.
5′-Azide–labeled RNA was prepared by in vitro transcription under conditions as above, except the concentration of ATP was reduced to 2 mM, and the mixture contained 5 mM 5′-γ-[2-azidoethyl]-ATP (Jena Biosciences) (27). In the first round of in vitro evolution, 160 μg (200 μg/mL) of dsDNA template was used, and the resulting RNA was purified by PAGE and ethanol precipitation. In all subsequent rounds, 2 μg (20 μg/mL) of dsDNA was used, and the RNA was purified using an RNeasy column (Qiagen) followed by five washes on an Amicon 30K spin filter (Millipore). For rounds 5–7, 5′-pCpC-3′-biotin was ligated to the 3′ end of the RNA in a reaction mixture containing 1 U/μL T4 RNA ligase I (New England Biolabs), 5 μM RNA, 10 μM Rev2s splint oligonucleotide, 30 μM pCpC-biotin, 1 mM ATP, 10 mM MgCl2, 1 mM DTT, 50 mM Tris (pH 7.5), and 10% (vol/vol) DMSO, which was incubated at 37 °C for 3 h (28). The ligated products were purified by five washes on an Amicon 30K spin filter. For all rounds, the 5′-azide–labeled RNA was joined to a 5′-hexynylated primer via a templated “click” reaction, using 15 μM RNA, 30 μM primer, 21 μM splint DNA, 2 mM Tris(3-hydroxypropyltriazolylmethyl)amine, 0.4 mM Cu(II)SO4, 10 mM sodium ascorbate, 100 mM NaOAc, 25 mM Na2HPO4 (pH 7.4), and 10% DMSO and incubating at 17 °C for 2 h (29, 30). The resulting ribozyme-primer conjugates were purified by PAGE and ethanol precipitation.
Primer Extension Reactions.
RNA-templated primer extension reactions used 3 nmol ribozyme-primer conjugate in the first round of in vitro evolution and 100 pmol conjugate in all subsequent rounds. For rounds 1–4 and 8–24, the template RNA was biotinylated by first adding 2´-azido-UTP (Tri-Link) to the 3′ end using yeast poly(A) polymerase (Affymetrix), as previously described (31), and then attaching biotin-alkyne (ThermoFisher) by click chemistry, as described above. The biotinylated template was purified by PAGE and ethanol precipitation. For rounds 5–7, the polymerase (rather than the template) was biotinylated, as described above. For all rounds, the ribozyme-primer conjugate first was annealed to the template, then primer extension was carried out in a mixture containing 4 mM each NTP, 200 mM MgCl2, 0.05% TWEEN20, and 50 mM Tris (pH 8.3), which was incubated at 17 °C for various times (Table S1). The reactions were quenched by adding 0.4 volumes of 500 mM EDTA.
Due to the high melting temperature of long RNA–RNA duplexes, the biotinylated double-stranded reaction products first were captured on streptavidin, and then the nonbiotinylated strand was removed by washing with alkali (25 mM NaOH, 1 mM EDTA, and 0.05% TWEEN20), immediately neutralized with 1 M Tris (pH 7.5), and ethanol precipitated. In the first round, the RNA was captured using 100 μL high-capacity streptavidin-agarose resin (ThermoFisher) and in all subsequent rounds was captured using 1 mg streptavidin-coated C1 Dynabeads (ThermoFisher). In the first round, the captured material was washed five times with 1 mL 1 M NaCl, 1 mM EDTA, 0.05% TWEEN20, and 10 mM Tris (pH 8.0) (TSE), washed five times with 1 mL 1 mM EDTA, 0.05% TWEEN20, and 10 mM Tris (pH 8.0) (TE), and then eluted with alkali. In rounds 2–4 and 8–24 (when the template was biotinylated), the captured material was washed twice with 200 μL TSE, washed twice with TE, and then eluted with alkali. In rounds 5–7 (when the ribozyme-primer conjugate was biotinylated), the template RNA was removed by washing three times with alkali, followed by three additional washes with TE plus 8 M urea, and then the extended products were recovered by eluting with 100 μL 98% formamide and 10 mM EDTA (pH 8.0) at 95 °C for 10 min, followed by ethanol precipitation.
Selection of Extended Products.
For gel-shift selection (rounds 1–4, 8–16, and 18–21), the reaction products were separated by PAGE (3.5%, 19:1 mono:bisacrylamide), identifying the full-length materials by comparison with authentic standards. These materials were excised from the gel, eluted, ethanol precipitated, and reverse transcribed. For aptamer-based selection with the cyanocobalamin aptamer (rounds 5–7), the reaction products were reverse transcribed, ethanol precipitated, and dissolved in binding buffer containing 3 M LiCl, 1 mM EDTA, 0.05% TWEEN20, and 50 mM Hepes (pH 7.4). This mixture was heated at 65 °C for 2 min, cooled to 4 °C over 5 min, and then applied to a 0.3-mL vitamin B12-agarose (Sigma-Aldrich) gravity-flow column that had been prewashed with binding buffer plus 0.1 mg/mL tRNA. The column and applied RNA were incubated at 4 °C for 1 h and then washed five times with binding buffer plus tRNA and once with binding buffer alone. The agarose resin was suspended in 1 mL binding buffer, and the ribozyme–cDNA heteroduplex was detached from the bound aptamer by photocleavage at 350 nm, collected in two 1-mL washes with binding buffer, and ethanol precipitated.
For combined gel-shift and aptamer-based selection with the GTP aptamer (rounds 17 and 22–24), gel-shift selection first was performed as described above, through to the reverse transcription step. The hybridized complexes were purified by five washes on an Amicon 30K spin filter to remove dGTP, and then the aptamer portion was captured and the ribozyme–cDNA portion was released as described above for the cyanocobalamin aptamer, except replacing the B12 binding buffer with a solution containing 5 mM MgCl2, 200 mM KCl, 0.1 mM EDTA, 0.05% TWEEN20, and 10 mM Na2HPO4 (pH 6.2) and replacing B12-agarose with GTP-agarose (Innova Biosciences). As described above, the immobilized complexes were photocleaved, and the released materials were collected and ethanol precipitated.
Amplification of Selected Materials.
Regardless of the selection method, the cDNA was amplified by PCR using primers Fwd2 and Rev2. Additional diversity was introduced after rounds 4, 12, 17, and 22 by mutagenic PCR (32). Following rounds 18 and 24, the amplified DNA was cloned into Escherichia coli using the TOPO-TA cloning kit (ThermoFisher), and the cells were grown at 37 °C for 16 h on LB agar plates containing 50 μg/mL kanamycin. DNA was isolated from individual colonies, PCR amplified, and sequenced by Genewiz. A shorter amplification product that first was apparent at round 15 and became prevalent by round 18 was found to result from selected mutations that created a partial internal priming site for Rev2. Accordingly, all subsequent reverse transcriptions were carried out using the truncated primer Rev3 and SuperScript III reverse transcriptase (ThermoFisher) at 55 °C. Rev3 also was used in the subsequent PCR amplifications.
Sequencing of Polymerization Products.
RNA polymerization was carried out with either the WT polymerase for 24 h or 24-3 polymerase for 30 min, in each case yielding ∼60% full-length material in the extension of primer P1 on template T1. The extended products were immobilized on streptavidin, and the template was removed as described above, and then the Universal miRNA Cloning Linker (New England Biolabs) was ligated to the 3′ end of the extended materials using K227Q T4 RNA Ligase 2 (New England Biolabs). The resulting material was washed twice with TE plus 8 M urea and then eluted with formamide as described above. The RNA was reverse transcribed using primer Rev4 and then PCR amplified using primers Fwd3 and Rev4. The resulting dsDNA, including all partially and fully extended products, was purified by 6% agarose gel electrophoresis and eluted from the gel using a Qiaex II gel extraction kit (Qiagen). The purified DNA was submitted to the High-Throughput Sequencing Core at The Scripps Research Institute and sequenced using an Illumina MiSeq with a 75-cycle, paired-end run.
Sequence data were processed as described previously (33). The adaptor sequences were removed with Trimmomatic (34), and paired reads were merged using FLASH (35), only retaining pairs with perfect complementarity. Further filtering discarded any reads that had a Phred score <36 at any position, that contained incorrect primer or tag sequences, that were shorter than the primer alone, or that were longer than the full-length material. For the remaining reads, the primer plus extended sequence was aligned to the target sequence using Bowtie2 (36) in end-to-end mode and with a minimum score of [–readlength – 2]. SAMtools (37) was used to prepare a compressed alignment file, removing any unaligned reads and sorting and indexing the remainder (>99%). A gapped alignment was generated using a module of Breseq (38, 39), converted to FASTA format, and the error frequencies were tabulated using a custom Python script that is available on request. Insertion mutations were rare (<0.05% per position) and disregarded in subsequent analyses. The average fidelity was calculated as the geometric mean of the fidelities for each templating base.
Analysis of Aptamer Binding.
The 24-3 polymerase was used to synthesize the cyanocobalamin aptamer using primer P2 or P3 and template T-B12 and to synthesize the GTP aptamer using primer P4 and template T-GTP. The templates were biotinylated before use, as described above. Polymerization was carried out under standard conditions for either 4 h (for primer P2) or 24 h (for primers P3 and P4). The reactions were quenched and the materials were captured on streptavidin C1 Dynabeads, washed twice with TSE and once with TE. The primers and extended products were eluted with alkali, immediately neutralized with 1 M Tris (pH 7.5), ethanol precipitated, and analyzed by PAGE. Full-length materials were eluted from the gel and ethanol precipitated.
Aptamer binding to 5 μL of either B12-agarose or GTP-agarose was measured using a MultiScreen filterplate (Millipore), which first was washed three times with the appropriate aptamer binding buffer (see above), and then 2 pmol of aptamer in 50 μL binding buffer was applied and allowed to bind under gentle agitation for 1 h. The filterplate was washed three times with binding buffer, with 5-min agitation for each wash, and then the remaining bound aptamer was eluted with alkali and neutralized; 2 M NaCl was included in the elution mixture for the cyanocobalamin aptamer to counteract the positive charge of B12-agarose. The fluorescently labeled RNA in each filtrate fraction was quantified using a FilterMax F5 plate reader (Molecular Devices). These data were compared with positive and negative controls using either cognate or noncognate synthetic aptamers (B12-std and GTP-std), respectively.
Analysis of Ligase Activity.
The 24-3 polymerase was used to synthesize the F1 ligase ribozyme, using primer P5 (labeled with fluorescein and photocleavable biotin) and template T-F1, carried out under standard conditions for 24 h. The primers and extended products were captured on streptavidin-agarose resin, and the template was removed by washing four times with alkali, twice with TE plus 8 M urea, twice with TSE plus 0.1 mg/mL tRNA, and once with TSE alone. The extended products were detached from the biotin by photocleavage, ethanol precipitated, and separated by PAGE. Full-length materials were eluted from the gel and ethanol precipitated.
Ligase substrate S1 was prepared by chemical synthesis, labeled with BODIPY TMR-X N-hydroxysuccinimidyl ester (ThermoFisher) according to the manufacturer’s instructions, and purified by PAGE and ethanol precipitation. Substrate S2 was prepared by in vitro transcription, as previously described (19). The F1 ligase also was prepared synthetically as a control (F1-std). RNA-catalyzed RNA ligation was carried out in a reaction mixture containing 1 μM ligase, 0.2 μM labeled S1, 10 μM S2, 25 mM MgCl2, and 50 mM EPPS (pH 8.5), which was incubated at 20 °C for 1 h, and then quenched by adding 98% formamide and 10 mM EDTA. The reaction products were analyzed by PAGE.
Sequencing of riboPCR Products.
Reactions were performed with primer pm1 (extended to make new complement RNA, unlabeled) and primer pm2-bf (extended to make new template RNA, labeled with biotin and fluorescein). This arrangement ensured that only newly made template RNA, requiring at least two cycles of riboPCR primer extension, was isolated for sequencing. The riboPCR products were captured on streptavidin C1 Dynabeads, washed four times with alkali and twice with TE plus 8 M urea, and then eluted with formamide. Full-length material was purified by PAGE and subsequent ethanol precipitation. The Universal miRNA Cloning Linker was ligated to the 3′ end of the purified RNA, and the products were immobilized again on streptavidin C1 Dynabeads, washed twice with TE plus 8 M urea, and again eluted with formamide. The eluted RNA was reverse transcribed using primer Rev4, then PCR amplified using primers Fwd4 and Rev4. The resulting dsDNAs were cloned and sequenced as described above.
Real-Time riboPCR.
RiboPCR amplification was performed as described in Materials and Methods, using varying concentrations of template and in three-way sets of reactions: only fluorescein-labeled, only Cy5-labeled, and both fluorescein- and Cy5-labeled primers. The reactions were carried out in a ThermoFisher Viaa7 thermocycler, monitoring in three separate channels: x1m1, excitation and emission of the donor (fluorescein); x1m5, excitation of the donor and emission of the acceptor (Cy5); and x5m5, excitation and emission of acceptor. Amplification was carried out during 72 cycles of denaturation at 68 °C for 1 s and then extension at 17 °C for 30 min, measuring fluorescence at the end of each extension step.
Raw data from riboPCR that was performed with either donor fluorophore or acceptor fluorophore alone was used to calculate correction factors for channel cross-talk (40), specific to each replicate sample and each cycle. Emission of the donor in channel x1m5 was normalized to emission of the donor in channel x1m1 to yield correction factor a; emission of the acceptor in channels x1m5 and x1m1 was normalized to emission of the acceptor in channel x5m5 to yield correction factors b and c, respectively. These factors were used to calculate donor emission due to donor fluorescence, D, and acceptor emission due to FRET, F, according to the equations
For each condition and replicate, the ratio of F/(F + D) was baseline corrected so that the minimum value over all cycles was 0. All traces were multiplied by the same constant to span the range of 0–100. Three replicates for each condition were averaged, and Ct was calculated for each as the last cycle before reaching the threshold value of 10. A plot of Ct vs. log[template] was fit by least-squares linear regression to calculate the slope m, which provides the per-cycle amplification as 10(–1/m). The results were not significantly affected by omitting corrections for cross-talk and instead using the ratio x1m5/(x1m5 + x1m1).
Acknowledgments
We thank Katherine Petrie for advice on DNA sequencing and Travis Young and Shoutian Zhou for assistance operating the Viaa7 thermocycler. This work was supported by NASA Grant NNX14AK15G and Simons Foundation Grant 287624. D.P.H. was supported by a Graduate Fellowship from the Fannie and John Hertz Foundation.
Footnotes
Conflict of interest statement: R.R.B. and G.F.J. appeared as coauthors of a paper published as Breaker RR, Joyce GF (2014) The expanding view of RNA and DNA function. Chem Biol 21(9):1059–1065. This paper was a Perspective commemorating the 20th anniversary of the journal.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1610103113/-/DCSupplemental.
References
- 1.Gilbert W. The RNA world. Nature. 1986;319(6055):618. [Google Scholar]
- 2.Joyce GF. The antiquity of RNA-based evolution. Nature. 2002;418(6894):214–221. doi: 10.1038/418214a. [DOI] [PubMed] [Google Scholar]
- 3.Attwater J, Holliger P. A synthetic approach to abiogenesis. Nat Methods. 2014;11(5):495–498. doi: 10.1038/nmeth.2893. [DOI] [PubMed] [Google Scholar]
- 4.Pressman A, Blanco C, Chen IA. The RNA world as a model system to study the origin of life. Curr Biol. 2015;25(19):R953–R963. doi: 10.1016/j.cub.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 5.Paul N, Joyce GF. A self-replicating ligase ribozyme. Proc Natl Acad Sci USA. 2002;99(20):12733–12740. doi: 10.1073/pnas.202471099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lincoln TA, Joyce GF. Self-sustained replication of an RNA enzyme. Science. 2009;323(5918):1229–1232. doi: 10.1126/science.1167856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue T, Orgel LE. A nonenzymatic RNA polymerase model. Science. 1983;219(4586):859–862. doi: 10.1126/science.6186026. [DOI] [PubMed] [Google Scholar]
- 8.Adamala K, Engelhart AE, Szostak JW. Generation of functional RNAs from inactive oligonucleotide complexes by non-enzymatic primer extension. J Am Chem Soc. 2015;137(1):483–489. doi: 10.1021/ja511564d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Been MD, Cech TR. RNA as an RNA polymerase: Net elongation of an RNA primer catalyzed by the Tetrahymena ribozyme. Science. 1988;239(4846):1412–1416. doi: 10.1126/science.2450400. [DOI] [PubMed] [Google Scholar]
- 10.Ekland EH, Bartel DP. RNA-catalysed RNA polymerization using nucleoside triphosphates. Nature. 1996;382(6589):373–376. doi: 10.1038/382373a0. [DOI] [PubMed] [Google Scholar]
- 11.Sczepanski JT, Joyce GF. A cross-chiral RNA polymerase ribozyme. Nature. 2014;515(7527):440–442. doi: 10.1038/nature13900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: Accurate and general RNA-templated primer extension. Science. 2001;292(5520):1319–1325. doi: 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- 13.Zaher HS, Unrau PJ. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA. 2007;13(7):1017–1026. doi: 10.1261/rna.548807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011;332(6026):209–212. doi: 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- 15.Attwater J, Wochner A, Holliger P. In-ice evolution of RNA polymerase ribozyme activity. Nat Chem. 2013;5(12):1011–1018. doi: 10.1038/nchem.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang QS, Cheng LKL, Unrau PJ. Characterization of the B6.61 polymerase ribozyme accessory domain. RNA. 2011;17(3):469–477. doi: 10.1261/rna.2495011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorsch JR, Szostak JW. In vitro selection of RNA aptamers specific for cyanocobalamin. Biochemistry. 1994;33(4):973–982. doi: 10.1021/bi00170a016. [DOI] [PubMed] [Google Scholar]
- 18.Carothers JM, Davis JH, Chou JJ, Szostak JW. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA. 2006;12(4):567–579. doi: 10.1261/rna.2251306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson MP, Joyce GF. Highly efficient self-replicating RNA enzymes. Chem Biol. 2014;21(2):238–245. doi: 10.1016/j.chembiol.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karimata H, Nakano S, Sugimoto N. The roles of cosolutes on the hammerhead ribozyme activity. Nucleic Acids Symp Ser (Oxf) 2006;50(1):81–82. doi: 10.1093/nass/nrl040. [DOI] [PubMed] [Google Scholar]
- 21.Desai R, Kilburn D, Lee H-T, Woodson SA. Increased ribozyme activity in crowded solutions. J Biol Chem. 2014;289(5):2972–2977. doi: 10.1074/jbc.M113.527861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins JM, Rogers KS. Melting point depression of DNA by tetraaklylammonium bromides. Chem Biol Interact. 1977;19(2):197–203. doi: 10.1016/0009-2797(77)90031-x. [DOI] [PubMed] [Google Scholar]
- 23.Golaś T, Miller M, Shugar D. The effects of tetraalkylammonium salts on helix-coil transition parameters in natural and synthetic ribo- and deoxyribo-polynucleotides. Chem Biol Interact. 1980;30(2):209–222. doi: 10.1016/0009-2797(80)90127-1. [DOI] [PubMed] [Google Scholar]
- 24.Kreysing M, Keil L, Lanzmich S, Braun D. Heat flux across an open pore enables the continuous replication and selection of oligonucleotides towards increasing length. Nat Chem. 2015;7(3):203–208. doi: 10.1038/nchem.2155. [DOI] [PubMed] [Google Scholar]
- 25.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289(5481):905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 26.Chaput JC, Ichida JK, Szostak JW. DNA polymerase-mediated DNA synthesis on a TNA template. J Am Chem Soc. 2003;125(4):856–857. doi: 10.1021/ja028589k. [DOI] [PubMed] [Google Scholar]
- 27.Fiammengo R, Musílek K, Jäschke A. Efficient preparation of organic substrate-RNA conjugates via in vitro transcription. J Am Chem Soc. 2005;127(25):9271–9276. doi: 10.1021/ja051179m. [DOI] [PubMed] [Google Scholar]
- 28.Hausch F, Jäschke A. Multifunctional dinucleotide analogs for the generation of complex RNA conjugates. Tetrahedron. 2001;57(7):1261–1268. [Google Scholar]
- 29.Hong V, Presolski SI, Ma C, Finn MG. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew Chem Int Ed Engl. 2009;48(52):9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar R, et al. Template-directed oligonucleotide strand ligation, covalent intramolecular DNA circularization and catenation using click chemistry. J Am Chem Soc. 2007;129(21):6859–6864. doi: 10.1021/ja070273v. [DOI] [PubMed] [Google Scholar]
- 31.Winz M-L, Samanta A, Benzinger D, Jäschke A. Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic Acids Res. 2012;40(10):e78. doi: 10.1093/nar/gks062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2(1):28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 33.Petrie KL, Joyce GF. Limits of neutral drift: Lessons from the in vitro evolution of two ribozymes. J Mol Evol. 2014;79(3-4):75–90. doi: 10.1007/s00239-014-9642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohse M, et al. RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012;40(Web Server issue) W1:W622–W267. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magoč T, Salzberg SL. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, et al. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrick JE, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461(7268):1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 39.Barrick JE, Lenski RE. Genome dynamics during experimental evolution. Nat Rev Genet. 2013;14(12):827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, Puhl HL, 3rd, Koushik SV, Vogel SS, Ikeda SR. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys J. 2006;91(5):L39–L41. doi: 10.1529/biophysj.106.088773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunn JJ, Studier FW. Nucleotide sequence from the genetic left end of bacteriophage T7 DNA to the beginning of gene 4. J Mol Biol. 1981;148(4):303–330. doi: 10.1016/0022-2836(81)90178-9. [DOI] [PubMed] [Google Scholar]