

A defining feature of heterochromatin is methylation of Lys9 of histone H3 (H3K9me), a binding site for heterochromatin protein 1 (HP1). Penke et al. generated and analyzed H3K9R mutant flies, separating the functions of H3K9 and nonhistone substrates of H3K9 methyltransferases.
Keywords: H3K9, heterochromatin, genomics, Drosophila, heterochromatin protein 1 (HP1), transposon
Abstract
A defining feature of heterochromatin is methylation of Lys9 of histone H3 (H3K9me), a binding site for heterochromatin protein 1 (HP1). Although H3K9 methyltransferases and HP1 are necessary for proper heterochromatin structure, the specific contribution of H3K9 to heterochromatin function and animal development is unknown. Using our recently developed platform to engineer histone genes in Drosophila, we generated H3K9R mutant flies, separating the functions of H3K9 and nonhistone substrates of H3K9 methyltransferases. Nucleosome occupancy and HP1a binding at pericentromeric heterochromatin are markedly decreased in H3K9R mutants. Despite these changes in chromosome architecture, a small percentage of H3K9R mutants complete development. Consistent with this result, expression of most protein-coding genes, including those within heterochromatin, is similar between H3K9R and controls. In contrast, H3K9R mutants exhibit increased open chromatin and transcription from piRNA clusters and transposons, resulting in transposon mobilization. Hence, transposon silencing is a major developmental function of H3K9.
The eukaryotic genome is organized and compacted within the nucleus through interaction with histones and other proteins to form chromatin. Cytological observation originally divided chromatin into two subgroups: euchromatin and heterochromatin. Euchromatin is gene-rich, transcriptionally active, and usually described as open or accessible. Conversely, heterochromatin is gene-poor and generally considered transcriptionally repressive and inaccessible. There are two major types of heterochromatin, facultative and constitutive, each of which has important cellular functions. The accessibility of facultative heterochromatin is regulated in order to control gene expression (Grewal and Jia 2007). In contrast, constitutive heterochromatin remains condensed throughout the cell cycle and functions in the repression of inappropriate recombination, transposons, and developmentally important genes (Yasuhara and Wakimoto 2006; Grewal and Jia 2007; Peng and Karpen 2008; Eissenberg and Elgin 2014). Despite its generally repressive role, a heterochromatic configuration is also required for expression of certain genes that reside within heterochromatin. Additionally, constitutive heterochromatin is implicated in the structural integrity of centromeres and therefore the promotion of faithful chromosome segregation during cell division (Kellum and Alberts 1995; Bernard et al. 2001). Given these critical functions, heterochromatin is generally considered essential for development.
A large body of evidence from fission yeast to humans has concluded that heterochromatin formation and function are accomplished by post-translational modification (PTM) of histones, particularly dimethylation and trimethylation (me2/me3) of Lys9 of histone H3 (H3K9). H3K9me2/me3 serves as a binding site for heterochromatin protein 1 (HP1) (Bannister et al. 2001; Lachner et al. 2001; Nakayama 2001), which is proposed to mediate chromatin condensation via multimerization of HP1 molecules on nearby nucleosomes (Canzio et al. 2011; Azzaz et al. 2014). HP1 multimers also serve as a scaffold to recruit various other chromatin remodelers associated with condensed chromatin, including H3K9 methyltransferases (Grewal and Jia 2007; Elgin and Reuter 2013). This compacted state is thought to prevent or limit access of proteins to DNA to achieve the repressive functions of heterochromatin. In support of this model, tethering HP1 is sufficient to render chromatin less accessible to nucleases (Danzer and Wallrath 2004). Thus, methylation of H3K9 is thought to serve as the foundation for constitutive heterochromatin formation and a repressive chromatin environment.
Determination of H3K9me's role in heterochromatin formation has primarily relied on mutations of the enzymes that methylate H3K9. Although these studies have greatly enhanced our understanding of heterochromatin biology, they are limited in two ways. First, cells contain multiple H3K9 methyltransferases. For example, at least three different, partially redundant, enzymes methylate H3K9 in Drosophila (Elgin and Reuter 2013). Analyzing phenotypes in single or double mutants is therefore complicated by genetic compensation. Second, enzymes that catalyze histone PTMs often have numerous nonhistone substrates, confounding analysis of the biological contribution of a given PTM (Huang and Berger 2008; Sims and Reinberg 2008; Biggar and Li 2014; Zhang et al. 2015). For example, the fission yeast H3K9 methyltransferase Clr4 modifies the nonhistone substrate Mlo3 to facilitate centromeric siRNA production, a process linked to heterochromatin formation (Gerace et al. 2010; Zhang et al. 2011). Preventing methylation of Mlo3 leads to a reduction in centromeric siRNAs, whereas preventing methylation of H3K9 via a K9R mutation does not. These findings underscore the importance of examining mutations in histone residues that leave other functions of histone-modifying enzymes intact. Until recently, the repetitive nature of histone genes in metazoans has prevented histone gene engineering. Consequently, despite being one of the most well-studied histone modifications, we do not know which aspects of heterochromatin structure and function require H3K9.
Recently, we and others have developed a method for functional replacement of replication-dependent histone genes in Drosophila (Günesdogan et al. 2010; McKay et al. 2015). To address the role of H3K9 in heterochromatin biology, we engineered flies that express only H3K9R mutant replication-dependent histones. In contrast to expectations, we found that H3K9R mutants express a relatively normal protein-coding transcriptome and can complete development, albeit with greatly reduced frequency compared with controls. However, we found that nucleosomes and HP1a are depleted from pericentromeric heterochromatin in H3K9R mutants, with HP1a redistributing along the largely euchromatic chromosome arms. We also found that most transposon families are derepressed, resulting in their mobilization. We propose that unrestricted transposition contributes to the reduced viability of H3K9R mutants.
Results
H3K9 is important, but not essential, for completion of Drosophila development
To test the role of H3K9 in Drosophila development and chromatin architecture, we generated lysine-to-arginine mutations at H3K9 using the histone replacement system that we recently developed (McKay et al. 2015). The replication-dependent histones genes in Drosophila melanogaster reside at a single locus that contains ∼100 copies of a tandemly repeated gene cluster (HisC). Each histone repeat unit contains one copy of a gene encoding histone H1, H2A, H2B, H3, and H4. This arrangement enables functional rescue of a HisC deletion with a single BAC-based transgene containing a synthetic tandem array of 12 histone repeat units (Supplemental Fig. 1). HisC deletion animals containing a histone wild-type or H3K9R transgenic array are referred to here as HWT or K9R, respectively. Importantly, Drosophila has two partially redundant histone H3 variants, H3.3A and H3.3B, encoded by single-copy genes located outside the HisC gene cluster (Henikoff and Ahmad 2005). H3.3 is enriched at active regions of the genome, contains modifications associated with euchromatin (Hake et al. 2006), and, in flies, is depleted from heterochromatin (Ahmad and Henikoff 2002). We therefore used the K9R mutant to investigate heterochromatin biology and focused our analyses on functions of replication-dependent H3K9.
We first characterized the developmental consequences of the K9R mutation. Surprisingly, we found that K9R flies occasionally survive to adulthood (∼2%), demonstrating that replication-dependent H3K9 is not absolutely essential for completion of development (Table 1). However, most K9R mutant progeny are developmentally delayed by 1–2 d and display a broad lethal phase, dying during larval development or pupation. This developmental delay prompted us to examine the proliferative capabilities of K9R cells by generating mitotic clones via the FLP–FRT system (Xu and Rubin 1993). Clones of HisC deletion cells containing an HWT transgene were equivalent in size to control clones containing endogenous histones (Fig. 1A,B). In contrast, K9R clones were approximately two-thirds the size of control clones, suggesting a modest growth defect. Western blots of whole third instar HWT and K9R larvae revealed similar amounts of H3 (Supplemental Fig. 1B) but substantially decreased H3K9me2 and H3K9me3 in K9R mutants (∼20-fold relative to HWT) (Fig. 1C). However, some residual H3K9me2/me3 signal remains in K9R mutants, perhaps representing H3K9 methylation of H3.3A and H3.3B. These results indicate that replication-dependent H3K9 is important but not absolutely necessary for completion of Drosophila development.
Table 1.
H3K9R flies can complete development
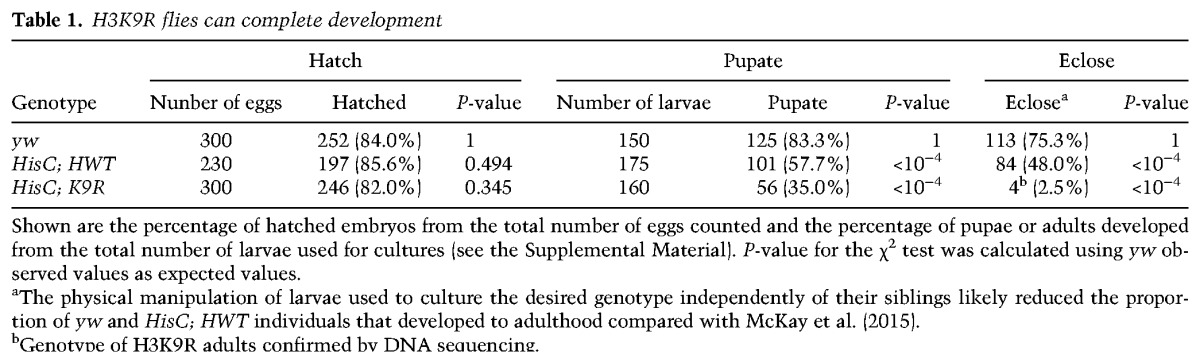
Figure 1.

H3K9R mutant cells proliferate with severely reduced H3K9me2 and me3 in pericentric heterochromatin. (A) Twin spot analysis of FLP–FRT-induced mitotic clones of HisC deletion wing disc cells rescued with an HWT transgene (top) or a K9R transgene (bottom). Wing discs were stained with DAPI to mark nuclei, anti-H3K9me2 to identify K9R cells, and anti-GFP to identify twin spots, (red lines). HisC deletion cells lack GFP, and control sister clones are homozygous HisC+ and express 2× GFP. Bar, 1000 µm. White boxes indicate magnified regions where the bar represents 20 µm. (B) Quantification of twin spot clone area. Each dot represents the area of the experimental (HWT or K9R) clone divided by the area of the control twin spot clone. (**) P < 0.005. (C) Western blot analysis of total cellular histone isolated from whole third instar larvae. (D) Polytene chromosome preparations from third instar larval salivary glands stained with DAPI and anti-H3K9me antibodies. Bar, 20 µm. Magnified images show H3K9me3 staining at the chromocenter (white box) and chromosome arms (yellow box). Bar, 10 µm. Note that H3K9me3 signal at the chromocenter is overexposed to reveal staining on chromosome arms.
H3K9 regulates chromocenter organization and nucleosome occupancy
Given the association of H3K9 methylation with heterochromatin formation, we next examined K9R mutants for defects in chromatin structure by examining salivary gland polytene chromosomes. Polytene chromosomes are generated by endoreduplication, in which cells become polyploid through repeated rounds of S phase without cell division. The pericentromeric heterochromatin of salivary gland cells is underreplicated relative to the euchromatin and coalesces into a structure known as the chromocenter. The polytene chromosome arms in both HWT and K9R displayed typical banded structures (Fig. 1D). In contrast, the chromocenter of K9R nuclei was highly disrupted compared with HWT. Chromosome arms frequently failed to meet in an organized structure, and the DNA appeared less condensed (Fig. 2A). We quantified this phenotype by blindly binning chromocenters into three categories: “organized,” “moderately disorganized,” and “severely disorganized.” Whereas 74% of HWT nuclei appeared organized, only 4% of K9R nuclei did, with 24% and 72% appearing moderately or severely disorganized, respectively (Fig. 2B). This cytological defect corresponds with a loss of H3K9me. Although the chromocenter of HWT polytene chromosomes has strong H3K9me2 and H3K9me3 signal, K9R chromocenters have severely diminished signal (Figs. 1D, 2A). In contrast, along the largely euchromatic arms, H3K9me3 signal is unchanged in K9R mutants (Fig. 1D). Because replication-independent H3.3 variants are typically found at transcriptionally active areas, we surmise that methylation of H3.3A and H3.3B might be the source of H3K9me3 along chromosome arms in K9R mutants.
Figure 2.

H3K9 regulates chromatin organization at pericentromeric heterochromatin. (A) HWT and K9R salivary gland polytene chromosomes stained with DAPI and H3K9me2. Bar, 20 µm. The bottom images show a magnified view (white squares) of the chromocenter (dashed lines). Bar, 10 µm. (B) Quantification of chromocenter organization from yw (contains endogenous histones), HWT, and K9R. (C) K9R/HWT ratio of normalized FAIRE (formaldehyde-assisted isolation of regulatory elements) signal from third instar imaginal wing discs at FAIRE peaks called by MACS2 (see also Supplemental Fig. 2). (CPM) Counts per million. The X-axis indicates the average HWT and K9R signal at each peak. Darker colors in the heat map indicate a higher number of peaks. Red peaks are statistically significant as determined by edgeR. P < 0.01. Lines indicate a twofold change. (D) Genome browser shot of FAIRE peaks near the euchromatic gene engrailed and the heterochromatic gene concertina. (Map) Read mappability. (E) K9R/HWT ratio of normalized FAIRE signal plotted versus genome coordinates of FAIRE peaks on chromosomes 2 and 3. Green regions in the chromosome schematic indicate approximate locations of pericentromeric heterochromatin (Riddle et al. 2011; Hoskins et al. 2015). Blue indicates largely euchromatic regions. Loess regression line of modENCODE K9me3 chromatin immunoprecipitation (ChIP) signal is shown in red. (F) Box plot of average ratio of FAIRE signal for FAIRE peaks assigned to one of nine chromatin states (Kharchenko et al. 2010). See also Supplemental Figure 3, C and D.
Current models posit that the “closed” nature of heterochromatin prevents access of transcription factors to DNA. Our cytological observations of salivary gland chromocenters suggested that pericentric heterochromatin may be more “open” in K9R mutants. We hypothesized that disruption of heterochromatin organization in K9R mutants might allow chromatin remodelers or other factors to access DNA, leading to changes in nucleosome occupancy. We therefore interrogated nucleosome occupancy throughout the genome of K9R mutants using formaldehyde-assisted isolation of regulatory elements (FAIRE) coupled with sequencing (FAIRE-seq). We performed FAIRE-seq on third instar larval imaginal wing discs, a tissue consisting of an epithelial sheet of diploid cells that can be uniformly cross-linked. For both HWT and K9R, characteristic peaks of FAIRE signal occurred near transcription start sites as expected and correlated well with previously published FAIRE data (Supplemental Fig. 2; McKay and Lieb 2013). To compare open chromatin between the two genotypes, we identified peaks of FAIRE signal and quantified the number of reads overlapping each peak, normalizing to the average read depth across the genome. Strikingly, of the 24,025 total FAIRE peaks, 5154 showed a significant increase in FAIRE signal in K9R mutants relative to HWT (P < 0.01) (Fig. 2C). In contrast, 642 peaks exhibited increased signal in HWT relative to K9R, and these peaks were characterized by much smaller differences in signal. Importantly, increases in FAIRE signal in K9R mutants were not due to changes in DNA copy number (Supplemental Fig. 3A).
We next determined the genomic location of peaks with differences in FAIRE signal. In concordance with our results using polytene chromosomes, genome browsing suggested that increases in FAIRE signal occurred in heterochromatic regions. For example, the euchromatic engrailed locus showed similar FAIRE signatures between K9R and HWT, whereas the heterochromatic concertina locus displayed increased signal in K9R mutants (Fig. 2D). Whole-genome analysis demonstrated an increase in FAIRE signal near centromeres in K9R mutants and that FAIRE signal was largely unchanged along chromosome arms (Fig. 2E; Supplemental Fig. S3B). The slight decrease in FAIRE signal along chromosome arms in K9R mutants is likely a by-product of the normalization procedure due to the vast number of reads redistributed to heterochromatic regions in K9R samples (Supplemental Fig. 2F). These results demonstrate that H3K9 is important for establishing or maintaining nucleosome occupancy at pericentromeric heterochromatin.
Previously, Kharchenko et al. (2010) used various combinations of histone PTMs and chromatin proteins to define nine distinct “chromatin states” in Drosophila (Fig. 2F). We therefore determined whether the changes in FAIRE signal that we observed in K9R mutants correlated with a particular chromatin state. We compared the average ratio of signal in K9R samples with HWT controls for each of the nine chromatin states. Regions of H3K9me2/me3, represented by chromatin state 7, on average had higher FAIRE signal in K9R samples (Fig. 2F). In contrast, the FAIRE signal was similar between the two genotypes at all other chromatin states, showing that regions of nucleosome depletion in K9R mutants occur where H3K9me is normally present. Taken together, our cytological and genome-wide data support an essential role for H3K9 in chromatin organization at pericentromeric regions.
H3K9 is required for HP1a localization to pericentromeric heterochromatin
HP1 family proteins are characterized by a chromodomain, which binds to H3K9me2/me3, and a chromo-shadow domain, which multimerizes to mediate internucleosomal interactions and recruit a variety of chromatin-regulating proteins (Eissenberg and Elgin 2014). In fission yeast, H3K9A/R mutations disrupt localization of the HP1 homolog Swi6 (Mellone et al. 2003). Therefore, one possible explanation for the altered FAIRE signatures in H3K9R mutants is the inability to recruit HP1 due to lack of H3K9me2/me3. The best-studied of the five Drosophila HP1 paralogs is HP1a, which primarily localizes to heterochromatin. We first examined HP1a localization via immunofluorescence in K9R salivary gland polytene chromosomes. Consistent with our earlier experiments, HP1a was severely depleted from the chromocenter in K9R mutants but remained localized to telomeres and chromosome arms (Fig. 3A). In fact, HP1a staining along chromosome arms in K9R mutants appeared stronger and was ectopically localized to regions that were undetectably stained in HWT (Fig. 3A, yellow boxes). Because HP1a protein levels were comparable in HWT and K9R salivary glands (Fig. 3B), this result suggests that HP1a relocalizes from pericentromeric heterochromatin to chromosome arms in the absence of H3K9.
Figure 3.

HP1a relocalizes from pericentromeres to chromosome arms in the absence of H3K9. (A) HWT and K9R salivary gland polytene chromosomes stained with anti-H3K9me2 and anti-HP1a antibodies. Bar, 20 µm. The bottom panels show a magnified view of the chromocenter (white box) and a chromosome arm (yellow box) for each genotype. Arrows indicate telomeres. Bar, 5 µm. (B) α-HP1a Western blot of 3, 6, and 12 µg of whole-cell extract from HWT and K9R salivary glands. (C) K9R/HWT ratio of HP1a ChIP-seq (ChIP combined with high-throughput sequencing) signal from whole third instar larvae within 1-kb windows tiled across the five autosome arms. The top 20% of 1-kb windows with the highest counts are shown (see Supplemental Fig. 4 for all windows). Pie charts show the percentage of significantly altered windows on pericentromeres or chromosome arms as called by edgeR. P < 0.01. (D) Scatter plot of HP1a signal at FAIRE peaks with higher signal in K9R samples (top) or a random selection of FAIRE peaks that are not significantly different between HWT and K9R (bottom).
To further explore the effect of H3K9 loss on HP1, we analyzed HP1a association with the genome in K9R mutants by performing ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) for HP1a using nuclei isolated from whole third instar larvae (Supplemental Fig. 4A,B). Given the HP1a immunofluorescence data in polytene chromosomes, we expected to detect relocalization of reads from repetitive heterochromatic to euchromatic regions in K9R mutants. As seen in the FAIRE-seq data, given a fixed sequencing depth, a large increase in reads that map to one region of the genome (e.g., pericentric heterochromatin) results in a corresponding reduction in read coverage throughout the rest of the genome (Fig. 2E). Consequently, normalizing to sequencing depth in this case misrepresents the relative signal between the two genotypes (Orlando et al. 2014). We therefore developed an alternative procedure that uses Drosophila virilis chromatin as an internal normalization control (see the Supplemental Material). The D. melanogaster HP1 antibody recognizes D. virilis HP1 (data not shown), and the D. virilis genome is sufficiently diverged from D. melanogaster to permit unambiguous mapping of high-throughput sequencing reads. We added D. virilis chromatin to the D. melanogaster chromatin preparations prior to HP1a immunoprecipitation and normalized D. melanogaster sequencing reads to uniquely mapping D. virilis reads (Supplemental Fig. 4C). To focus on HP1a-enriched regions, we quantified the total number of reads within 1-kb windows across the genome and selected for analysis those windows with the highest number of reads (top 20%). Similar to results from polytene chromosomes, HP1a was significantly depleted at pericentromeric heterochromatin in K9R mutants but showed higher signal along chromosome arms (Fig. 3C; Supplemental Fig. S4E). For example, 99% of pericentromeric windows with significantly different HP1a signal showed more signal in HWT than in K9R samples. In contrast, 66% of significantly different windows in chromosome arms showed more signal in K9R than in HWT samples. We also carried out HP1a ChIP from wing discs; however, the signal to noise ratios from these experiments were too low to make confident conclusions, presumably due to low-input DNA. Nevertheless, the ChIP-seq read patterns described above were also seen in the wing disc data set (Supplemental Fig. 4E).
To examine how HP1a redistribution to chromosome arms occurs, we asked whether any specific genomic feature correlated with increased HP1a in K9R mutants. Using modENCODE data, we did not find a correlation between relocalized HP1 signal and any particular histone PTM. For example, we did not observe an increase in HP1 signal at regions of H3K27me3 despite the similarity in peptide sequence surrounding the H3K9 and H3K27 residues. Rather, metagene analysis demonstrated that HP1a is enriched across gene bodies along chromosome arms (Supplemental Fig. 4D). In addition to the pericentromeric regions, we observed a significant decrease in HP1a binding to the largely heterochromatic fourth chromosome (Fig. 3C). These data indicate that H3K9 is necessary for recruitment of HP1a to pericentromeres and chromosome 4, and, in the absence of these strong binding sites, HP1a relocalizes to chromosome arms.
Interestingly, both FAIRE and HP1a signals are altered at the pericentromeric heterochromatin of K9R mutants relative to HWT. To determine whether this correlation might have a functional basis, we examined HP1a signal at FAIRE peaks that have greater signal in K9R relative to HWT samples. In K9R mutants, we found a decrease in HP1a signal at those regions of the genome with increased FAIRE signal (Fig. 3D). In contrast, HP1a signal at a random selection of peaks was equivalent between K9R and HWT. This result suggests that HP1a functions to promote nucleosome occupancy or maintain a “closed” chromatin environment. However, loss of HP1a from a subset of pericentric peaks (Fig. 3D) and the fourth chromosome (Supplemental Fig. 3B) resulted in few changes in nucleosome occupancy, indicating that HP1 depletion does not always change nucleosome occupancy.
H3K9 prevents accumulation of transcripts from heterochromatic regions of the genome
Analyses in a variety of organisms have indicated that HP1 and H3K9 methyltransferases are important regulators of transcription of both protein-coding and noncoding RNAs (Grewal and Jia 2007; Elgin and Reuter 2013). We therefore investigated whether the changes in chromatin organization and HP1a localization that we observed in K9R mutant animals impact the transcriptome. To examine the effects of H3K9R on gene regulation, we performed total RNA sequencing (RNA-seq) in wing discs from HWT and K9R third instar larvae and assembled a transcriptome using Cufflinks (Supplemental Fig. 5A,B; Trapnell et al. 2012). Of the transcripts that were differentially expressed between the two genotypes, 999 were higher in K9R discs relative to HWT compared with 175 that were lower (P< 0.05) (Fig. 4A). These data suggest that H3K9 generally functions to repress gene expression.
Figure 4.

Differential expression of transcripts between HWT and K9R correlates with changes in FAIRE signal and HP1a localization. (A) K9R/HWT ratio of RNA-seq signal from third instar imaginal wing discs. Statistically different ratios identified using DESeq2 are indicated in red. P < 0.05. Blue lines indicate a twofold change. (B) Normalized FAIRE signal at all transcripts differentially expressed between HWT and K9R (red dots in A). Signal is expressed as counts per million (CPM). Black dots indicate transcripts annotated in the RefSeq reference transcriptome. Green dots here and in C indicate transcripts identified in Cufflinks transcriptome assembly but not in the reference transcriptome. (C) Scatter plot of normalized HP1a ChIP signal within transcripts differentially expressed between K9R and HWT (left) or a random selection of transcripts that are not significantly different between the two genotypes (right).
The majority of elevated transcripts in K9R mutants was unannotated in the reference transcriptome but identified in our transcriptome assembly (Supplemental Fig. 6F). Surprisingly, significantly fewer than expected protein-coding genes were differentially expressed (Fisher's exact test, P< 10−15). Some protein-coding genes reside in heterochromatin and require a heterochromatic environment to be properly expressed (Lu et al. 2000; Yasuhara and Wakimoto 2006; Corradini et al. 2007). However, we found only two of 46 heterochromatic genes on chromosome 2L (Supplemental Fig. 5C) and a small subset of genes on the fourth chromosome (Supplemental Fig. 6E) that exhibited altered expression in K9R mutants. The majority of these genes, including the two on chromosome 2L whose expression changed, exhibited increased FAIRE signal and decreased HP1a binding in K9R mutants, demonstrating that these particular changes to chromatin are not sufficient to cause gene expression changes in wing discs. In addition to blocking methylation, the H3K9R mutation also prevents H3K9 acetylation, a modification found near transcription start sites and associated with gene activation (Wang et al. 2008; Kharchenko et al. 2010). However, we found that H3K9ac is not a predictor of altered gene expression in K9R mutants (Supplemental Fig. 5D). These data suggest that H3K9, in contrast to HP1, is not essential for expression of most protein-coding genes (Grewal and Jia 2007; Elgin and Reuter 2013).
As with our FAIRE-seq and HP1a ChIP-seq data, transcripts differentially expressed between HWT and K9R were preferentially found in regions of heterochromatin (Supplemental Fig. 6A). To examine this correlation more closely, we compared our FAIRE-seq and RNA-seq data sets to establish the relationship between nucleosome occupancy and changes in gene expression. A scatter plot of HWT versus K9R FAIRE-seq data identified two classes of transcripts: those with equal FAIRE signal in the two genotypes and those with higher FAIRE signal in K9R samples. The majority of the second class consisted of unannotated transcripts (Fig. 4B). These transcripts were enriched in K9R samples (Supplemental Fig. 6B) and were composed of simple or interspersed repeats.
Similarly, in K9R mutants, HP1a was depleted at the majority of differentially expressed transcripts (Fig. 4C). This result is specific, as HP1a signal was essentially equivalent at a random selection of transcripts that do not significantly differ between K9R and HWT (Fig. 4C). Moreover, differentially expressed transcripts were more likely to be near distinct HP1a and FAIRE regions than were a random selection of transcripts (Supplemental Fig. 6C,D). The specificity of these transcripts to heterochromatic regions and their correlation to altered nucleosome occupancy and HP1a localization suggest that H3K9 plays a direct role in their regulation.
H3K9 represses transposon activation and mobilization
The repetitive nature of the unannotated transcripts identified in the preceding analyses prompted us to look more specifically at transposons. In Drosophila, >130 different transposon families have been identified, with most families consisting of multiple insertions throughout the genome (Rahman et al. 2015). This repetition complicates mapping of reads and consequently the analysis of high-throughput sequencing data. We therefore used the piPipes program, which circumvents these difficulties by mapping reads to transposon families instead of unique transposon insertions in the genome (Han et al. 2015). In both FAIRE-seq and RNA-seq data sets, transposons showed higher signal in K9R samples compared with HWT, whereas control euchromatic regions or coding RNAs were largely equivalent between K9R and HWT (Fig. 5A,B). Transcripts from piRNA clusters can be processed into small piRNAs or endo-siRNAs that function with Argonaute proteins to repress transposons (Brennecke et al. 2007; Czech et al. 2008). We therefore examined FAIRE and RNA signatures at piRNA clusters. Similar to transposon analyses, both data sets showed increased signal in K9R samples relative to HWT (Fig. 5A–C). We next examined HP1a localization at transposon families and piRNA clusters using a similar strategy. Whereas transposons and piRNA clusters from HWT samples showed HP1a enrichment, K9R samples lacked detectable signal above input (Fig. 5D,E; Supplemental Fig. S7A). Transposon expression in K9R mutants is therefore correlated with an increase in FAIRE signal and a decrease in HP1a localization at both transposons and piRNA clusters.
Figure 5.

H3K9 represses transposon and piRNA cluster expression. (A) Scatter plot of FAIRE-seq signal ([RPKM] reads per kilobase per million) at individual transposon families (red) or piRNA clusters (blue). H3K4me2/me3-enriched promoter regions (grey) are shown for comparison. (B) Scatter plot of RNA-seq signal at transposons (red) and piRNA clusters (blue). A random selection of protein-coding RNAs was selected for comparison (gray). (C) Genome browser shot of FAIRE and RNA signal at the 42AB piRNA cluster. FAIRE signal is shown for both uniquely and multiple mapping reads. RNA signal contains uniquely mapping reads only. Highlighted areas indicate regions of increased FAIRE and RNA signal in K9R samples. (D,E) Scatter plot of HP1a ChIP-seq signal at transposon families (D) or piRNA clusters (E) comparing input and immunoprecipitated (IP) samples.
Finally, the derepression of transposon expression in K9R mutants suggests that transposons may be activated, resulting in higher frequency of mobilization. To test this hypothesis, we took advantage of a gypsy-TRAP reporter that activates GFP expression upon de novo integration of gypsy transposons into a reporter transgene (Fig. 6A; Li et al. 2013). Cell proliferation subsequent to de novo gypsy integration results in clones of GFP+ cells. GFP expression has been shown previously to coincide with molecularly confirmed gypsy integration events (Li et al. 2013). We therefore counted the number of GFP+ clones in pupae (Fig. 6C) or larvae (Fig. 6D) to assess the relative number of transposition events between K9R and HWT. The fraction of pupae that contained a transposition event in K9R animals was 75% compared with only 15% in HWT (Fig. 6C). Similar results were obtained in larvae (Fig. 6D). Additionally, significantly more K9R animals had multiple transposition events. These results demonstrate that gypsy transposons mobilize at an elevated frequency in K9R animals relative to controls. Herz et al. (2014) also observed activation of a gypsy-lacZ reporter after expression of H3K9M, which likely inhibits H3K9 methyltransferases via sequestration.
Figure 6.

H3K9 represses gypsy transposon mobilization. (A) Schematic of the gypsy transposon mobilization assay. Ovo-binding site-dependent (green bars) gypsy insertion disrupts Gal80, resulting in Gal4-directed expression of yellow fluorescent protein (YFP) (Li et al. 2013). (B) Pupae showing one, two, or three or more YFP-positive clones representing gypsy mobilization events, which are limited to the mesoderm due to the twi-Gal4 driver and thus likely underestimate the total number of mobilization events in each animal. (C,D) Histogram of the average number of pupae (C; n = 200 for each genotype in six independent experiments) or larvae (D; n = 100 for each genotype in three independent experiments) with zero, one, two, and three or more mobilization events. Error bars represent standard deviation. (*) P < 0.05; (**) P < 0.005; (***) P < 0.0005.
We also searched for evidence of transposon insertion and depletion events in our whole-genome sequencing data sets using the program TIDAL, which uses split read analysis to identify junction reads that span both transposon and unique sequences (Rahman et al. 2015). We examined input DNA from both our wing disc FAIRE experiment and our whole larvae HP1a ChIP experiment. In wing discs, TIDAL detected a similar number of insertion and depletion events in K9R and HWT samples (154 vs. 130 insertions and 99 vs. 90 depletions, respectively) (Supplemental Fig. 7D). In whole larvae, however, we detected a twofold increase in insertion and depletion events in K9R samples relative to HWT (200 vs. 94 insertions and 100 vs. 59 depletions, respectively) (Supplemental Fig. 7E; see also the Supplemental Material). Combined, these data support a model in which the absence of H3K9 prevents recruitment of HP1a and the formation of a repressive chromatin environment, leading to the activation and mobilization of transposons.
Discussion
In diverse organisms from fission yeast to humans, recruitment of HP1 by H3K9me2/me3 serves as a paradigm for histone PTM-mediated chromatin regulation. Here, we performed a set of genetic, cytological, and whole-genome sequencing experiments in Drosophila to directly interrogate the role of H3K9 in animal development.
H3K9 and metazoan development
H3K9me function is typically inferred through H3K9 methyltransferase loss-of-function studies, including by mutation or sequestration of the enzymes (Herz et al. 2014). However, such studies are unable to directly test the contribution of H3K9 to heterochromatin biology due to complexities and redundancies among H3K9 methyltransferases as well as their ability to methylate nonhistone substrates. Although the three Drosophila methyltransferases Su(var)3-9, dG9a, and SetDB1/Eggless generally function at different regions of the genome, the enzymes are partially redundant and show complex genetic interactions. Su(var)3-9 and dG9a single mutants are viable and fertile, but double mutants have reduced viability (Schotta et al. 2003; Mis et al. 2006). Interestingly, SetDB1 single mutants display reduced viability and fertility, and a loss of Su(var)3-9 in this background counterintuitively increases viability (Seum et al. 2007; Brower-Toland et al. 2009). To our knowledge, triple mutants have not been studied and would be technically challenging to create. These complexities have hindered our understanding of the true function of H3K9 methylation. Although the K9R mutation that we engineered in histone H3 cannot specifically test the role of K9 methylation, K9R mutant flies nonetheless provide an important new resource for studying metazoan heterochromatin biology.
Given that many previous studies have concluded that H3K9me is essential for heterochromatin establishment and function, preventing this modification should have severe developmental consequences. Indeed, the viability of H3K9R mutants is greatly reduced. However, we were surprised to find that H3K9 is not absolutely essential for the completion of Drosophila development. Notably, C. elegans H3K9 methyltransferase mutants that lack detectable H3K9 methylation are both viable and fertile (Towbin et al. 2012). One possibility is that modification of Drosophila H3.3K9 compensates for the absence of H3K9. The converse may also be true, as H3.3K9R mutants are viable in Drosophila (Sakai et al. 2009). However, our data indicate that H3.3 does not compensate for H3 in pericentric heterochromatin, as HP1 and nucleosomes are depleted from pericentric heterochromatin, and we were unable to detect H3K9me2 or H3K9me3 signal at the chromocenter of polytene chromosomes. These data are in concordance with previous results showing that H3.3 is depleted from heterochromatin in wild-type flies (Ahmad and Henikoff 2002). In other organisms, there may be more redundancy between H3.3 and H3 at pericentric heterochromatin. In mouse embryonic stem cells, H3.3 is present at telomeres as well as pericentric repeats (Goldberg et al. 2010; Lewis et al. 2010) and is necessary for silencing a subset of transposable elements (Elsässer et al. 2015).
H3K9 regulation of chromatin architecture and composition
Recent studies have illuminated the diversity of heterochromatin that exists throughout the genome (Haynes et al. 2007; Riddle et al. 2011; Wang et al. 2014). These different types of heterochromatin may rely on H3K9me to differing extents. Indeed, in Drosophila embryos, H3K9me plays less of a role in heterochromatin formation at the 359-base-pair (bp) repeat than at other heterochromatic regions (Yuan and Farrell 2016). Additionally, HP1 levels at certain promoters (Figueiredo et al. 2012) and recruitment to telomeres (for review, see Raffa et al. 2011) are reported to be independent of H3K9me. Accordingly, we detected HP1a association with telomeres in K9R mutants (Fig. 3A).
Our cytological data from polytene chromosomes and genome-wide analyses of diploid cells both indicate a critical role for H3K9 in regulating chromatin architecture of the pericentromeres and, to a lesser extent, the fourth chromosome. Multimerization of HP1 molecules on neighboring or interstrand nucleosomes has been proposed to mediate higher-order chromatin folding (Canzio et al. 2011; Azzaz et al. 2014). Loss of HP1a may therefore lead to unfolding of condensed structures and result in a more open chromatin environment. Such decondensation might explain the disorganized chromocenter that we observed in polytene chromosome spreads and could allow nucleosome remodelers or transcription factors access to previously inaccessible chromatin, leading to the changes in nucleosome occupancy that we measured by FAIRE. HP1 has also been implicated in regulating replication timing (Quivy et al. 2008; Schwaiger et al. 2010). Pericentromeric regions in polyploid salivary glands are underreplicated, and disruption of this process might also contribute to the disorganized chromocenters that we observed in K9R polytene chromosomes.
In both cytological and ChIP-seq experiments, the loss of HP1a from pericentromeric regions in K9R mutants was accompanied by an increase in HP1a along chromosome arms. Previous studies demonstrated that HP1a normally associates with numerous euchromatic sites on chromosome arms (De Wit et al. 2007) in an RNA-dependent manner (Piacentini et al. 2003). Additionally, other HP1 isoforms, including Drosophila HP1c and mammalian HP1γ, are primarily euchromatic and found at transcriptionally active domains (Kwon and Workman 2011). An intriguing possibility is that H3K9-mediated binding of HP1a to pericentromeric regions may prevent its spread to inappropriate areas such as regions of HP1c enrichment. Along these lines, the Y chromosome's capacity to act as a suppressor of variegation has been attributed to its potential function as a sink for a limited pool of HP1 (Dorer and Henikoff 1994).
Our metagene analysis revealed that the increase of HP1a on chromosome arms occurs along gene bodies (Supplemental Fig. 4D). The presence of HP1 at some of these sites is required for proper expression of genes within these regions (Cryderman et al. 2005) and may be involved in RNA processing or transcriptional elongation (Vakoc et al. 2005; Piacentini et al. 2009). Although we identified a handful of cases in K9R mutants where ectopic HP1a signal correlated with antisense transcription in the middle of a gene, the majority of genes showed no change in expression levels (data not shown).
H3K9 regulation of gene expression and transposons
One of our most striking observations was an increase in transposon-derived RNA in K9R mutants that is associated with an increase in open chromatin and loss of HP1 at almost all transposon families. Since transposons make up 15%–20% of the D. melanogaster genome, loss of H3K9 has a substantial impact on global chromatin organization (Kaminker et al. 2002). HP1 is thought to limit RNA polymerase occupancy by facilitating formation of closed chromatin structures or recruiting additional silencing factors (Danzer and Wallrath 2004; Grewal and Jia 2007). The absence of HP1 and increased DNA accessibility could therefore permit inappropriate transcription. Active transposons can lead to illegitimate recombination, DNA breaks, and the disruption of genes (Slotkin and Martienssen 2007; Levin and Moran 2011). Organisms therefore have mechanisms that repress transposons, including the piRNA pathway, which is predominantly active in germline cells, and the endo-siRNA pathway, which functions primarily in nongonadal somatic cells (Brennecke et al. 2007; Czech et al. 2008; Ghildiyal and Zamore 2008; Kawamura et al. 2008). In both pathways, small RNAs direct Argonaute family proteins to silence targets (Saito and Siomi 2010; Senti and Brennecke 2010; Mani and Juliano 2013). These small RNAs are derived in part from piRNA clusters, which are composed of numerous inactive fragments of transposons that can be hundreds of kilobases long. In K9R mutant wing discs, transposon activation is correlated with an increase in the levels of piRNA clusters. Moreover, HP1a association with both transposons and piRNA clusters is reduced in K9R mutants. Because the endo-siRNA pathway is predominantly active in nongonadal somatic cells, we speculate that the absence of H3K9 disrupts this pathway. Previous studies have demonstrated that mutations in H3K9 methyltransferases and HP1 result in transposon activation (Brower-Toland et al. 2009; Lundberg et al. 2013). Increased piRNA levels could therefore be a response to this activation. Interestingly, the germline requires the H3K9 methyltransferase SetDB1 and the HP1 family protein Rhino for piRNA cluster transcription (Klattenhoff et al. 2009; Rangan et al. 2011; Mohn et al. 2014). Other mechanisms for endo-siRNA production likely exist in nongonadal cells, as Rhino is not expressed outside the germline. Our results suggest that neither methylation of H3K9 nor HP1a is necessary for piRNA expression in somatic cells. Alternatively, H3K9 and HP1a may be required for correct processing of piRNA cluster transcripts into endo-siRNAs. Preventing this processing may lead to a buildup of piRNA cluster transcripts and a decrease in endo-siRNAs available for transposon silencing.
We were surprised to find that protein-coding gene expression was similar between HWT and K9R mutants. In particular, expression of most protein-coding genes residing within heterochromatin on chromosome 2L was unchanged, suggesting that H3K9 does not play a critical role in regulating expression of these genes. However, because these data were derived from wing discs only, H3K9 may regulate heterochromatin protein-coding genes in other tissues. Expression of protein-coding genes associated with H3K9ac was also relatively unchanged by mutating H3K9. One possibility is that the major function of H3K9ac is to prevent H3K9me and subsequent gene repression. Thus, in K9R mutants, the absence of H3K9ac may be inconsequential in the absence of H3K9me. Alternatively, other acetylation marks may compensate for the loss of H3K9ac, including H3.3K9ac.
Our studies demonstrate an important role for H3K9 in the regulation of pericentromeric chromatin architecture and the maintenance of transposon repression. Future analyses of other heterochromatin-associated processes, including mitosis, DNA replication, and DNA damage, in K9R mutants will be of great interest. Given the conservation of H3K9 methyltransferases and HP1 proteins, our observations should be informative to studies of heterochromatin biology in other eukaryotes.
Materials and methods
Culture conditions
All stocks were maintained on standard corn medium. For cross scheme, see Supplemental Figure 1A. Fifty ΔHisC, twi-GAL4/CyO females and 20 ΔHisC, UAS-2xEYFP/CyO; HWT/HWT or K9R/K9R males were placed in a cage at 25°C and allowed to lay eggs on a grape juice agar plate. To measure the completion of embryogenesis, GFP-positive eggs from a 4-h collection were moved to a separate plate and aged 24 h prior to counting hatchings. The number of hatched eggs (observed) and the total number of eggs scored are indicated in Table 1. For all other developmental assays, overnight collections were used. To measure the completion of development from egg hatching to adult eclosion, ∼50 GFP-positive larvae were moved to a corn medium vial 48 h after egg laying to separate ΔHisC, UAS-2xEYFP/ΔHisC, twi-GAL4 mutants from their siblings. For each group of ∼50 larvae, the number that pupated and the number that eclosed as adults were determined and summed, as was the total number of larvae scored. An identical procedure was used to follow development of randomly selected yw (i.e., essentially wild type) progeny. Expected values for χ2 tests were based on the observed value of yw animals. For genomic or molecular analyses of salivary gland, wing disc, or whole-larvae samples, culture vials were cleared of wandering third instar larvae, and, after 4–6 h, newly wandering larvae were selected. HWT and K9R wandering third instar larvae contained similarly sized wing discs at the same developmental stage, as measured by expression of the Wingless morphogen (data not shown). K9R larvae took 1–2 d longer than HWT to reach the wandering third instar stage. In addition, culturing K9R mutants in isolation is necessary for development to the third larval instar stage because the survival of K9R animals is severely reduced when cocultured with heterozygous siblings.
Immunofluorescence
Mitotic recombination experiments and salivary gland polytene chromosome preparations were performed as previously described (Cai et al. 2010; McKay et al. 2015). Salivary glands from HWT and K9R genotypes were squashed and stained on the same slide to control for variations in individual preparations. Classifications in Figure 2B were performed blindly before identifying genotype. Only chromocenters that could be unambiguously identified by the convergence of chromosome arms were scored. K9R mutant nuclei were identified by lack of anti-K9me2 (Abcam, 1220) antibody staining. K9me3 and HP1a were stained with anti-K9me3 (Active Motif, 39161) and anti-HP1a (Developmental Studies Hybridoma Bank, C1A9), respectively. Images shown are single confocal images taken at a constant gain on a Leica TCS SP5 AOBS UV/spectral confocal laser-scanning system mounted on an inverted DM IRE2 microscope. gypsy mobilization assay was performed with adaptations from Li et al. (2013). w, ovo; ΔHisC,UAS-2xEYFP/CyO, Act-GFP females were crossed to either ΔHisC, twi-GAL4/CyO, Act-GFP; HWT/HWT or K9R/K9R males. Embryos were collected on grape juice plates overnight and aged 36–48 h. Fifty GFP-negative larvae were moved to a corn medium vial and allowed to wander or pupate. Gal80 from the w, ovo chromosome represses fluorescence in ΔHisC, twi-GAL4/ΔHisC, UAS-2xEYFP progeny but does not repress the distinct fluorescent pattern from the CyO, Act-GFP chromosome because Act-GFP expression is not Gal4-dependent. The number of larvae or pupae with zero, one, two, or three or more YFP-positive clones was then counted to assay cells in which Gal80 repression of twi-GAL4-driven YFP is disrupted.
Sample preparation for sequencing
FAIRE-seq and RNA-seq samples from third instar imaginal discs were prepared as described previously (McKay and Lieb 2013). HP1a ChIP-seq samples from whole third instar larvae were prepared essentially as described (Soruco et al. 2013), with an equal amount of third instar larval D. virilis chromatin added to each HWT and K9R replicate before immunoprecipitation (see the Supplemental Material for detailed procedure). Libraries were prepared with the TruSeq DNA kit (FAIRE), Total RNA TruSeq-Stranded Ribo-Zero Gold kit (RNA), and ThruPLEX DNA-seq kit (ChIP). Sequencing was performed on an Illumina HiSeq2500.
Sequence data analysis
Expression data were deposited in the Gene Expression Omnibus (GEO) repository under accession number GSE85374. FAIRE-seq, and ChIP-seq samples were aligned to the dm6 reference genome (release 6.04) using Bowtie2 default parameters (Langmead and Salzberg 2012). Analysis of only uniquely mapping reads (MAPQ ≥ 10) provided results similar to those reported here. FAIRE peaks were called using MACS2 with a shift size of 110 bp and a stringency cutoff of 0.01 (Zhang et al. 2008). FAIRE-seq signal at FAIRE peaks was normalized to sequencing depth. Differential signal analysis was performed with edgeR (Robinson et al. 2009). HP1a ChIP-seq signal within 1-kb windows was normalized to the number of uniquely mapping D. virilis reads. For ChIP analysis, windows were used instead of peaks due to broad redistribution of HP1a that precluded peak calling along chromosome arms. RNA-seq reads were aligned to the dm6 reference genome using TopHat and assembled into transcripts with Cufflinks (Trapnell et al. 2012). DESeq2 was used for differential expression analysis for both RNA and HP1a ChIP data sets (Love et al. 2014). Data were visualized using the Integrative Genomics Viewer (Robinson et al. 2011). The following modENCODE third instar larval ChIP-seq data sets were used: K9ac (GSE48510), K27me3 (GSE49490), K9me2 (GSE47260), K9me3 (GSE47258), and HP1a (GSE47243). Peaks were called using the above parameters except broad peak calling was implemented for K9me2, K9me3, and HP1a data sets. Analysis of transposons and piRNA clusters was performed with piPipes using the dm3 reference genome (Han et al. 2015).
Supplementary Material
Acknowledgments
We thank Dr. Corbin Jones for D. virilis and next-generation sequencing advice, Brian Jones from Dr. Stephen Helfand's laboratory for gypsy mobilization assay stocks, and Robin Armstrong for critical reading of the manuscript. This work was supported by 5T32GM007092-39 and F31GM115194 to T.J.R.P., and R01DA036897 to D.J.M., B.D.S., A.G.M., and R.J.D.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.286278.116.
References
- Ahmad K, Henikoff S. 2002. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 9: 1191–1200. [DOI] [PubMed] [Google Scholar]
- Azzaz AM, Vitalini MW, Thomas AS, Price JP, Blacketer MJ, Cryderman DE, Zirbel LN, Woodcock CL, Elcock AH, Wallrath LL, et al. 2014. Human heterochromatin protein 1α promotes nucleosome associations that drive chromatin condensation. J Biol Chem 289: 6850–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124. [DOI] [PubMed] [Google Scholar]
- Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. 2001. Requirement of heterochromatin for cohesion at centromeres. Science 294: 2539–2542. [DOI] [PubMed] [Google Scholar]
- Biggar KK, Li SS-C. 2014. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol 16: 5–17. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. [DOI] [PubMed] [Google Scholar]
- Brower-Toland B, Riddle NC, Jiang H, Huisinga KL, Elgin SCR. 2009. Multiple SET methyltransferases are required to maintain normal heterochromatin domains in the genome of Drosophila melanogaster. Genetics 181: 1303–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Jin Y, Girton J, Johansen J, Johansen KM. 2010. Preparation of Drosophila polytene chromosome squashes for antibody labeling. J Vis Exp 36: 1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, Madhani HD, Narlikar GJ, Al-Sady B. 2011. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol Cell 41: 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini N, Rossi F, Giordano E, Caizzi R, Verní F, Dimitri P. 2007. Drosophila melanogaster as a model for studying protein-encoding genes that are resident in constitutive heterochromatin. Heredity (Edinb) 98: 3–12. [DOI] [PubMed] [Google Scholar]
- Cryderman DE, Grade SK, Li Y, Fanti L, Pimpinelli S, Wallrath LL. 2005. Role of Drosophila HP1 in euchromatic gene expression. Dev Dyn 232: 767–774. [DOI] [PubMed] [Google Scholar]
- Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel JA, Sachidanandam R, et al. 2008. An endogenous small interfering RNA pathway in Drosophila. Nature 453: 798–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer JR, Wallrath LL. 2004. Mechanisms of HP1-mediated gene silencing in Drosophila. Development 131: 3571–3580. [DOI] [PubMed] [Google Scholar]
- De Wit E, Greil F, Van Steensel B. 2007. High-resolution mapping reveals links of HP1 with active and inactive chromatin components. PLoS Genet 3: 0346–0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer DR, Henikoff S. 1994. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 77: 993–1002. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Elgin SCR. 2014. HP1a: a structural chromosomal protein regulating transcription. Trends Genet 30: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin SCR, Reuter G. 2013. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb Perspect Biol 5: a017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsässer SJ, Noh K-M, Diaz N, Allis CD, Banaszynski LA. 2015. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522: 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo MLA, Philip P, Stenberg P, Larsson J. 2012. HP1a recruitment to promoters is independent of H3K9 methylation in Drosophila melanogaster. PLoS Genet 8: e1003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerace EL, Halic M, Moazed D. 2010. The methyltransferase activity of Clr4Suv39h triggers RNAi independently of histone H3K9 methylation. Mol Cell 39: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. 2008. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 320: 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh K, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. 2010. Distinct factors control histone at specific genomic regions. Cell 140: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, Jia S. 2007. Heterochromatin revisited. Nat Rev Genet 8: 35–46. [DOI] [PubMed] [Google Scholar]
- Günesdogan U, Jäckle H, Herzig A. 2010. A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO Rep 11: 772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, Bazett-jones DP, Allis CD, Hunt DF. 2006. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J Biol Chem 281: 559–568. [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Zamore PD, Weng Z. 2015. PiPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome-and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 31: 593–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes KA, Gracheva E, Elgin SCR. 2007. A distinct type of heterochromatin within Drosophila melanogaster chromosome 4. Genetics 175: 1539–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K. 2005. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol 21: 133–153. [DOI] [PubMed] [Google Scholar]
- Herz H-M, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. 2014. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345: 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, Booth BW, Pfeiffer BD, George RA, Svirskas R, et al. 2015. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res 25: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Berger SL. 2008. The emerging field of dynamic lysine methylation of non-histone proteins. Curr Opin Genet Dev 18: 152–8. [DOI] [PubMed] [Google Scholar]
- Kaminker J, Bergman C, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler D, Lewis S, Rubin G, et al. 2002. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol 3: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Saito K, Kin T, Ono Y, Asai K, Sunohara T, Okada TN, Siomi MC, Siomi H. 2008. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature 453: 793–797. [DOI] [PubMed] [Google Scholar]
- Kellum R, Alberts BM. 1995. Heterochromatin protein 1 is required for correct chromosome segregation in Drosophila embryos. J Cell Sci 108: 1419–1431. [DOI] [PubMed] [Google Scholar]
- Kharchenko P, Alekseyenko A, Schwartz YB, Minoda A, Riddle NC, Sabo PJ, Larschan EN, Gorchakov AA, Karpen GH, Park PJ. 2010. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Xi H, Li C, Lee S, Xu J, Khurana JS, Zhang F, Schultz N, Koppetsch BS, Nowosielska A, et al. 2009. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell 138: 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Workman JL. 2011. HP1c casts light on dark matter. Cell Cycle 10: 625–630. [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV. 2011. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12: 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Elsaesser SJ, Noh K, Stadler SC, Allis CD. 2010. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci 107: 14075–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Prazak L, Chatterjee N, Grüninger S, Krug L, Theodorou D, Dubnau J. 2013. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16: 529–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu BY, Emtage PC, Duyf BJ, Hilliker AJ, Eissenberg JC. 2000. Heterochromatin protein 1 is required for the normal expression of two heterochromatin genes in Drosophila. Genetics 155: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg LE, Stenberg P, Larsson J. 2013. HP1a, Su(var)3-9, SETDB1 and POF stimulate or repress gene expression depending on genomic position, gene length and expression pattern in Drosophila melanogaster. Nucleic Acids Res 41: 4481–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SR, Juliano CE. 2013. Untangling the web: the diverse functions of the PIWI/piRNA pathway. Mol Reprod Dev 80: 632–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay DJ, Lieb JD. 2013. A common set of DNA regulatory elements shapes Drosophila appendages. Dev Cell 27: 306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay DJ, Klusza S, Penke TJR, Meers MP, Curry KP, McDaniel SL, Malek PY, Cooper SW, Tatomer DC, Lieb JD, et al. 2015. Interrogating the function of metazoan histones using engineered gene clusters. Dev Cell 32: 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone BG, Ball L, Suka N, Grunstein MR, Partridge JF, Allshire RC. 2003. Centromere silencing and function in fission yeast is governed by the amino terminus of histone H3. Curr Biol 13: 1748–1757. [DOI] [PubMed] [Google Scholar]
- Mis J, Ner SS, Grigliatti TA. 2006. Identification of three histone methyltransferases in Drosophila: dG9a is a suppressor of PEV and is required for gene silencing. Mol Genet Genomics 275: 513–526. [DOI] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, Brennecke J. 2014. The rhino-deadlock-cutoff complex licenses noncanonical transcription of dual-strand piRNA clusters in Drosophila. Cell 157: 1364–1379. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SIS. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113. [DOI] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, Guenther MG. 2014. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9: 1163–1170. [DOI] [PubMed] [Google Scholar]
- Peng JC, Karpen GH. 2008. Epigenetic regulation of heterochromatic DNA stability. Curr Opin Genet Dev 18: 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentini L, Fanti L, Berloco M, Perrini B, Pimpinelli S. 2003. Heterochromatin protein 1 (HP1) is associated with induced gene expression in Drosophila euchromatin. J Cell Biol 161: 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentini L, Fanti L, Negri R, Del Vescovo V, Fatica A, Altieri F, Pimpinelli S. 2009. Heterochromatin protein 1 (HP1a) positively regulates euchromatic gene expression through RNA transcript association and interaction with hnRNPs in Drosophila. PLoS Genet 5: e1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivy J-P, Gérard A, Cook AJL, Roche D, Almouzni G. 2008. The HP1–p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat Struct Mol Biol 15: 972–979. [DOI] [PubMed] [Google Scholar]
- Raffa GD, Ciapponi L, Cenci G, Gatti M. 2011. Terminin: a protein complex that mediates epigenetic maintenance of Drosophila telomeres. Nucleus 2: 383–391. [DOI] [PubMed] [Google Scholar]
- Rahman R, Chirn G-W, Kanodia A, Sytnikova YA, Brembs B, Bergman CM, Lau NC. 2015. Unique transposon landscapes are pervasive across Drosophila melanogaster genomes. Nucleic Acids Res 43: 10655–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P, Malone CD, Navarro C, Newbold SP, Hayes PS, Sachidanandam R, Hannon GJ, Lehmann R. 2011. piRNA production requires heterochromatin formation in Drosophila. Curr Biol 21: 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, et al. 2011. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21: 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2009. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander E, Getz G, Mesirov J. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Siomi MC. 2010. Small RNA-mediated quiescence of transposable elements in animals. Dev Cell 19: 687–697. [DOI] [PubMed] [Google Scholar]
- Sakai A, Schwartz BE, Goldstein S, Ahmad K. 2009. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr Biol 19: 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Reuter G. 2003. SU(VAR)3-9 is a conserved key function in heterochromatic gene silencing. Genetica 117: 149–158. [DOI] [PubMed] [Google Scholar]
- Schwaiger M, Kohler H, Oakeley EJ, Stadler MB, Schübeler D. 2010. Heterochromatin protein 1 (HP1) modulates replication timing of the Drosophila genome. Genome Res 20: 771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senti K-A, Brennecke J. 2010. The piRNA pathway: a fly's perspective on the guardian of the genome. Trends Genet 26: 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seum C, Reo E, Peng H, Rauscher FJ, Spierer P, Bontron S. 2007. Drosophila SETDB1 is required for chromosome 4 silencing. PLoS Genet 3: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, Reinberg D. 2008. Is there a code embedded in proteins that is based on post-translational modifications? Nat Rev Mol Cell Biol 9: 815–820. [DOI] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272–285. [DOI] [PubMed] [Google Scholar]
- Soruco MML, Chery J, Bishop EP, Siggers T, Tolstorukov MY, Leydon AR, Sugden AU, Goebel K, Feng J, Xia P, et al. 2013. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev 27: 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. 2012. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150: 934–947. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. 2005. Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol Cell 19: 381–391. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh T-Y, Peng W, Zhang MQ, et al. 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Nan R, Accardo MC, Sentmanat M, Dimitri P, Elgin SCR. 2014. A distinct type of heterochromatin at the telomeric region of the Drosophila melanogaster Y chromosome. PLoS One 9: e86451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Rubin GM. 1993. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117: 1223–1237. [DOI] [PubMed] [Google Scholar]
- Yasuhara JC, Wakimoto BT. 2006. Oxymoron no more: the expanding world of heterochromatic genes. Trends Genet 22: 330–338. [DOI] [PubMed] [Google Scholar]
- Yuan K, Farrell PHO. 2016. TALE-light imaging reveals maternally emergence of functional heterochromatin in Drosophila embryos. Genes Dev 30: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Open access model-based analysis of ChIP-Seq (MACS). Genome Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Fischer T, Porter RL, Dhakshnamoorthy J, Zofall M, Zhou M, Veenstra T, Grewal SIS. 2011. Clr4/Suv39 and RNA quality control factors cooperate to trigger RNAi and suppress antisense RNA. Science 331: 1624–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang Y, Shi X. 2015. Emerging roles of lysine methylation on non-histone proteins. Cell Mol Life Sci 72: 4257–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.