

This Outlook discusses the finding by Bent et al. that PI3K/AKT/mTOR mediates a senescence secretory switch that controls chemoprotective endothelial secretory responses.
Keywords: tumor microenvironment, chemoresistance, paracrine, senescence, endothelial cell, SASP
Abstract
The tumor microenvironment influences cancer progression and therapy outcome by mechanisms not yet fully understood. In this issue of Genes & Development, Bent and colleagues (pp. 1811–1821) show how chemotherapy causes endothelial senescence. Interestingly, senescent endothelial cells do not mount a typical senescence-associated secretory phenotype but instead acutely secrete IL-6, promoting chemoresistance. This study unveils a physiological switch involving PI3K/AKT/mTOR signaling that restrains the senescence secretory responses to limit the detrimental consequences of persistent inflammation.
Senescence is a cellular response that limits the replication of aged or damaged cells. It is often accompanied by secretion of various factors collectively known as the senescence-associated secretory phenotype (SASP). The SASP attracts immune cells to the damaged tissue, which is beneficial at first glance but can also have adverse side effects through persistent tissue inflammation or recruitment of immature myeloid progenitors (Coppe et al. 2010).
In previous work, Gilbert and Hemann (2010) had studied the response of Eμ-myc p19Arf−/− B-cell lymphomas to doxorubicin treatment. Accumulation of IL-6 and TIMP-1 in the thymus of treated mice provided a prosurvival microenvironment that slowed lymphoma regression. This was suggested to be the result of nonmalignant endothelial cells undergoing senescence upon chemotherapy treatment. Here, Bent et al. (2016) used an endothelial-specific knockout to confirm that hypothesis: Depletion of endothelial IL-6 resulted in decreased thymic tumor burden upon chemotherapy.
Senescence is thought to have evolved as a mechanism facilitating tissue remodeling to maintain homeostasis and prevent damage. Senescence also limits cancer progression. However, the aberrant accumulation of senescent cells can be detrimental not just during aging but also in cancer. Factors secreted by senescent cells exert a range of protumorigenic effects, including facilitating angiogenesis, invasion, and metastasis (Coppe et al. 2010). The role of the SASP in chemoresistance has not been so well documented, but earlier work also reported that chemotherapy-induced damage of stromal fibroblasts can promote therapy resistance via production of WNT16B (Sun et al. 2012).
We often refer to the secretory responses of senescent cells with the umbrella term SASP. However, SASP composition depends on the senescence inducer or the cell type undergoing senescence. While many SASP components are conserved across different senescent cells, it is clear that senescent cells can mount different secretory responses. Interestingly, the paracrine response described here by Bent et al. (2016) is self-restrained. In spite of the fact that doxorubicin triggers endothelial senescence, the secretory response of endothelial cells is limited in duration and composition when compared with the SASP. This atypical transient senescence-associated secretory response has been termed acute stress-associated phenotype (ASAP).
Beyond the canonical SASP and the acute ASAP response, senescence induced by knocking out CKIα in colorectal cells induces a so-called senescence inflammatory response (SIR) (Pribluda et al. 2013), more like parainflammation than canonical SASP. Dysfunctional mitochondria also trigger senescence with a distinct secretory phenotype termed MIDAS (mitochondrial dysfunction-associated secretome) (Wiley et al. 2015). A systematic approach is needed to understand the commonalities and differences of the various secretomes of senescent cells.
To understand how ASAP is established and restricted, the investigators used an in vitro system. Human umbilical vein endothelial cells (HUVECs) treated with doxorubicin acutely produced IL-6 and a restricted set of secreted factors followed by cell cycle arrest and the up-regulation of conventional markers of senescence. Induction of ASAP in HUVECs was dependent on reactive oxygen species (ROS)-induced activation of p38 but not on NF-κB, a key regulator of the SASP. Two recent studies showed that mTOR controls the translation of IL-1α and MAPKAPK2 (a kinase downstream from p38) to regulate the SASP (Herranz et al. 2015; Laberge et al. 2015). Moreover, PI3K/AKT/mTOR signaling has also been shown to regulate a paracrine response responsible for melanoma resistance to BRAF inhibitors (Obenauf et al. 2015). In light of these findings, Bent et al. (2016) explored the role of the PI3K/AKT/mTOR pathway on ASAP regulation. Surprisingly, they found that the pathway was repressed as cells became senescent (Fig. 1). The investigators hypothesized that this physiological mTOR inactivation underscores a shielding mechanism restraining the duration of IL-6 secretion to avoid the long-term side effects of chronic inflammation. Consistent with this, the mTORC1 inhibitor rapamycin did not abolish ASAP. It would be interesting to test whether p38 inhibition or ROS scavengers would mimic genetic depletion of IL-6 and prevent resistance to chemotherapy.
Figure 1.
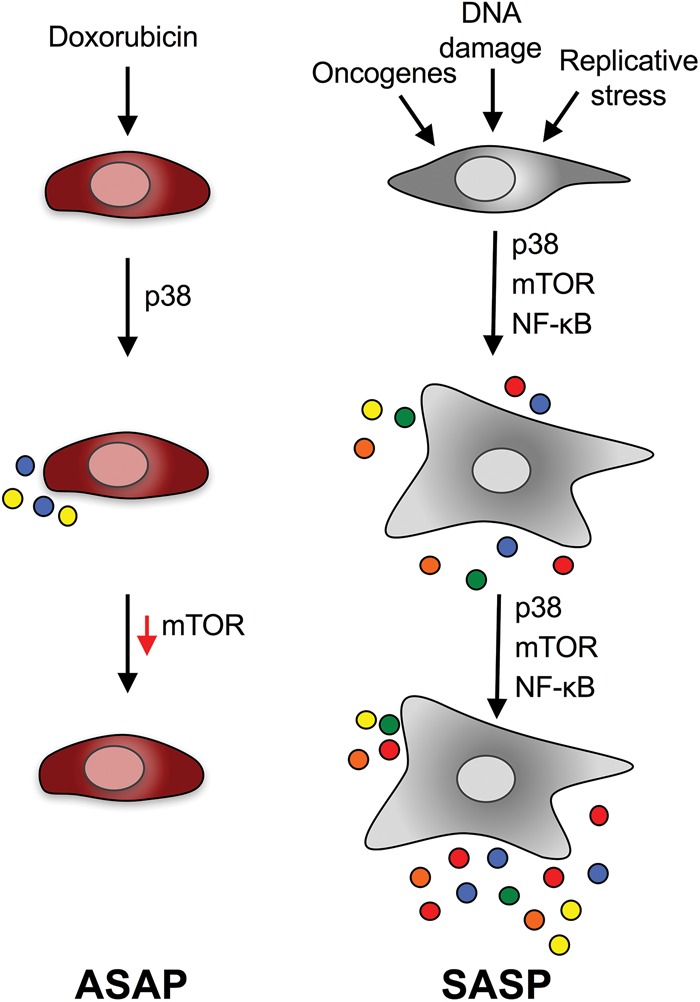
The ASAP is a self-limited senescence secretory phenotype. Bent et al. (2016) described an acute senescence secretory phenotype resulting from chemotherapy and termed it the ASAP. ASAP induction relies on p38 and is innately resolved by mTOR inhibition. The SASP is a self-sustained response that relies also on NF-κB activation. In contrast to the ASAP, the SASP is a chronic response and involves secretion of a wider range of factors. Multicolored dots represent secreted factors.
The benefit of inflammatory responses upon damage is restricted in time, and, consequently, organisms have evolved mechanisms to counteract overt inflammation and maintain tissue homeostasis. The accumulation of senescent cells with age results in persistent inflammation that contributes to a tumor-permissive environment. Very recently, it was shown that sFRP2 secreted from aged senescent fibroblasts drives therapy resistance of melanoma cells and promotes metastasis (Kaur et al. 2016). Whether this is because aged cells have lost their ability to restrain the SASP merits further investigation.
The secretomes of senescent cells can mediate many functions (either beneficial or detrimental). Recent evidence suggests that they can also contribute to acquired chemoresistance. In this regard, the present study shows that doxorubicin-induced endothelial cell senescence in the thymus is the main culprit of IL-6–driven protection of lymphomas against chemotherapy. This response is restrained by the gradual down-regulation of the mTOR pathway. Previously, inhibition of the mTOR pathway has been proposed as a potential strategy to repress the SASP (Herranz et al. 2015; Laberge et al. 2015). The present study suggests that the PI3K/AKT/mTOR pathway might be a physiological switch that controls the duration of paracrine responses to avoid the detrimental effects caused by chronic low-grade inflammation.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.288571.116.
References
- Bent EH, Gilbert LA, Hemann MT. 2016. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev (this issue). 10.1101/gad.284851.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Hemann MT. 2010. DNA damage-mediated induction of a chemoresistant niche. Cell 143: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al. 2015. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17: 1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH III, Dang VM, Appleton J, O'Connell MP, Cheng P, et al. 2016. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532: 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. 2015. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 520: 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, Burstain I, Morgenstern Y, Brachya G, Billauer H, et al. 2013. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24: 242–256. [DOI] [PubMed] [Google Scholar]
- Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, True L, Nelson PS. 2012. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 18: 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, et al. 2015. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23: 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]